
Cis-Element Engineering Promotes the Expression of Bacillus subtilis Type I L-Asparaginase and Its Application in Food


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
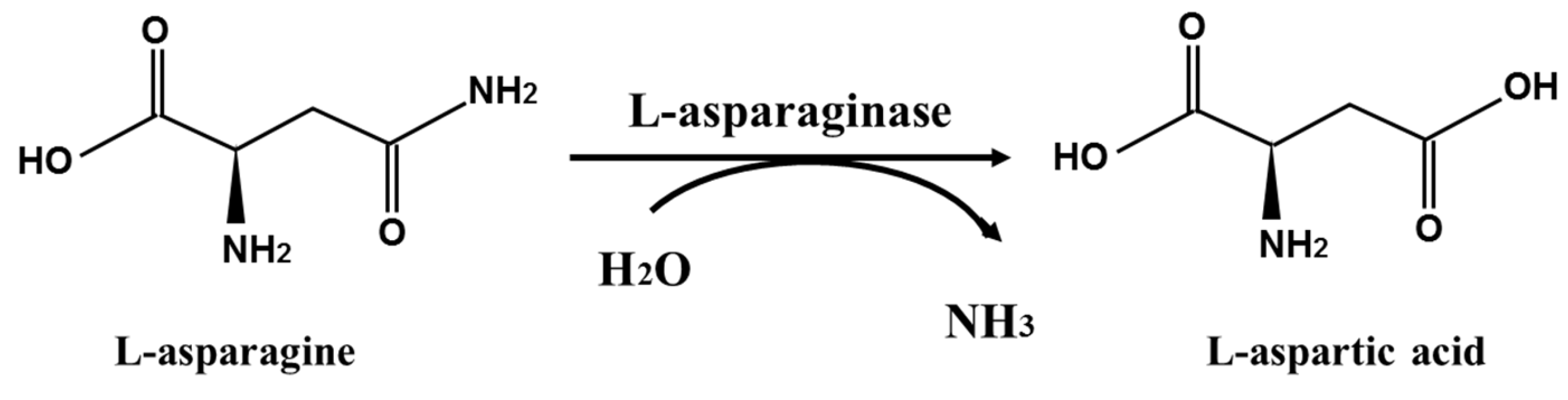
:1. Introduction
2. Results and Discussion
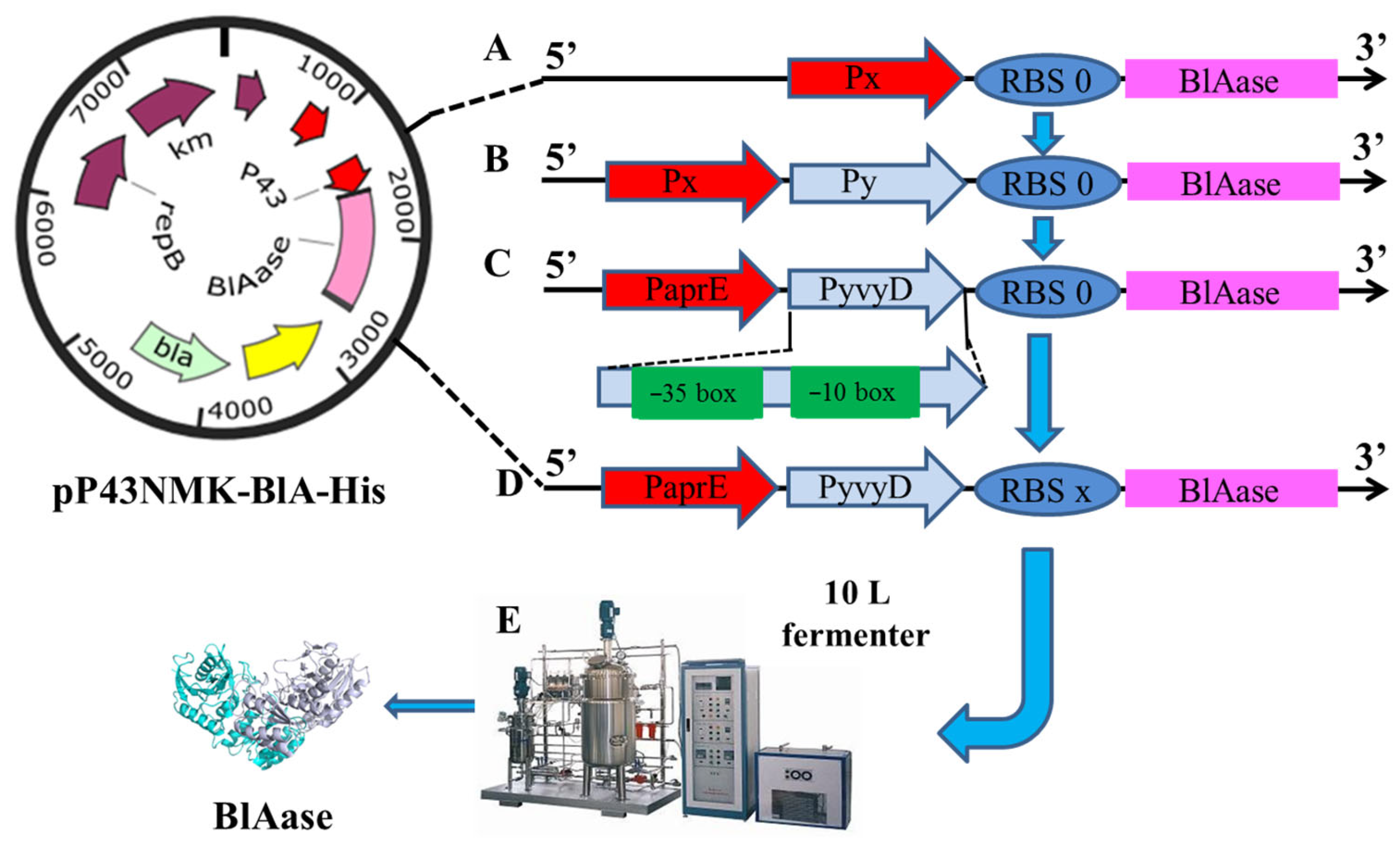
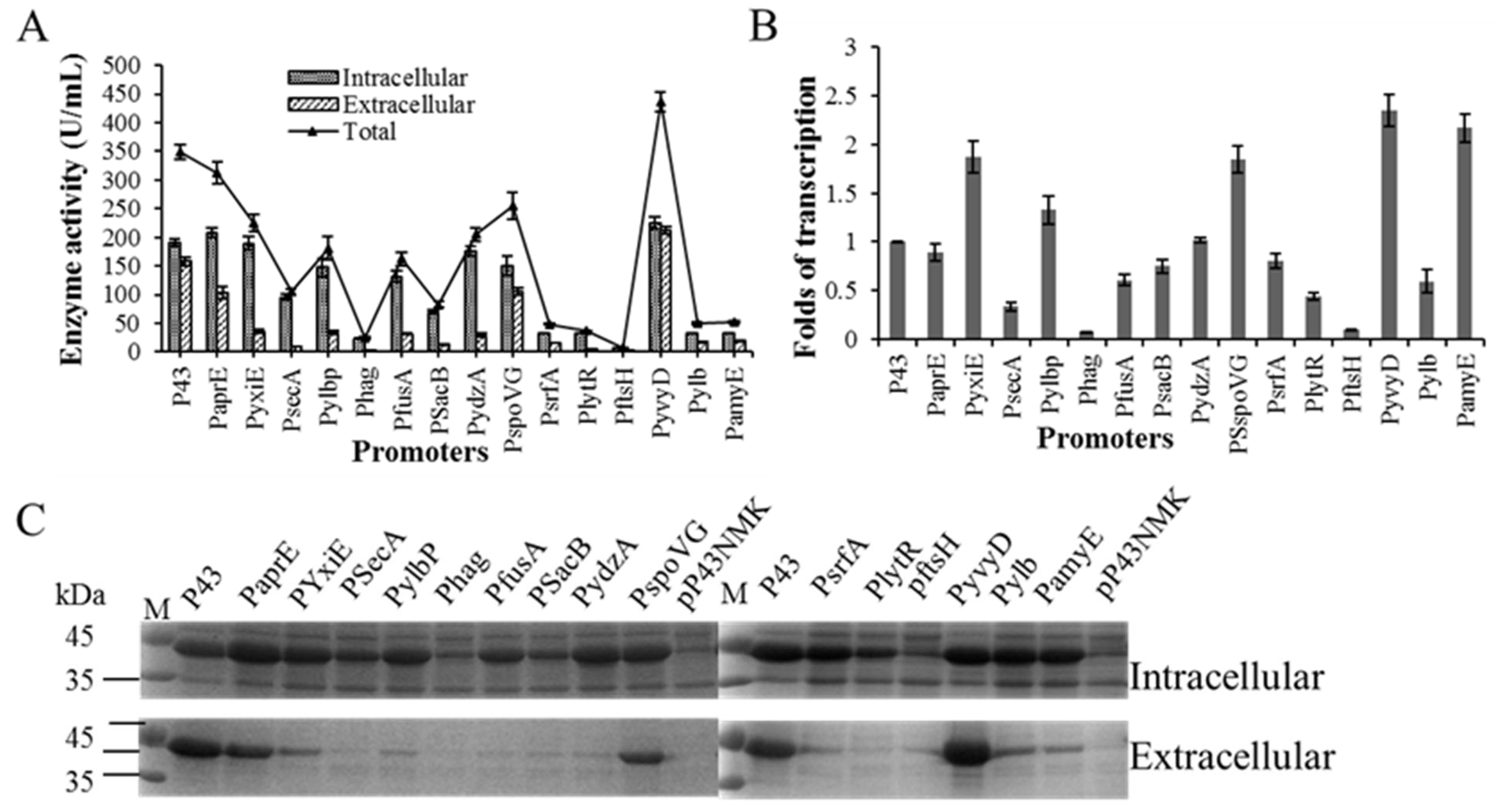
2.1. Enhance BlAase Production with a Single-Initiator Subsystem
2.2. Enhance BlAase Production with a Dual-Promoter System
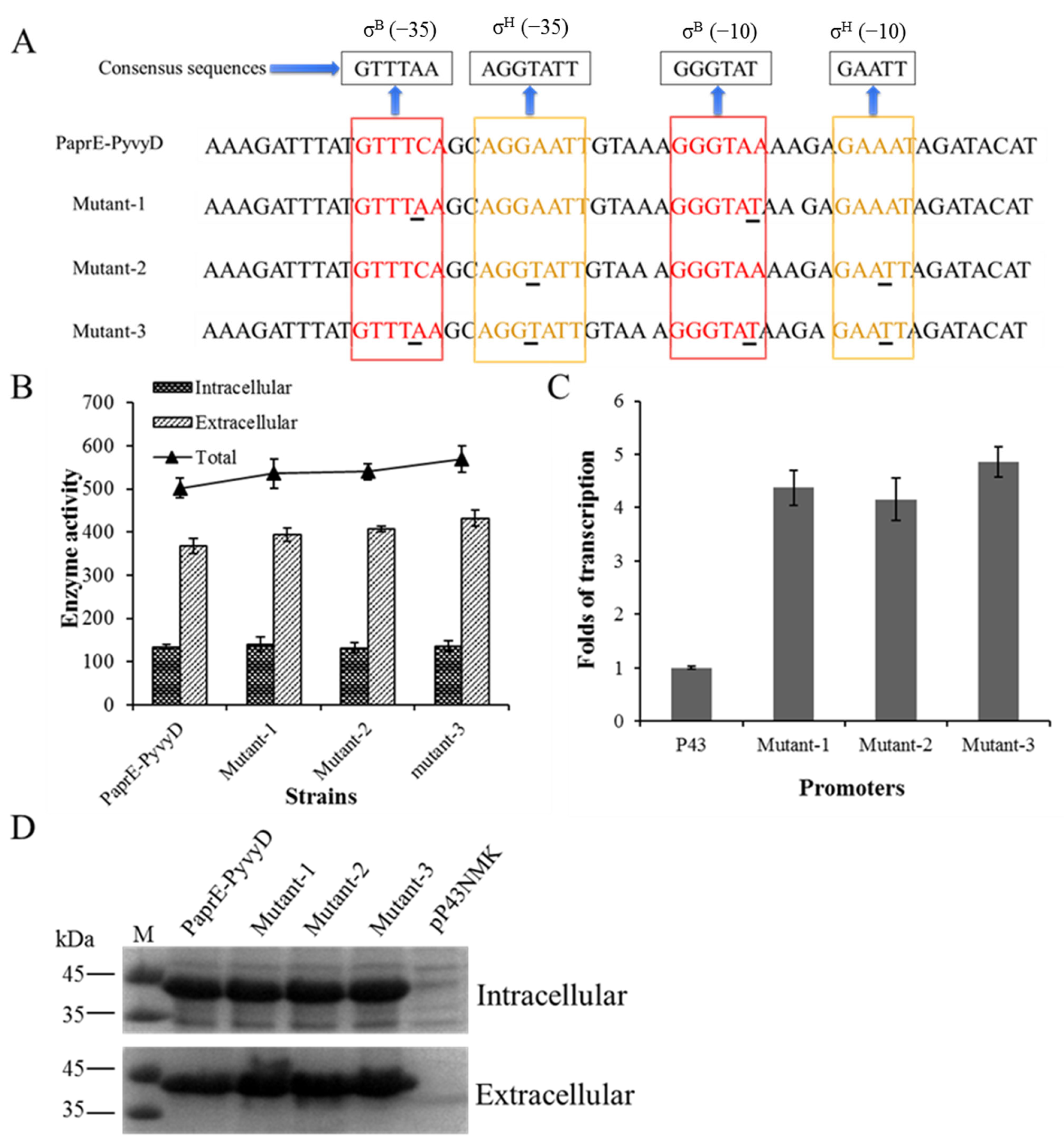
2.3. Optimization of the Core Region of Promoter PyvyD to Increase Transcription Intensity
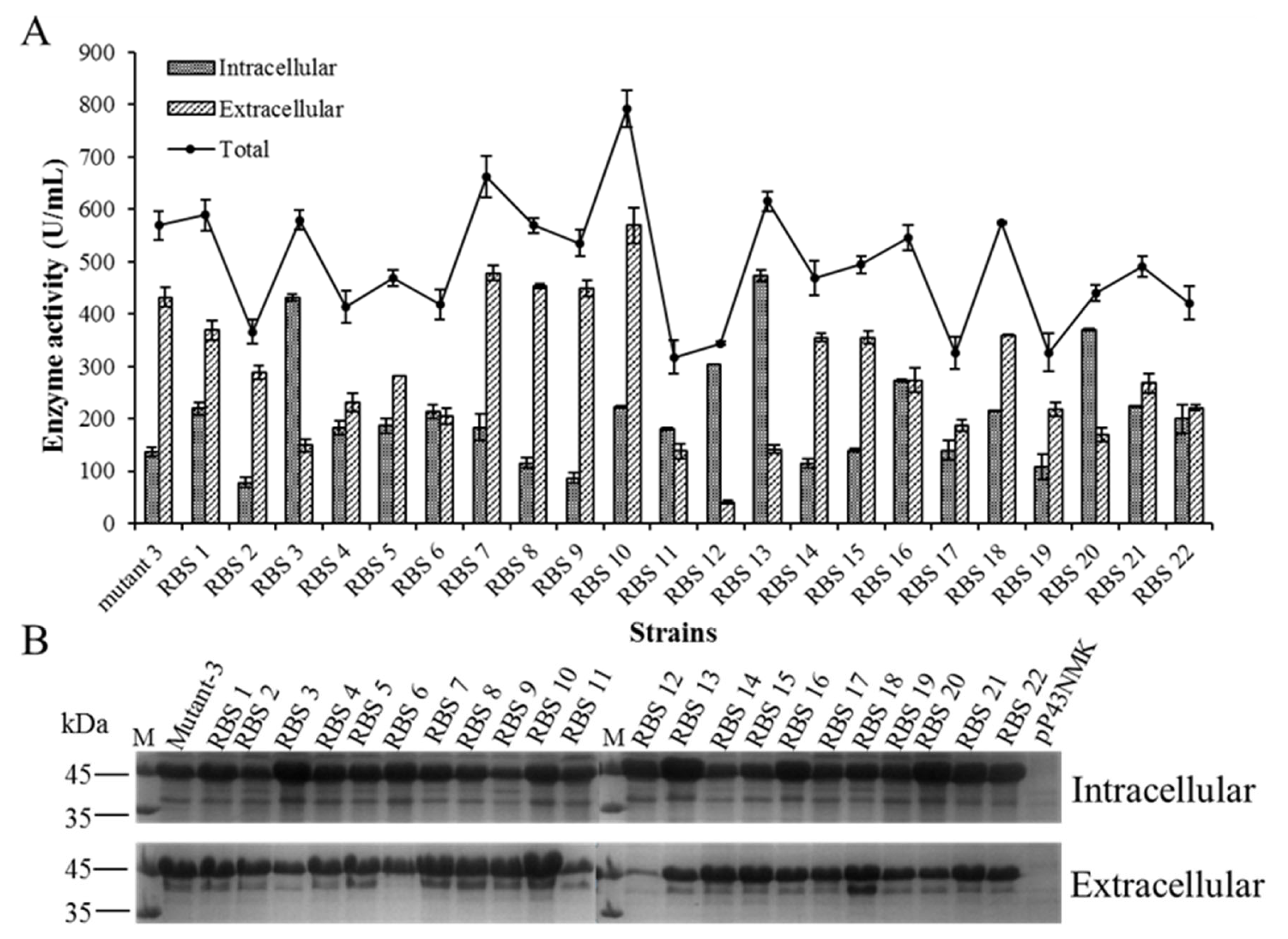
2.4. Enhanced BlAase Production by Optimizing RBS
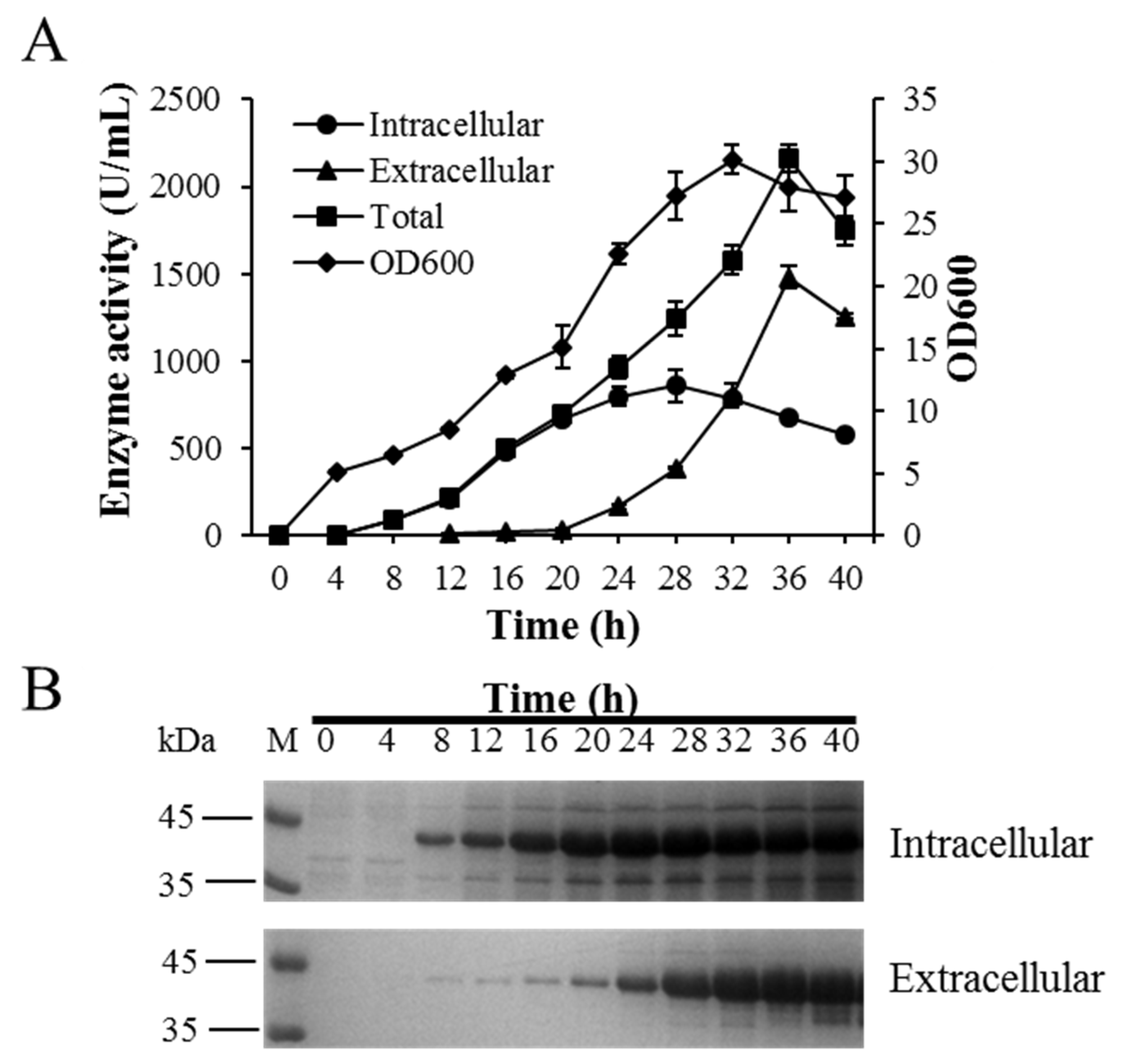
2.5. BlAase Production in a 10 L Fermentation Machine
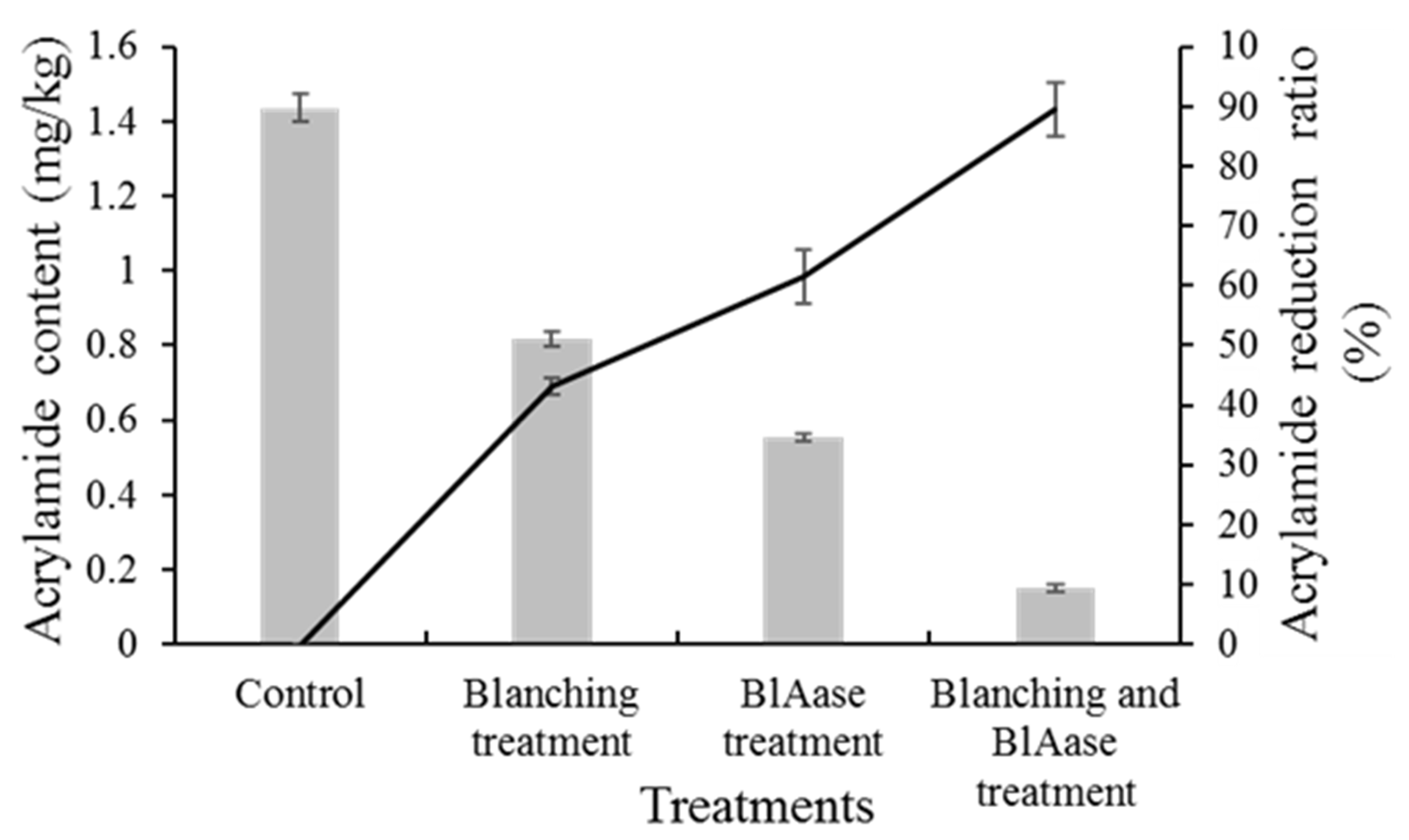
2.6. Food Application of BlAase
3. Materials and Methods
3.1. Strains and Plasmids
3.2. Plasmid Construction
3.3. Expression of the BlAase in B. subtilis
3.4. RNA Extraction and qRT-PCR
3.5. Measurement of BlAase Activity
3.6. Production of BlAase in a 10 L Fermenter
3.7. Reduction in Acrylamide in Potato Chips
3.7.1. Sample Preparation
- (I)
- They were rinsed in distilled water without any further processing at 37 °C for 2 h;
- (II)
- Potato slices were blanched at 85 °C for 10 min and then rinsed in distilled water at 37 °C for 2 h;
- (III)
- They were immersed in a 40 U/mL BlAase solution at 37 °C for 2 h;
- (IV)
- They were blanched at 85 °C for 10 min and then immersed in a 40 U/mL BlAase solution at 37 °C for 2 h.
3.7.2. Acrylamide Determination
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sun, Z.; Qin, R.; Li, D.; Ji, K.; Wang, T.; Cui, Z.; Huang, Y. A novel bacterial type II L-asparaginase and evaluation of its enzymatic acrylamide reduction in French fries. Int. J. Biol. Macromol. 2016, 92, 232–239. [Google Scholar] [CrossRef]
- Moguel, I.S.; Yamakawa, C.K.; Pessoa, A.; Mussatto, S.I. L-asparaginase production by Leucosporidium scottii in a Bench-Scale bioreactor with Co-production of lipids. Front. Bioeng. Biotech. 2020, 8, 576511. [Google Scholar] [CrossRef] [PubMed]
- Ran, T.; Jiao, L.; Wang, W.; Chen, J.; Chi, H.; Lu, Z.; Zhang, C.; Xu, D.; Lu, F. Structures of L-asparaginase from Bacillus licheniformis reveal an essential residue for its substrate stereoselectivity. J. Agr. Food. Chem. 2021, 69, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Krishnapura, P.R.; Belur, P.D.; Subramanya, S. A critical review on properties and applications of microbial L-asparaginases. Crit. Rev. Microbiol. 2016, 42, 720–737. [Google Scholar]
- Elmore, J.S.; Parker, J.K.; Halford, N.G.; Muttucumaru, N.; Mottram, D.S. Effects of plant sulfur nutrition on acrylamide and aroma compounds in cooked wheat. J. Agr. Food. Chem. 2008, 56, 6173–6179. [Google Scholar] [CrossRef]
- da Cunha, M.C.; Aguilar, J.G.D.; de Melo, R.R.; Nagamatsu, S.T.; Ali, F.; de Castro, R.J.S.; Sato, H.H. Fungal L-asparaginase: Strategies for production and food applications. Food. Res. Int. 2019, 126, 108658. [Google Scholar] [CrossRef] [PubMed]
- Nunes, J.C.F.; Cristovao, R.O.; Freire, M.G.; Santos-Ebinuma, V.C.; Faria, J.L.; Silva, C.G.; Tavares, A.P.M. Recent strategies and applications for L-Asparaginase confinement. Molecules 2020, 25, 5827. [Google Scholar] [CrossRef] [PubMed]
- Chityala, S.; Dasu, V.V.; Ahmad, J.; Prakasham, R.S. High yield expression of novel glutaminase free L-asparaginase II of Pectobacterium carotovorum MTCC 1428 in Bacillus subtilis WB800N. Bioproc. Biosyst. Eng. 2015, 38, 2271–2284. [Google Scholar] [CrossRef]
- Yao, D.; Su, L.; Li, N.; Wu, J. Enhanced extracellular expression of Bacillus stearothermophilus alpha-amylase in Bacillus subtilis through signal peptide optimization, chaperone overexpression and alpha-amylase mutant selection. Microb. Cell Fact. 2019, 18, 69. [Google Scholar] [CrossRef]
- Harwood, C.R.; Cranenburgh, R. Bacillus protein secretion: An unfolding story. Trends. Microbiol. 2008, 16, 73–79. [Google Scholar] [CrossRef]
- Sauer, C.; van Themaat, E.V.L.; Boender, L.G.M.; Groothuis, D.; Cruz, R.; Hamoen, L.W.; Harwood, C.R.; van Rij, T. Exploring the nonconserved sequence space of synthetic expression modules in Bacillus subtilis. Acs. Synth. Biol. 2018, 7, 1773–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Liu, S.; Jiao, Y.; Gao, H.; Wang, M.; Du, G.; Chen, J. Enhanced extracellular production of L-asparaginase from Bacillus subtilis 168 by B-subtilis WB600 through a combined strategy. Appl. Microbiol. Bio. 2017, 101, 1509–1520. [Google Scholar] [CrossRef]
- Liu, X.; Wang, H.; Wang, B.; Pan, L. High-level extracellular protein expression in Bacillus subtilis by optimizing strong promoters based on the transcriptome of Bacillus subtilis and Bacillus megaterium. Protein Expr. Purif. 2018, 151, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Su, L.; Duan, X.; Liu, L.; Wu, J. High-level extracellular protein production in Bacillus subtilis using an optimized dual-promoter expression system. Microb. Cell Fact. 2017, 16, 32. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Zhu, X.; Nie, T.; Lu, F.; Bie, X.; Lu, Y.; Trouth, F.; Lu, Z. Enhanced expression of pullulanase in Bacillus subtilis by new strong promoters mined from transcriptome data, both alone and in combination. Front. Microbiol. 2018, 9, 2638. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.H.; Shi, C.S.; Li, D.K.; Chen, X.J.; Li, J.L.; Zhang, Y.W.; Yuan, H.; Li, Y.; Lu, F.P. Engineering a highly efficient expression system to produce BcaPRO protease in Bacillus subtilis by an optimized promoter and signal peptide. Int. J. Biol. Macromol. 2019, 138, 903–911. [Google Scholar] [CrossRef]
- Sun, H.; Yang, J.; Song, H. Engineering mycobacteria artificial promoters and ribosomal binding sites for enhanced sterol production. Biochem. Eng. J. 2020, 162, 107739. [Google Scholar] [CrossRef]
- Jin, P.; Kang, Z.; Yuan, P.; Du, G.; Chen, J. Production of specific-molecular-weight hyaluronan by metabolically engineered Bacillus subtilis 168. Metab. Eng. 2016, 35, 21–30. [Google Scholar] [CrossRef]
- Pan, M.; Li, J.; Lv, X.; Du, G.; Liu, L. Molecular engineering of chitinase from Bacillus sp. DAU101 for enzymatic production of chitooligosaccharides. Enzyme. Microb. Tech. 2019, 124, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Bjerknes, M.; Kumar, R.; Jay, E. Determination of the optimal aligned spacing between the Shine-Dalgarno sequence and the translation initiation codon of Escherichia-coli messenger-RNAs. Nucleic. Acids. Res. 1994, 22, 4953–4957. [Google Scholar] [CrossRef] [Green Version]
- Pang, B.; Zhou, L.; Cui, W.J.; Liu, Z.M.; Zhou, Z.M. Production of a thermostable pullulanase in Bacillus subtilis by optimization of the expression elements. Starch-Starke 2020, 72, 2000018. [Google Scholar] [CrossRef]
- Niu, J.; Meng, F.; Zhou, Y.; Zhang, C.; Lu, Z.; Lu, F.; Chen, M. Non-classical secretion of a type I L-asparaginase in Bacillus subtilis. Int. J. Biol. Macromol. 2021, 180, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Niu, T.F.; Lv, X.Q.; Liu, Y.F.; Li, J.H.; Lu, W.; Du, G.C.; Chen, J.; Liu, L. Secretory expression Fine-Tuning and directed evolution of diacetylchitobiose deacetylase by Bacillus subtilis. Appl. Environ. Microb. 2019, 85, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.Y.; Wang, W.S.; Wang, H.Z.; Yuan, F.; Xu, Z.; Yang, K.Q.; Li, Z.L.; Chen, Y.H.; Fan, K.Q. Improvement of stress tolerance and riboflavin production of Bacillus subtilis by introduction of heat shock proteins from thermophilic Bacillus strains. Appl. Microbiol. Biot. 2019, 103, 4455–4465. [Google Scholar] [CrossRef]
- Pan, X.L.; Yang, Y.L.; Liu, X.W.; Li, D.; Li, J.; Guo, X.Z.; Zhou, Z.G. Secretory expression of a heterologous protein, Aiio-AIO6BS, in Bacillus subtilis via a non-classical secretion pathway. Biochem. Bioph. Res. Commun. 2016, 478, 881–886. [Google Scholar] [CrossRef]
- Cai, D.; Wei, X.; Qiu, Y.; Chen, Y.; Chen, J.; Wen, Z.; Chen, S. High-level expression of nattokinase in Bacillus licheniformis by manipulating signal peptide and signal peptidase. J. Appl. Microbiol. 2016, 121, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Vellanoweth, R.L.; Rabinowitz, J.C. The influence of ribosome-binding-site elements on translational efficiency in Bacillus-subtilis and Escherichia-coli in vivo. Mol. Microbiol. 1992, 6, 1105–1114. [Google Scholar] [CrossRef]
- Han, L.; Cui, W.; Suo, F.; Miao, S.; Hao, W.; Chen, Q.; Guo, J.; Liu, Z.; Zhou, L.; Zhou, Z. Development of a novel strategy for robust synthetic bacterial promoters based on a stepwise evolution targeting the spacer region of the core promoter in Bacillus subtilis. Microb. Cell. Fact. 2019, 18, 96. [Google Scholar] [CrossRef] [Green Version]
- Borkowski, O.; Goelzer, A.; Schaffer, M.; Calabre, M.; Maeder, U.; Aymerich, S.; Jules, M.; Fromion, V. Translation elicits a growth rate-dependent, genome-wide, differential protein production in Bacillus subtilis. Mol. Syst. Biol. 2016, 12, 870. [Google Scholar] [CrossRef]
- Seo, S.W.; Yang, J.-S.; Cho, H.-S.; Yang, J.; Kim, S.C.; Park, J.M.; Kim, S.; Jung, G.Y. Predictive combinatorial design of mRNA translation initiation regions for systematic optimization of gene expression levels. Sci. Rep. 2014, 4, 4515. [Google Scholar] [CrossRef] [Green Version]
- Levin-Karp, A.; Barenholz, U.; Bareia, T.; Dayagi, M.; Zelcbuch, L.; Antonovsky, N.; Noor, E.; Milo, R. Quantifying translational coupling in E. coli synthetic operons using RBS modulation and fluorescent reporters. Acs. Synth. Biol. 2013, 2, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Luan, M.Y.; Li, Y.F. Ribosomal binding site sequences and promoters for expressing glutamate decarboxylase and producing gamma-aminobutyrate in Corynebacterium glutamicum. AMB Express 2018, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Salis, H.M.; Mirsky, E.A.; Voigt, C.A. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 2009, 27, 946–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, C.R.; Cui, W.J.; Cheng, J.T.; Liu, R.; Liu, Z.M.; Zhou, L.; Zhou, Z.M. Construction of a highly active secretory expression system via an engineered dual promoter and a highly efficient signal peptide in Bacillus subtilis. New Biotechnol. 2016, 33, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Ye, B.; Cheng, S.; Zhao, L.; Liu, Y.; Jiang, J.; Yan, X. Promoter engineering enables overproduction of foreign proteins from a single copy expression cassette in Bacillus subtilis. Microb. Cell Fact. 2019, 18, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.-J.; Pan, J.-G.; Park, S.-H.; Choi, S.-K. Development of a stationary phase-specific autoinducible expression system in Bacillus subtilis. J. Biotechnol. 2010, 149, 16–20. [Google Scholar] [CrossRef]
- Drzewiecki, K.; Eymann, C.; Mittenhuber, G.; Hecker, M. The yvyD gene of Bacillus subtilis is under dual control of sigma(B) and sigma(H). J. Bacteriol 1998, 180, 6674–6680. [Google Scholar] [CrossRef] [Green Version]
- Jan, J.; Valle, F.; Bolivar, F.; Merino, E. Construction of protein overproducer strains in Bacillus subtilis by an integrative approach. Appl. Microbiol. Biot 2001, 55, 69–75. [Google Scholar] [CrossRef]
- Rao, Y.; Cai, D.; Wang, H.; Xu, Y.; Xiong, S.; Gao, L.; Xiong, M.; Wang, Z.; Chen, S.; Ma, X. Construction and application of a dual promoter system for efficient protein production and metabolic pathway enhancement in Bacillus licheniformis. J. Biotechnol. 2020, 312, 1–10. [Google Scholar] [CrossRef]
- Li, X.; Xu, S.; Zhang, X.; Xu, M.; Yang, T.; Wang, L.; Zhang, H.; Fang, H.; Osire, T.; Yang, S.; et al. Design of a high-efficiency synthetic system for l-asparaginase production in Bacillus subtilis. Eng. Life. Sci. 2019, 19, 229–239. [Google Scholar] [CrossRef] [Green Version]
- Shi, F.; Fan, Z.; Zhang, S.; Wang, Y.; Tan, S.; Li, Y. Optimization of ribosomal binding site sequences for gene expression and 4-hydroxyisoleucine biosynthesis in recombinant Corynebacterium glutamicum. Enzyme. Microb. Technol. 2020, 140, 109622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhou, N.; Liu, Y.-M.; Liu, C.; Lou, C.-B.; Jiang, C.-Y.; Liu, S.-J. Ribosome binding site libraries and pathway modules for shikimic acid synthesis with Corynebacterium glutamicum. Microb. Cell Fact. 2015, 14, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliver, J.W.K.; Machado, I.M.P.; Yoneda, H.; Atsumi, S. Combinatorial optimization of cyanobacterial 2,3-butanediol production. Metab. Eng. 2014, 22, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.M.; Xu, M.J.; He, B.B.; Rao, Z.M. Cloning, expression, and characterization of L-asparaginase from a newly isolated Bacillus subtilis B11-06. J. Agr. Food. Chem. 2013, 61, 9428–9434. [Google Scholar] [CrossRef]
- Feng, Y.; Liu, S.; Jiao, Y.; Wang, Y.L.; Wang, M.; Du, G.C. Gene cloning and expression of the L-asparaginase from Bacillus cereus BDRD-ST26 in Bacillus subtilis WB600. J. Biosci. Bioeng. 2019, 127, 418–424. [Google Scholar] [CrossRef]
- Pedreschi, F.; Mariotti, S.; Granby, K.; Risum, J. Acrylamide reduction in potato chips by using commercial asparaginase in combination with conventional blanching. LWT-Food Sci. Technol. 2011, 44, 1473–1476. [Google Scholar] [CrossRef]
- Gryczan, T.J.; Contente, S.; Dubnau, D. Characterization of Staphylococcus aureus plasmids introduced by transformation into Bacillus subtilis. J. Bacteriol. 1978, 134, 318–329. [Google Scholar] [CrossRef] [Green Version]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Zhang, S.; Xie, Y.; Zhang, C.; Bie, X.; Zhao, H.; Lu, F.; Lu, Z. Biochemical characterization of a novel L-asparaginase from Bacillus megaterium H-1 and its application in French fries. Food. Res. Int. 2015, 77, 527–533. [Google Scholar] [CrossRef]
- Chi, H.; Chen, M.; Jiao, L.; Lu, Z.; Bie, X.; Zhao, H.; Lu, F. Characterization of a novel L-Asparaginase from Mycobacterium gordonae with acrylamide mitigation potential. Foods 2021, 10, 2819. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Hwang, B.Y.; Roh, J.; Lee, J.K.; Kim, K.; Wong, S.L.; Yun, H.; Lee, S.G.; Kim, B.G. Camparison of P-aprE, P-amyE, and P-P43 promoter strength for beta-galactosidase and staphylokinase expression in Bacillus subtilis. Biotechnol. Bioproc. Eng. 2008, 13, 313–318. [Google Scholar] [CrossRef]
- Liu, Z.M.; Zheng, W.H.; Ge, C.L.; Cui, W.J.; Zhou, L.; Zhou, Z.M. High-level extracellular production of recombinant nattokinase in Bacillus subtilis WB800 by multiple tandem promoters. BMC Microbiol. 2019, 19, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.X.; Zhang, G.Q.; Zhou, J.W.; Li, J.H.; Du, G.C. Combinatorial engineering for efficient production of protein-glutaminase in Bacillus subtilis. Enzyme. Microb. Tech. 2021, 150, 109863. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.Y.; Shao, J.; Tang, M.W.; Xin, Y.; Zhang, L. Promote the expression and corrected folding of an extremely stable N-demethylase by promoter reconstruction, native environment simulation and surface design. Int. J. Biol. Macromol. 2021, 178, 434–443. [Google Scholar] [CrossRef]
- Jiao, S.; Li, X.; Yu, H.M.; Yang, H.; Li, X.; Shen, Z.Y. In situ enhancement of surfactin biosynthesis in Bacillus subtilis using novel artificial inducible promoters. Biotechnol. Bioeng. 2017, 114, 832–842. [Google Scholar] [CrossRef]
- Kang, X.-M.; Cai, X.; Huang, Z.-H.; Liu, Z.-Q.; Zheng, Y.-G. Construction of a highly active secretory expression system in Bacillus subtilis of a recombinant amidase by promoter and signal peptide engineering. Int. J. Biol. Macromol. 2020, 143, 833–841. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, J.; Yan, R.; Shen, J.; Zhu, X.; Meng, F.; Lu, Z.; Lu, F. Cis-Element Engineering Promotes the Expression of Bacillus subtilis Type I L-Asparaginase and Its Application in Food. Int. J. Mol. Sci. 2022, 23, 6588. https://doi.org/10.3390/ijms23126588
Niu J, Yan R, Shen J, Zhu X, Meng F, Lu Z, Lu F. Cis-Element Engineering Promotes the Expression of Bacillus subtilis Type I L-Asparaginase and Its Application in Food. International Journal of Molecular Sciences. 2022; 23(12):6588. https://doi.org/10.3390/ijms23126588
Chicago/Turabian StyleNiu, Jiafeng, Ruxue Yan, Juan Shen, Xiaoyu Zhu, Fanqiang Meng, Zhaoxin Lu, and Fengxia Lu. 2022. "Cis-Element Engineering Promotes the Expression of Bacillus subtilis Type I L-Asparaginase and Its Application in Food" International Journal of Molecular Sciences 23, no. 12: 6588. https://doi.org/10.3390/ijms23126588