
The Transcriptional Landscape of BRAF Wild Type Metastatic Melanoma: A Pilot Study

 , , , and
, , , and 
Abstract
:1. Introduction
2. Results
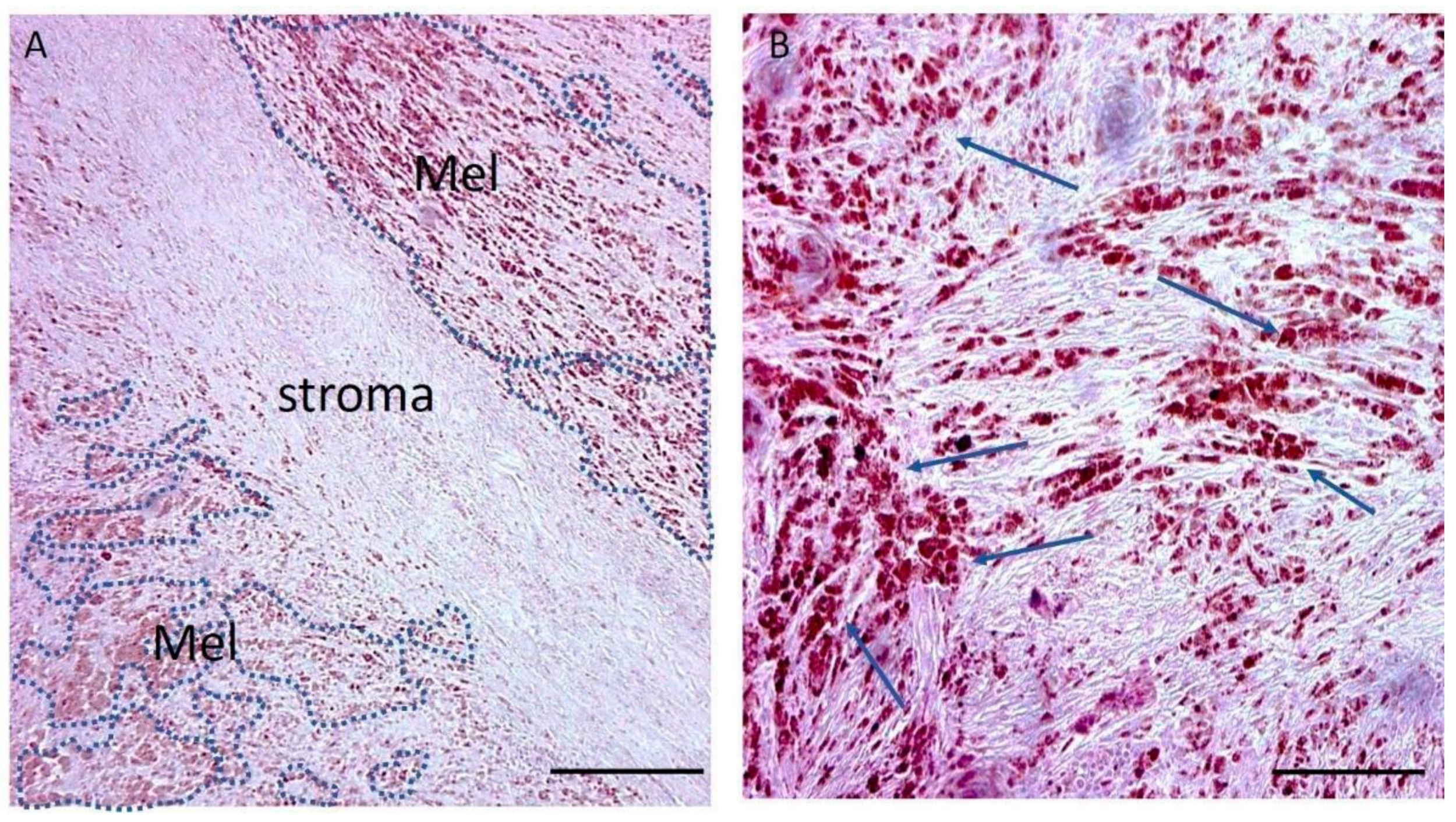
2.1. RNA Extraction and Array Hybridisation
2.2. Differential Expression Analysis
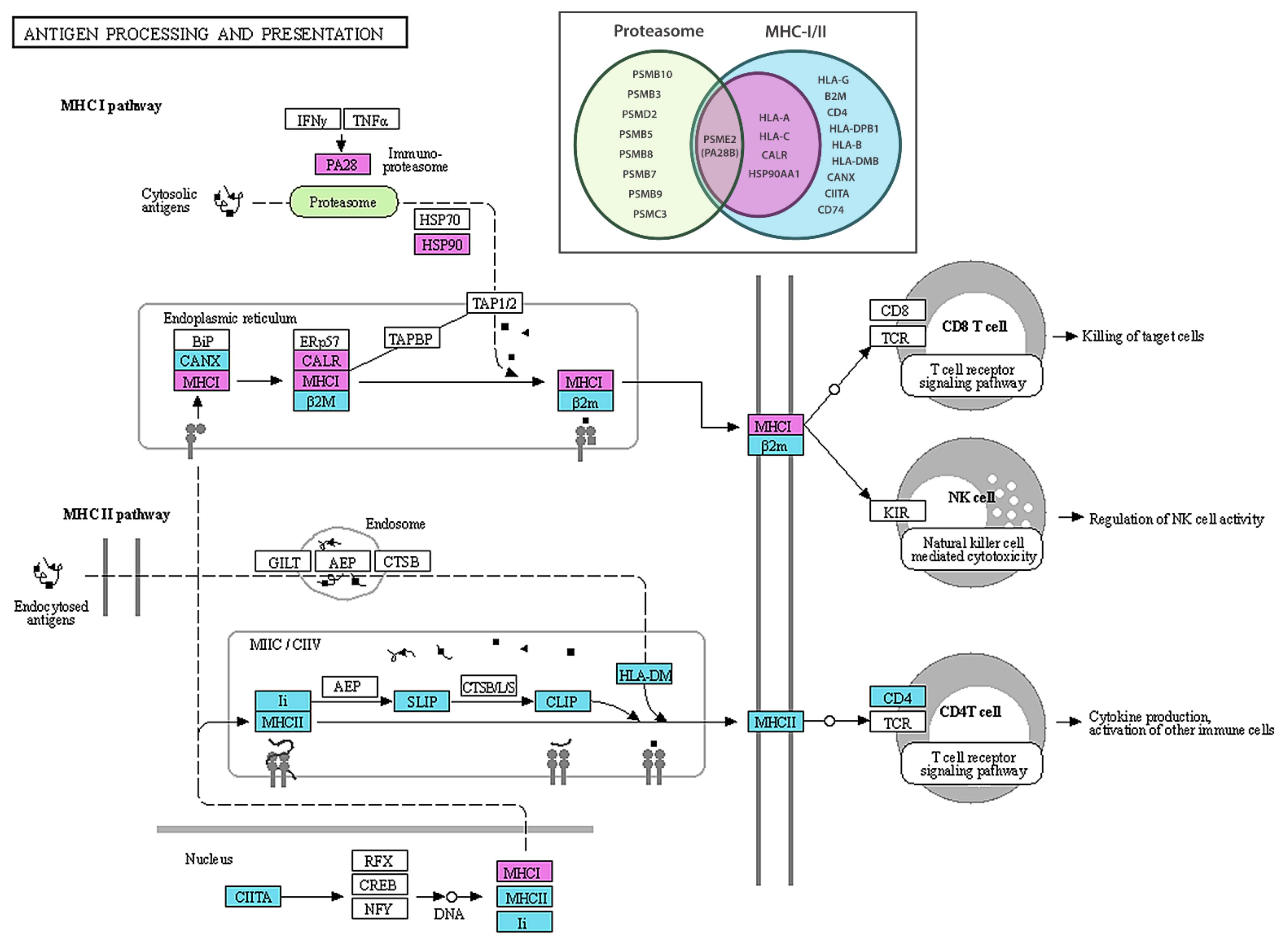
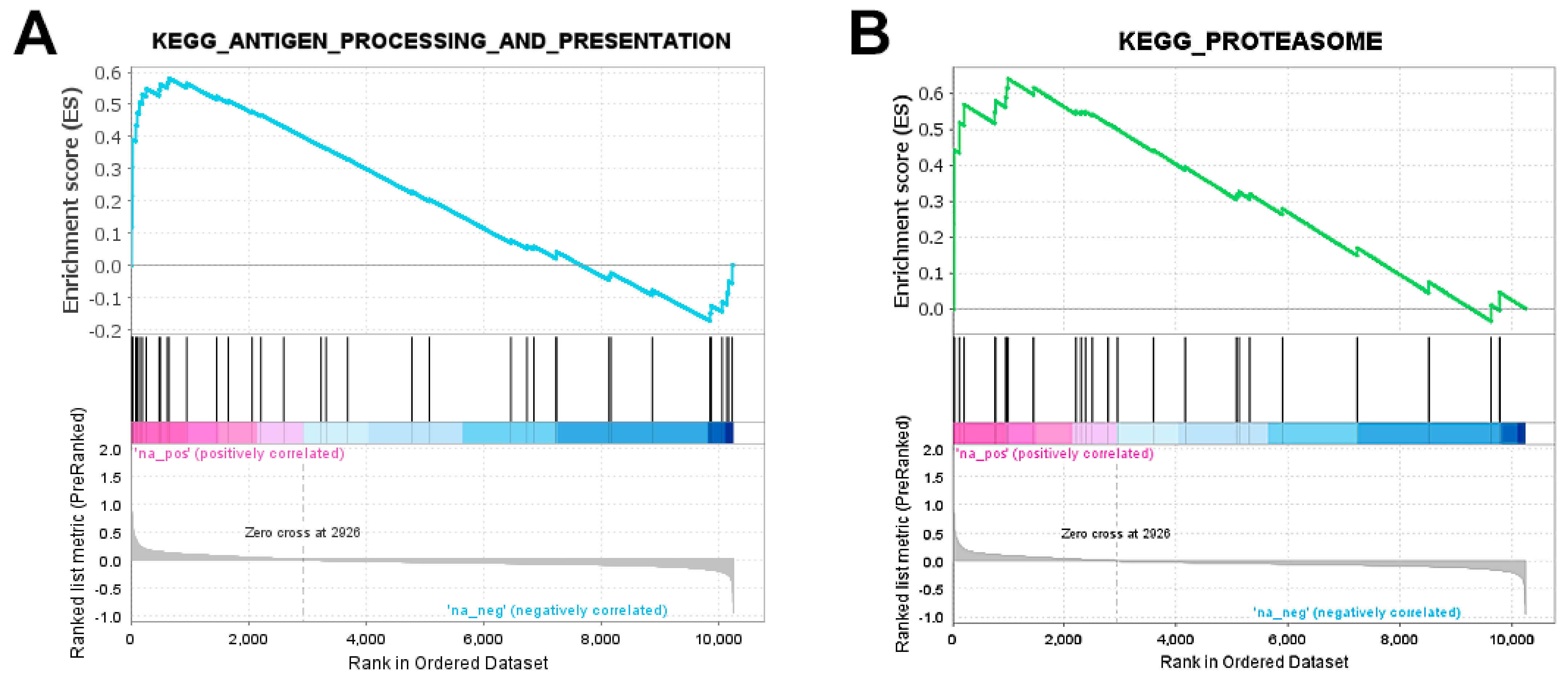
2.3. Genes Related to Antigen Processing and Presentation Are Upregulated in Tumour vs. Control
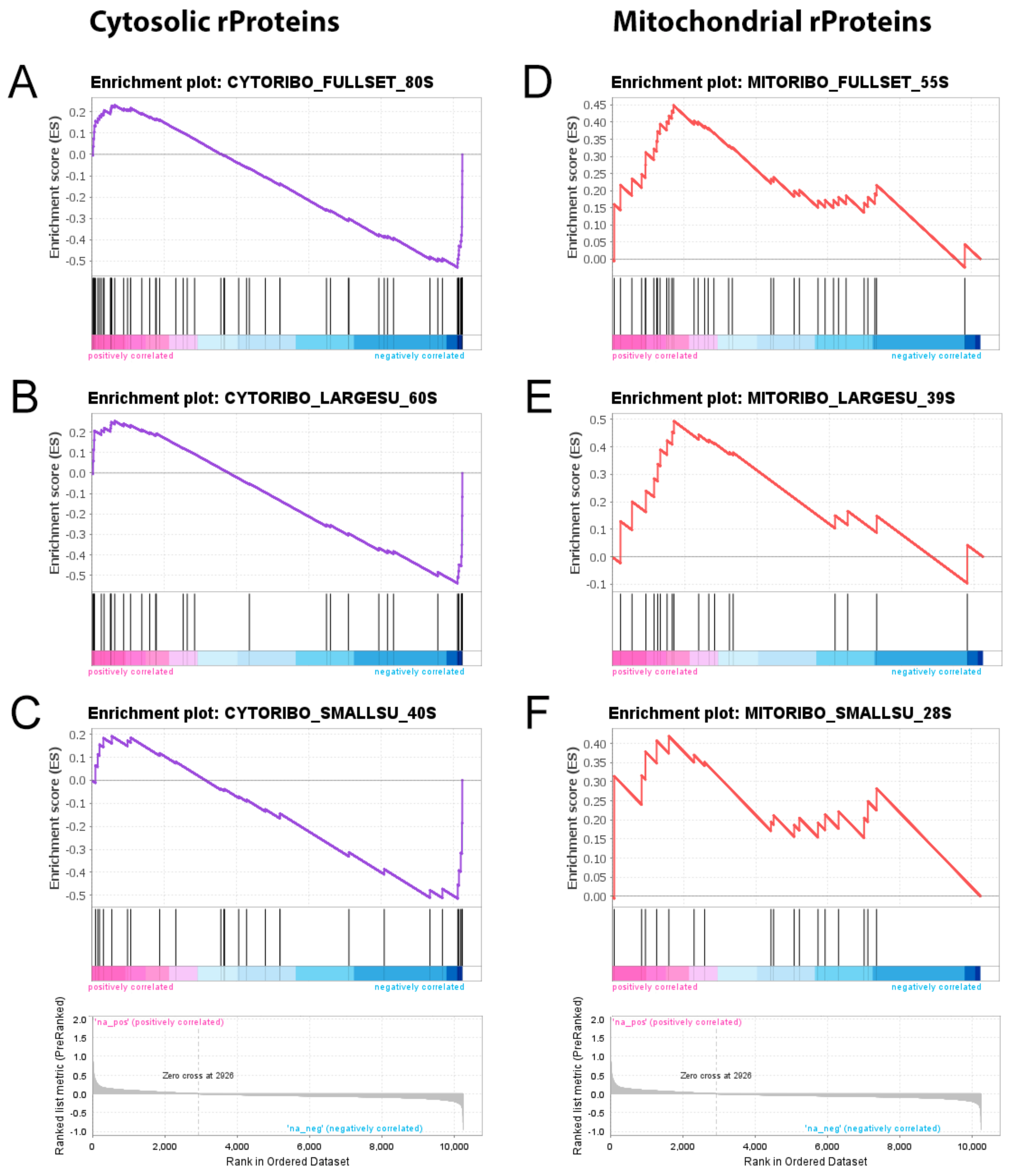
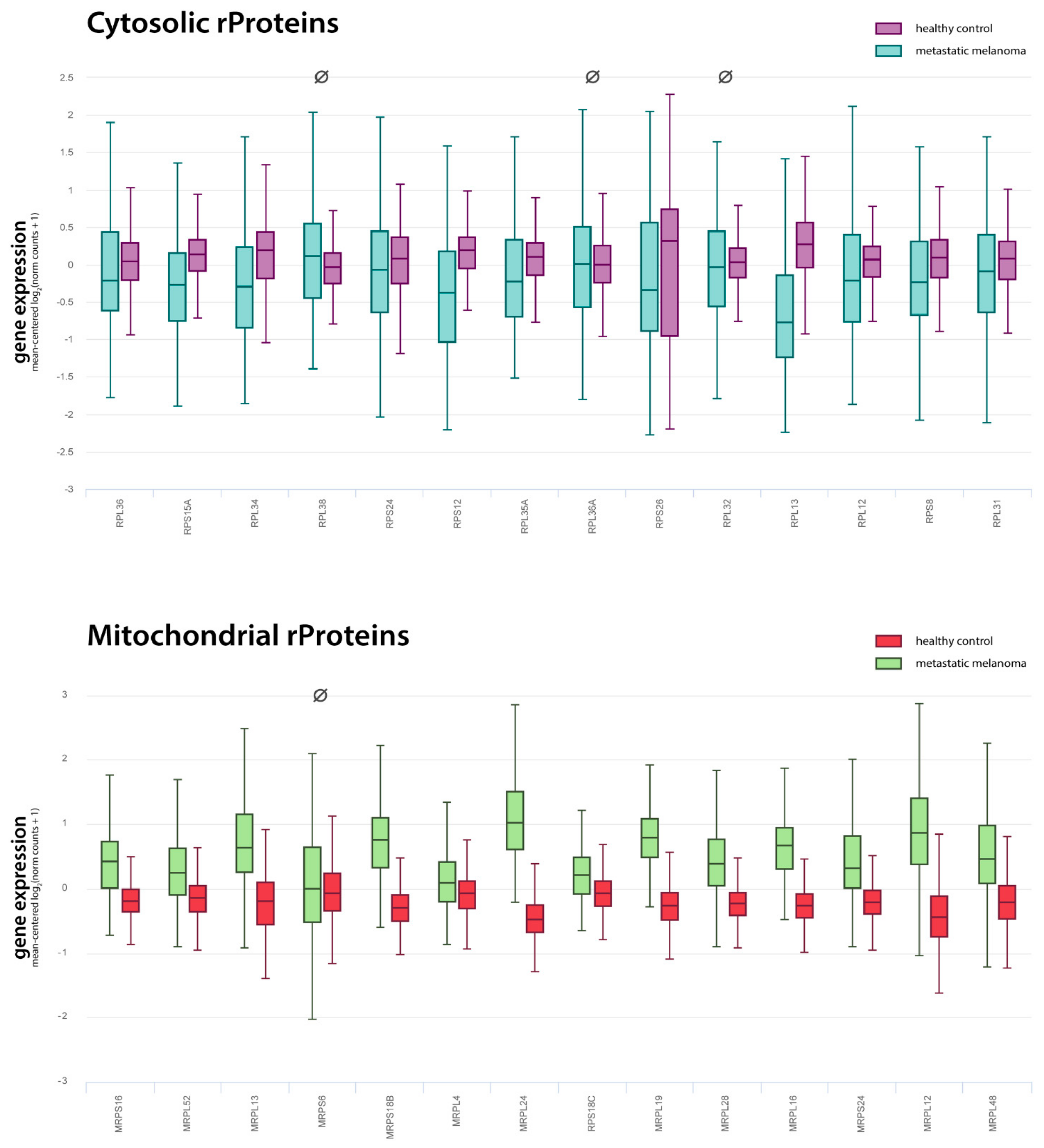
2.4. Cytosolic Ribosome Proteins Are Downregulated in Tumour vs. Control
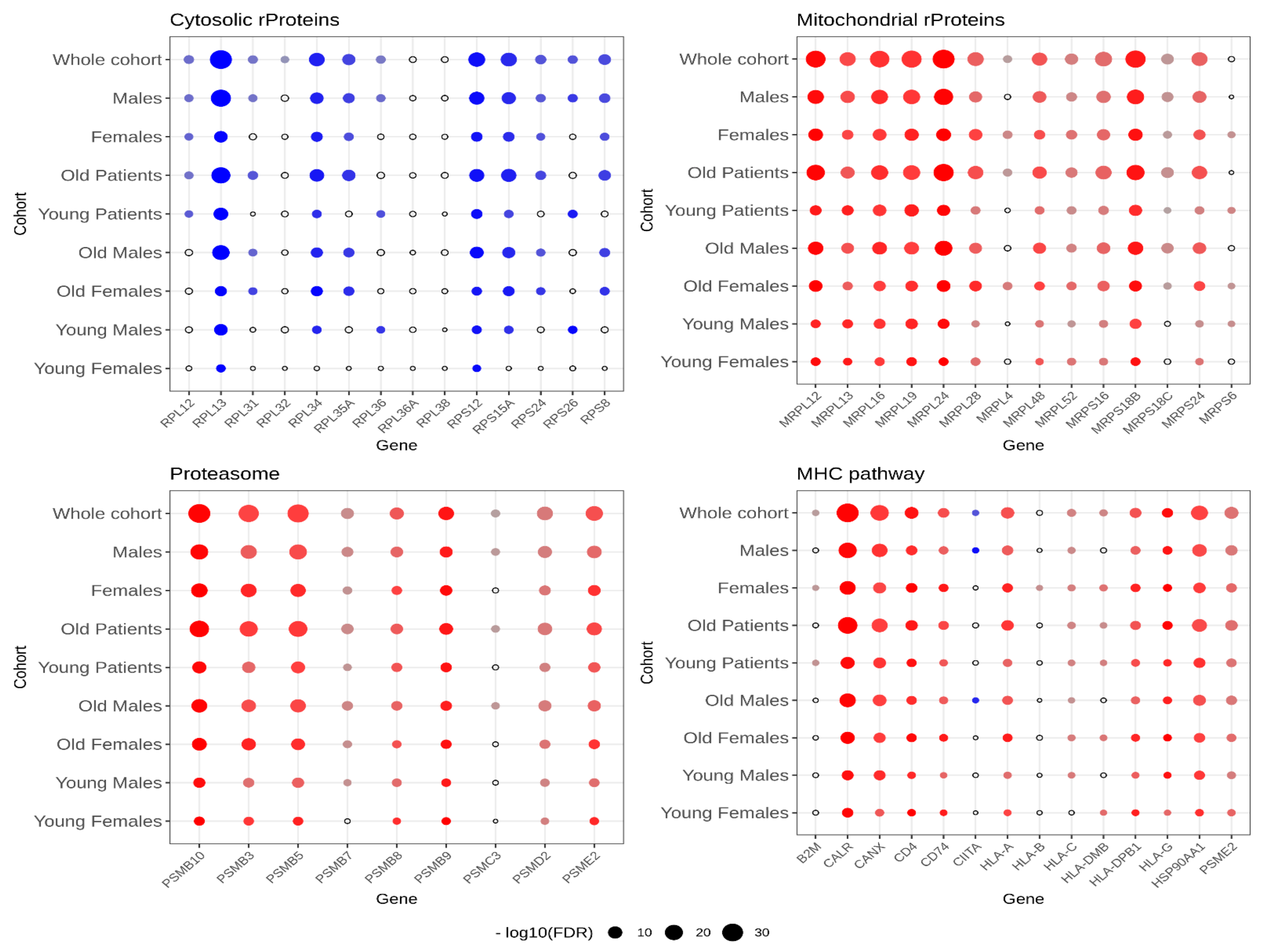
2.5. External Validation through TCGA vs. GTEx Cohorts
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Sample Preparation
4.3. RNA Extraction and Quality Control
4.4. Microarray Hybridisation
4.5. Differential Expression Analysis
4.6. Enrichment Analysis
4.7. TCGA/GTEx Validation
4.8. Real-Time PCR Validation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tseng, H.W.; Shiue, Y.L.; Tsai, K.W.; Huang, W.C.; Tang, P.L.; Lam, H.C. Risk of skin cancer in patients with diabetes mellitus. Medicine 2016, 95, e4070. [Google Scholar] [CrossRef] [PubMed]
- Laurenzana, A.; Biagioni, A.; D’Alessio, S.; Bianchini, F.; Chillà, A.; Margheri, F.; Luciani, C.; Mazzanti, B.; Pimpinelli, N.; Torre, E.; et al. Melanoma cell therapy: Endothelial progenitor cells as shuttle of the MMP12 uPAR-degrading enzyme. Oncotarget 2014, 5, 3711–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcangeli, A.; Lastraioli, E.; Piccini, B.; D’Amico, M.; Lenzi, L.; Pillozzi, S.; Calabrese, M.; Toni, S.; Arcangeli, A. Circulating endothelial progenitor cells in type 1 diabetic patients: Relation with patients’ age and disease duration. Front. Endocrinol. 2017, 8, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keung, E.Z.; Gershenwald, J.E. The eighth edition American Joint Committee on Cancer (AJCC) melanoma staging system: Implications for melanoma treatment and care. Expert Rev. Anticancer Ther. 2018, 18, 775. [Google Scholar] [CrossRef]
- Dickson, P.V.; Gershenwald, J.E. Staging and prognosis of cutaneous melanoma. Surg. Oncol. Clin. 2011, 20, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, L.E.; Shalin, S.C.; Tackett, A.J. Current state of melanoma diagnosis and treatment. Cancer Biol. Ther. 2019, 20, 1366–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, W.H.; From, L.; Bernardino, E.A.; Mihm, M.C. The Histogenesis and Biologic Behavior of Primary Human Malignant Melanomas of the Skin. Cancer Res. 1969, 29, 705–727. [Google Scholar]
- Scolyer, R.A.; Busam, K.J. Prognosis, Staging, and Reporting of Melanomas. In Pathology of Melanocytic Tumors; Elsevier: Amsterdam, The Netherlands, 2019; pp. 386–396. [Google Scholar] [CrossRef]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proietti, I.; Skroza, N.; Michelini, S.; Mambrin, A.; Balduzzi, V.; Bernardini, N.; Marchesiello, A.; Tolino, E.; Volpe, S.; Maddalena, P.; et al. BRAF Inhibitors: Molecular Targeting and Immunomodulatory Actions. Cancers 2020, 12, 1823. [Google Scholar] [CrossRef]
- Chapman, P.B. Mechanisms of resistance to RAF inhibition in melanomas harboring a BRAF mutation. Soc. Clin. Oncol. Educ. Book 2013, 33, e80–e82. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Schachter, J.; Ribas, A.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab for advanced melanoma: Final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 2017, 390, 1853–1862. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Di Giacomo, A.M.; Mortier, L.; Rutkowski, P.; Hassel, J.C.; McNeil, C.M.; Kalinka, E.A.; et al. Five-year outcomes with nivolumab in patients with wild-type BRAF advanced melanoma. J. Clin. Oncol. 2020, 38, 3937–3946. [Google Scholar] [CrossRef]
- Iorio, J.; Lastraioli, E.; Tofani, L.; Petroni, G.; Antonuzzo, L.; Messerini, L.; Perrone, G.; Caputo, D.; Francesconi, M.; Amato, M.M.; et al. hERG1 and HIF-2α Behave as Biomarkers of Positive Response to Bevacizumab in Metastatic Colorectal Cancer Patients. Transl. Oncol. 2020, 13, 100740. [Google Scholar] [CrossRef]
- Lastraioli, E.; Ruffinatti, F.A.; Di Costanzo, F.; Sala, C.; Munaron, L.; Arcangeli, A. A Transcriptomic Approach Reveals Selective Ribosomal Remodelling in the Tumour Versus the Stromal Compartment of Metastatic Colorectal Cancer. Cancers 2021, 13, 4188. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- The Gtex Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Rodig, S.J.; Gusenleitner, D.; Jackson, D.G.; Gjini, E.; Giobbie-Hurder, A.; Jin, C.; Chang, H.; Lovitch, S.B.; Horak, C.; Weber, J.S.; et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci. Transl. Med. 2018, 10, eaar3342. [Google Scholar] [CrossRef] [Green Version]
- Shklovskaya, E.; Lee, J.H.; Lim, S.Y.; Stewart, A.; Pedersen, B.; Ferguson, P.; Saw, R.P.M.; Thompson, J.F.; Shivalingam, B.; Carlino, M.S.; et al. Tumor MHC Expression Guides First-Line Immunotherapy Selection in Melanoma. Cancers 2020, 12, 3374. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Estrada, M.V.; Salgado, R.; Sanchez, V.; Doxie, D.B.; Opalenik, S.R.; Vilgelm, A.E.; Feld, E.; Johnson, A.S.; Greenplate, A.R.; et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat. Commun. 2016, 7, 10582. [Google Scholar] [CrossRef] [Green Version]
- Gadeyne, L.; Van Herck, Y.; Milli, G.; Atak, Z.K.; Bolognesi, M.M.; Wouters, J.; Marcelis, L.; Minia, A.; Pliaka, V.; Roznac, J.; et al. A Multi-Omics Analysis of Metastatic Melanoma Identifies a Germinal Center-Like Tumor Microenvironment in HLA-DR-Positive Tumor Areas. Front. Oncol. 2021, 11, 787. [Google Scholar] [CrossRef]
- Degenhardt, Y.; Huang, J.; Greshock, J.; Horiates, G.; Nathanson, K.; Yang, X.; Herlyn, M.; Weber, B. Distinct MHC Gene Expression Patterns During Progression of Melanoma. Genes. Chromosomes Cancer 2010, 49, 144. [Google Scholar] [CrossRef] [Green Version]
- Wolter, K.G.; Verhaegen, M.; Fernández, Y.; Nikolovska-Coleska, Z.; Riblett, M.; Martin de la Vega, C.; Wang, S.; Soengas, M.S. Therapeutic window for melanoma treatment provided by selective effects of the proteasome on Bcl-2 proteins. Cell Death Differ. 2007, 14, 1605–1616. [Google Scholar] [CrossRef]
- Sidor-Kaczmarek, J.; Cichorek, M.; Spodnik, J.H.; Wójcik, S.; Moryś, J. Proteasome inhibitors against amelanotic melanoma. Cell Biol. Toxicol. 2017, 33, 557. [Google Scholar] [CrossRef] [Green Version]
- Yerlikaya, A.; Erin, N. Differential sensitivity of breast cancer and melanoma cells to proteasome inhibitor Velcade. Int. J. Mol. Med. 2008, 22, 817–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastide, A.; David, A. The ribosome, (slow) beating heart of cancer (stem) cell. Oncogenesis 2018, 7, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artero-Castro, A.; Kondoh, H.; Fernández-Marcos, P.J.; Serrano, M.; y Cajal, S.R.; LLeonart, M.E. Rplp1 bypasses replicative senescence and contributes to transformation. Exp. Cell Res. 2009, 315, 1372–1383. [Google Scholar] [CrossRef] [PubMed]
- Fancello, L.; Kampen, K.R.; Hofman, I.J.F.; Verbeeck, J.; De Keersmaecker, K. The ribosomal protein gene RPL5 is a haploinsufficient tumor suppressor in multiple cancer types. Oncotarget 2017, 8, 14462. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; You, K.R.; Kim, I.H.; Cho, B.H.; Kim, C.Y.; Kim, D.G. Over-expression of the ribosomal protein L36a gene is associated with cellular proliferation in hepatocellular carcinoma. Hepatology 2004, 39, 129–138. [Google Scholar] [CrossRef]
- Li, X.; Cai, H.; Zheng, W.; Tong, M.; Li, H.; Ao, L.; Li, J.; Hong, G.; Li, M.; Guan, Q.; et al. An individualized prognostic signature for gastric cancer patients treated with 5-Fluorouracil-based chemotherapy and distinct multi-omics characteristics of prognostic groups. Oncotarget 2016, 7, 8743–8755. [Google Scholar] [CrossRef] [Green Version]
- Beck-Engeser, G.B.; Monach, P.A.; Mumberg, D.; Yang, F.; Wanderling, S.; Schreiber, K.; Espinosa, R.; Le Beau, M.M.; Meredith, S.C.; Schreiber, H. Point Mutation in Essential Genes with Loss or Mutation of the Second AlleleRelevance to the Retention of Tumor-Specific Antigens. J. Exp. Med. 2001, 194, 285–300. [Google Scholar] [CrossRef] [Green Version]
- Wan, Q.; Jin, L.; Wang, Z. Comprehensive analysis of cancer hallmarks in cutaneous melanoma and identification of a novel unfolded protein response as a prognostic signature. Aging 2020, 12, 20684–20701. [Google Scholar] [CrossRef]
- Parmenter, T.J.; Kleinschmidt, M.; Kinross, K.M.; Bond, S.T.; Li, J.; Kaadige, M.R.; Rao, A.; Sheppard, K.E.; Hugo, W.; Pupo, G.M.; et al. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov. 2014, 4, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Tarrado-Castellarnau, M.; de Atauri, P.; Cascante, M. Oncogenic regulation of tumor metabolic reprogramming. Oncotarget 2016, 7, 62726–62753. [Google Scholar] [CrossRef] [Green Version]
- Haq, R.; Fisher, D.E.; Widlund, H.R. Molecular pathways: BRAF induces bioenergetic adaptation by attenuating oxidative phosphorylation. Clin. Cancer Res. 2014, 20, 2257–2263. [Google Scholar] [CrossRef] [Green Version]
- Jose, C.; Bellance, N.; Rossignol, R. Choosing between glycolysis and oxidative phosphorylation: A tumor’s dilemma? Biochim. Biophys. Acta 2011, 1807, 552–561. [Google Scholar] [CrossRef] [Green Version]
- Barbi de Moura, M.; Vincent, G.; Fayewicz, S.L.; Bateman, N.W.; Hood, B.L.; Sun, M.; Suhan, J.; Duensing, S.; Yin, Y.; Sander, C.; et al. Mitochondrial respiration--an important therapeutic target in melanoma. PLoS ONE 2012, 7, e40690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, G.M.; Vashisht Gopal, Y.N.; McQuade, J.L.; Peng, W.; DeBerardinis, R.J.; Davies, M.A. Metabolic strategies of melanoma cells: Mechanisms, interactions with the tumor microenvironment, and therapeutic implications. Pigment Cell Melanoma Res. 2018, 31, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Park, J.H.; Jung, K.H.; Levine, H.; Kaipparettu, B.A. Elucidating the Metabolic Plasticity of Cancer: Mitochondrial Reprogramming and Hybrid Metabolic States. Cells 2018, 7, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.R.; Moore, J.A.; Bowles, K.M.; Rushworth, S.A.; Moncrieff, M.D. Mitochondrial oxidative phosphorylation in cutaneous melanoma. Br. J. Cancer 2021, 124, 115–123. [Google Scholar] [CrossRef]
- Scott, D.A.; Richardson, A.D.; Filipp, F.V.; Knutzen, C.A.; Chiang, G.G.; Ronai, Z.A.; Osterman, A.L.; Smith, J.W. Comparative Metabolic Flux Profiling of Melanoma Cell Lines: BEYOND THE WARBURG EFFECT*. J. Biol. Chem. 2011, 286, 42626. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.; de Moura, M.B.; Lin, Y.; Vincent, G.; Thorne, S.; Duncan, L.M.; Hui-Min, L.; Kirkwood, J.M.; Becker, D.; Van Houten, B.; et al. Importance of glycolysis and oxidative phosphorylation in advanced melanoma. Mol. Cancer 2012, 11, 76. [Google Scholar] [CrossRef] [Green Version]
- Pavlides, S.; Vera, I.; Gandara, R.; Sneddon, S.; Pestell, R.G.; Mercier, I.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; et al. Warburg meets autophagy: Cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid. Redox Signal. 2012, 16, 1264–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Shang, Q.; Li, W.; Guo, W.; Stojadinovic, A.; Mannion, C.; Man, Y.G.; Chen, T. Antibiotics for cancer treatment: A double-edged sword. J. Cancer 2020, 11, 5135. [Google Scholar] [CrossRef]
- Del Carratore, F.; Jankevics, A.; Eisinga, R.; Heskes, T.; Hong, F.; Breitling, R. RankProd 2.0: A refactored bioconductor package for detecting differentially expressed features in molecular profiling datasets. Bioinformatics 2017, 33, 2774–2775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, F.; Breitling, R.; McEntee, C.W.; Wittner, B.S.; Nemhauser, J.L.; Chory, J. RankProd: A bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics 2006, 22, 2825–2827. [Google Scholar] [CrossRef] [Green Version]
- Heskes, T.; Eisinga, R.; Breitling, R. A fast algorithm for determining bounds and accurate approximate-values of the rank product statistic for replicate experiments. BMC Bioinform. 2014, 15, 367. [Google Scholar] [CrossRef] [Green Version]
- Eisinga, R.; Breitling, R.; Heskes, T. The exact probability distribution of the rank product statistics for replicated experiments. FEBS Lett. 2013, 587, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Breitling, R.; Armengaud, P.; Amtmann, A.; Herzyk, P. Rank products: A simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett. 2004, 573, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, E45. [Google Scholar] [CrossRef]
- Christensen, J.N.; Schmidt, H.; Steiniche, T.; Madsen, M. Identification of robust reference genes for studies of gene expression in FFPE melanoma samples and melanoma cell lines. Melanoma Res. 2020, 30, 26–38. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Number (Percentage) | |
|---|---|---|
| Age (mean, range) | 60.3 (range 46–70) | |
| Gender | Male | 4 (66.7) |
| Female | 2 (33.3) | |
| TNM stage at diagnosis | I | 1 (16.7) |
| II | 2 (33.3) | |
| III | 2 (33.3) | |
| IV | 1 (16.7) | |
| Metastatic site | Skin | 3 (50.0) |
| Lung | 3 (50.0) | |
| Histology | Nodular | 2 (33.3) |
| Superficial spreading | 1 (16.7) | |
| Desmoplastic | 2 (33.3) | |
| Naevoid | 1 (16.7) | |
| Clark’s level | 1 | 0 (0.0) |
| 2 | 0 (0.0) | |
| 3 | 0 (0.0) | |
| 4 | 5 (83.3) | |
| 5 | 0 (0.0) | |
| undefined | 1 (16.7) | |
| Breslow’s depth | I | 0 (0.0) |
| II | 0 (0.0) | |
| III | 5 (83.3) | |
| IV | 1 (16.7) | |
| Ulceration | No | 5 (83.3) |
| Yes | 1 (16.7) | |
| Regression | No | 3 (50.0) |
| Yes | 1 (16.7) | |
| Undefined | 2 (33.3) | |
| Vascular involvement | No | 2 (33.3) |
| Yes | 3 (50.0) | |
| Undefined | 1 (16.7) | |
| Perineural involvement | No | 3 (50.0) |
| Yes | 1 (16.7) | |
| Undefined | 2 (33.3) | |
| NRAS status | Wild type | 1 (16.7) |
| Mutated | 2 (33.3) | |
| Undefined | 3 (50.0) | |
| Best response | CR | 0 (0.0) |
| PR | 3 (50.0) | |
| SD | 1 (16.7) | |
| PD | 1 (16.7) | |
| Undefined | 1 (16.7) |
| Probe ID | Gene Symbol | Description | log2FC | q-Value BH-FDR | p-Value |
|---|---|---|---|---|---|
| A_32_P137939 | ACTB | actin beta | 1.295 | 9.18 × 10−8 | 5.46 × 10−11 |
| A_33_P3223592 | APOE | apolipoprotein E | 1.903 | 2.36 × 10−9 | 3.51 × 10−13 |
| A_33_P3378531 | AS3MT | arsenite methyltransferase | 1.071 | 9.02 × 10−6 | 1.07 × 10−8 |
| A_33_P3296198 | C5orf63 | chromosome 5 open reading frame 63 | 1.375 | 3.40 × 10−6 | 2.53 × 10−9 |
| A_33_P3292854 | CALR | calreticulin | 0.642 | 4.77 × 10−4 | 1.74 × 10−6 |
| A_33_P3280066 | CAVIN1 | caveolae associated protein 1 | 0.918 | 2.92 × 10−5 | 5.63 × 10−8 |
| A_33_P3284508 | CD14 | CD14 molecule | 1.829 | 4.27 × 10−9 | 1.27 × 10−12 |
| A_33_P3229196 | CD151 | CD151 molecule (Raph blood group) | 0.633 | 5.93 × 10−4 | 2.38 × 10−6 |
| A_33_P3252612 | CYP2W1 | cytochrome P450 family 2 subfamily W member 1 | 0.546 | 1.77 × 10−3 | 9.09 × 10−6 |
| A_24_P100673 | EMC4 | ER membrane protein complex subunit 4 | 0.676 | 4.38 × 10−3 | 2.83 × 10−5 |
| A_33_P3333455 | EMILIN1 | elastin microfibril interfacer 1 | 0.535 | 3.99 × 10−4 | 1.31 × 10−6 |
| A_33_P3379436 | FAM74A4 | family with sequence similarity 74 member A4 | 0.847 | 3.36 × 10−5 | 6.99 × 10−8 |
| A_32_P342064 | FTH1 | ferritin heavy chain 1 | 0.505 | 5.50 × 10−3 | 3.80 × 10−5 |
| A_23_P13899 | GAPDH | glyceraldehyde-3-phosphate dehydrogenase | 0.613 | 3.96 × 10−4 | 1.33 × 10−6 |
| A_33_P3585268 | GNAI2 | G protein subunit alpha i2 | 1.332 | 1.01 × 10−7 | 5.25 × 10−11 |
| A_24_P108451 | GPI | glucose-6-phosphate isomerase | 0.834 | 5.52 × 10−6 | 4.51 × 10−9 |
| A_33_P3354322 | GPX1 | glutathione peroxidase 1 | 0.841 | 4.53 × 10−5 | 1.04 × 10−7 |
| A_33_P3287218 | GSTK1 | glutathione S-transferase kappa 1 | 0.588 | 2.01 × 10−4 | 5.36 × 10−7 |
| A_33_P3379962 | HLA-A | major histocompatibility complex, class I, A | 0.863 | 1.30 × 10−3 | 6.17 × 10−6 |
| A_33_P3424803 | HLA-C | major histocompatibility complex, class I, C | 0.764 | 1.06 × 10−3 | 4.73 × 10−6 |
| A_23_P162874 | HSP90AA1 | heat shock protein 90 alpha family class A member 1 | 0.616 | 3.78 × 10−4 | 1.21 × 10−6 |
| A_23_P72737 | IFITM1 | interferon induced transmembrane protein 1 | 0.807 | 5.98 × 10−6 | 5.77 × 10−9 |
| A_24_P605563 | IGLC1 | immunoglobulin lambda constant 1 | 0.517 | 5.77 × 10−4 | 2.23 × 10−6 |
| A_23_P167168 | JCHAIN | joining chain of multimeric IgA and IgM | 0.529 | 2.26 × 10−3 | 1.22 × 10−5 |
| A_32_P452655 | LGALS9C | galectin 9C | 0.605 | 1.38 × 10−4 | 3.58 × 10−7 |
| A_23_P91619 | MIF | macrophage migration inhibitory factor | 0.957 | 9.65 × 10−6 | 1.22 × 10−8 |
| A_23_P1904 | MS4A2 | membrane spanning 4-domains A2 | 0.630 | 2.62 × 10−5 | 4.47 × 10−8 |
| A_23_P106844 | MT2A | metallothionein 2A | 0.552 | 4.39 × 10−4 | 1.53 × 10−6 |
| A_33_P3239879 | NAA38 | N-alpha-acetyltransferase 38, NatC auxiliary subunit | 0.782 | 4.66 × 10−5 | 1.04 × 10−7 |
| A_23_P33022 | POLR2L | RNA polymerase II, I and III subunit L | 0.837 | 1.24 × 10−5 | 1.75 × 10−8 |
| A_33_P3377199 | PRDX1 | peroxiredoxin 1 | 0.723 | 3.41 × 10−4 | 1.07 × 10−6 |
| A_33_P3234899 | PSMB3 | proteasome 20S subunit beta 3 | 0.794 | 9.37 × 10−5 | 2.30 × 10−7 |
| A_23_P65427 | PSME2 | proteasome activator subunit 2 | 0.701 | 1.03 × 10−5 | 1.38 × 10−8 |
| A_23_P434301 | PTMA | prothymosin alpha | 0.582 | 4.56 × 10−4 | 1.63 × 10−6 |
| A_33_P3382595 | RN7SK | RNA component of 7SK nuclear ribonucleoprotein | 0.680 | 2.80 × 10−5 | 4.37 × 10−8 |
| A_23_P69431 | RPL4 | ribosomal protein L4 | 0.509 | 9.25 × 10−4 | 4.06 × 10−6 |
| A_23_P106708 | RPS2 | ribosomal protein S2 | 0.664 | 4.08 × 10−3 | 2.46 × 10−5 |
| A_23_P372874 | S100A13 | S100 calcium binding protein A13 | 1.043 | 2.04 × 10−5 | 3.04 × 10−8 |
| A_24_P261169 | SEMA4D | semaphorin 4D | 0.574 | 3.85 × 10−5 | 8.30 × 10−8 |
| A_33_P3413989 | SERPING1 | serpin family G member 1 | 1.286 | 6.05 × 10−7 | 4.05 × 10−10 |
| A_23_P95213 | SFTPC | surfactant protein C | 0.854 | 5.55 × 10−3 | 3.87 × 10−5 |
| A_33_P3481987 | SLC16A12 | solute carrier family 16 member 12 | 0.850 | 3.45 × 10−05 | 6.93 × 10−8 |
| A_33_P3388491 | SLC66A1 | solute carrier family 66 member 1 | 1.440 | 6.62 × 10−6 | 7.38 × 10−9 |
| A_33_P3587376 | SNAR-A3 | small NF90 (ILF3) associated RNA A3 | 1.423 | 8.95 × 10−5 | 2.13 × 10−7 |
| A_33_P3370461 | SUZ12P1 | SUZ12 pseudogene 1 | 0.916 | 3.03 × 10−5 | 5.63 × 10−8 |
| A_33_P3332690 | SUZ12P1 | SUZ12 pseudogene 1 | 0.597 | 2.65 × 10−4 | 7.69 × 10−7 |
| A_33_P3274199 | TP53I13 | tumor protein p53 inducible protein 13 | 0.579 | 4.75 × 10−4 | 1.76 × 10−6 |
| A_23_P325654 | TRIM42 | tripartite motif containing 42 | 0.925 | 4.70 × 10−2 | 9.40 × 10−4 |
| A_33_P3409062 | TYROBP | transmembrane immune signaling adaptor TYROBP | 1.452 | 6.77 × 10−8 | 2.52 × 10−11 |
| A_24_P101391 | YBX1 | Y-box binding protein 1 | 0.714 | 2.99 × 10−4 | 9.10 × 10−7 |
| Probe ID | Gene Symbol | Description | log2FC | q-Value BH-FDR | p-Value |
|---|---|---|---|---|---|
| A_23_P114445 | MAGEE1 | MAGE family member E1 | −0.508 | 5.47 × 10−5 | 1.30 × 10−7 |
| A_23_P112774 | PTP4A3 | protein tyrosine phosphatase 4A3 | −0.513 | 2.71 × 10−5 | 4.23 × 10−8 |
| A_33_P3332348 | RN7SL1 | RNA component of signal recognition particle 7SL1 | −0.803 | 2.86 × 10−6 | 4.25 × 10−10 |
| A_33_P3244165 | RNA28SN5 | RNA, 28S ribosomal N5 | −1.378 | 6.92 × 10−6 | 2.06 × 10−9 |
| A_33_P3346552 | RNA28SN5 | RNA, 28S ribosomal N5 | −1.012 | 2.05 × 10−5 | 2.89 × 10−8 |
| A_33_P3279708 | RNU2−2P | RNA, U2 small nuclear 2, pseudogene | −0.954 | 7.28 × 10−7 | 5.41 × 10−11 |
| A_23_P217068 | RPL12 | ribosomal protein L12 | −0.753 | 5.96 × 10−4 | 3.32 × 10−6 |
| A_24_P142228 | RPL13 | ribosomal protein L13 | −0.578 | 2.01 × 10−6 | 4.48 × 10−10 |
| A_32_P184518 | RPL21 | ribosomal protein L21 | −0.807 | 6.08 × 10−6 | 3.62 × 10−9 |
| A_32_P118258 | RPL21 | ribosomal protein L21 | −0.861 | 2.75 × 10−5 | 4.09 × 10−8 |
| A_24_P213783 | RPL31 | ribosomal protein L31 | −0.944 | 6.65 × 10−6 | 4.45 × 10−9 |
| A_23_P18142 | RPL32 | ribosomal protein L32 | −0.568 | 1.53 × 10−3 | 1.20 × 10−5 |
| A_33_P3329916 | RPL6 | ribosomal protein L6 | −0.803 | 7.19 × 10−6 | 3.21 × 10−9 |
| A_32_P857658 | RPLP1 | ribosomal protein lateral stalk subunit P1 | −0.735 | 3.44 × 10−4 | 1.48 × 10−6 |
| A_23_P147888 | RPLP2 | ribosomal protein lateral stalk subunit P2 | −0.512 | 9.10 × 10−4 | 6.02 × 10−6 |
| A_24_P418418 | RPS17 | ribosomal protein S17 | −0.931 | 1.74 × 10−5 | 2.07 × 10−8 |
| A_23_P116694 | RPS26 | ribosomal protein S26 | −0.539 | 1.76 × 10−5 | 1.96 × 10−8 |
| A_33_P3221680 | RPS28 | ribosomal protein S28 | −0.750 | 1.48 × 10−5 | 1.43 × 10−8 |
| A_23_P46182 | RPS8 | ribosomal protein S8 | −0.757 | 9.37 × 10−5 | 2.86 × 10−7 |
| Pathway Name | GSEA Leading Edge | Xena Opposite FC | Xena Not Significant | Xena Concordant | Validation Score |
|---|---|---|---|---|---|
| MHC pathway | 14 | 1 | 0 | 13 | 92.9% |
| proteasome | 9 | 0 | 0 | 9 | 100% |
| cytosolic rProteins | 14 | 1 | 2 | 11 | 78.6% |
| mitochondrial rProteins | 14 | 0 | 1 | 13 | 92.9% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lastraioli, E.; Ruffinatti, F.A.; Bagni, G.; Visentin, L.; di Costanzo, F.; Munaron, L.; Arcangeli, A. The Transcriptional Landscape of BRAF Wild Type Metastatic Melanoma: A Pilot Study. Int. J. Mol. Sci. 2022, 23, 6898. https://doi.org/10.3390/ijms23136898
Lastraioli E, Ruffinatti FA, Bagni G, Visentin L, di Costanzo F, Munaron L, Arcangeli A. The Transcriptional Landscape of BRAF Wild Type Metastatic Melanoma: A Pilot Study. International Journal of Molecular Sciences. 2022; 23(13):6898. https://doi.org/10.3390/ijms23136898
Chicago/Turabian StyleLastraioli, Elena, Federico Alessandro Ruffinatti, Giacomo Bagni, Luca Visentin, Francesco di Costanzo, Luca Munaron, and Annarosa Arcangeli. 2022. "The Transcriptional Landscape of BRAF Wild Type Metastatic Melanoma: A Pilot Study" International Journal of Molecular Sciences 23, no. 13: 6898. https://doi.org/10.3390/ijms23136898