
TNMplot.com: A Web Tool for the Comparison of Gene Expression in Normal, Tumor and Metastatic Tissues

Abstract
:1. Introduction
2. Results
2.1. Integrated Database
2.2. TNMplot.com Analysis Platform
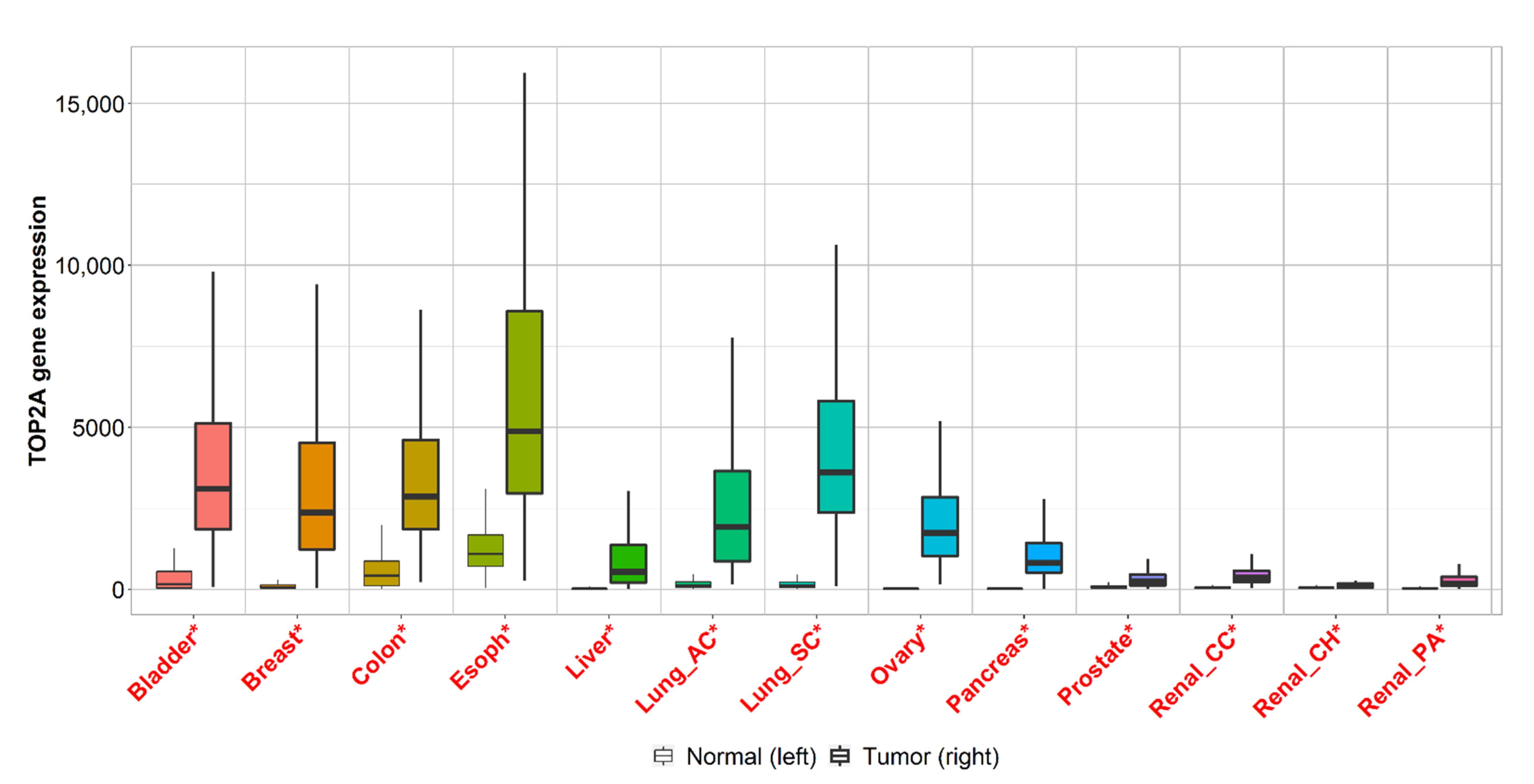
2.3. Gene Expression Analysis of Cancers with the Highest Mortality
2.4. Linking the Most Significant Genes to Cancer Hallmarks
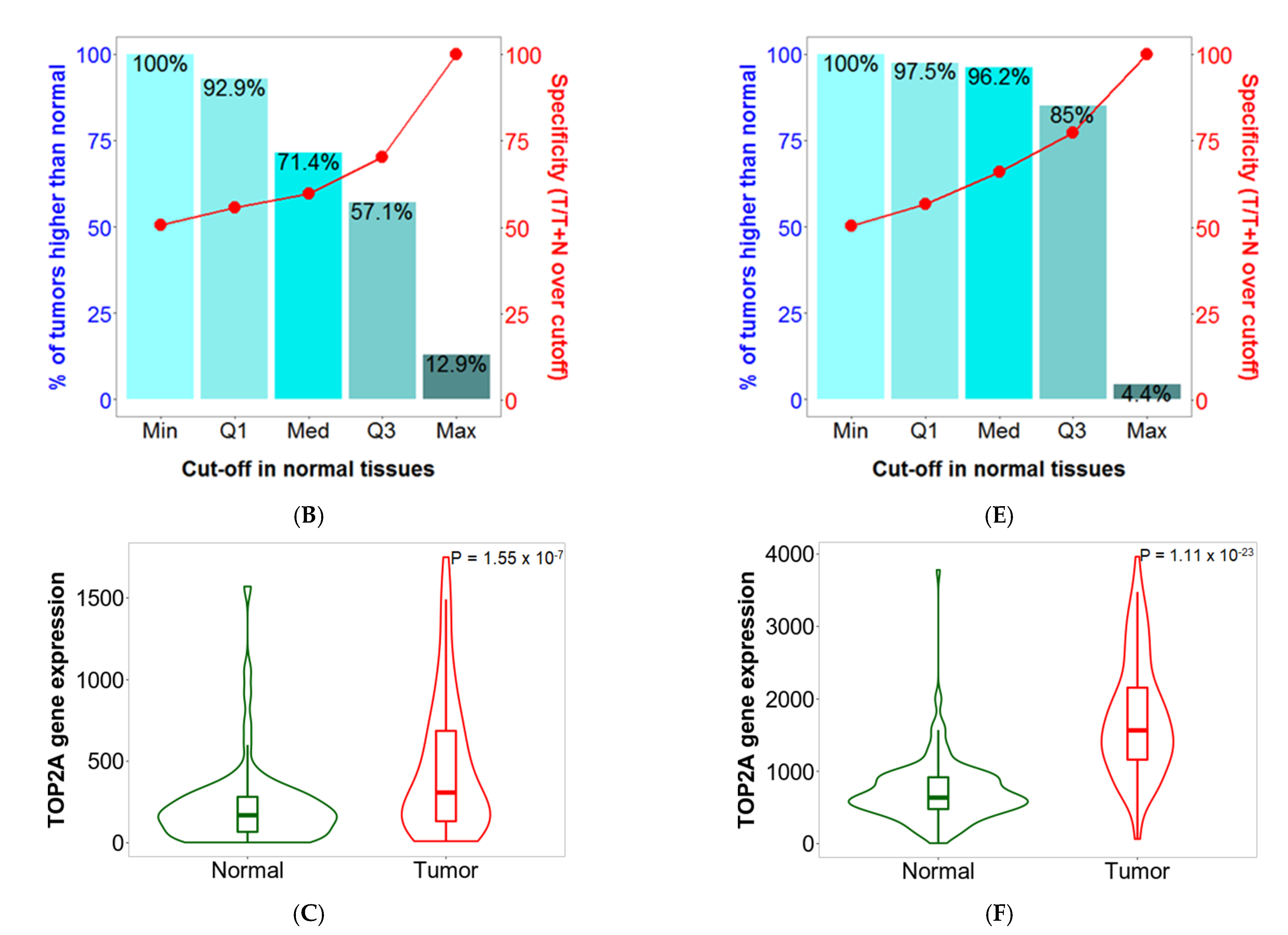
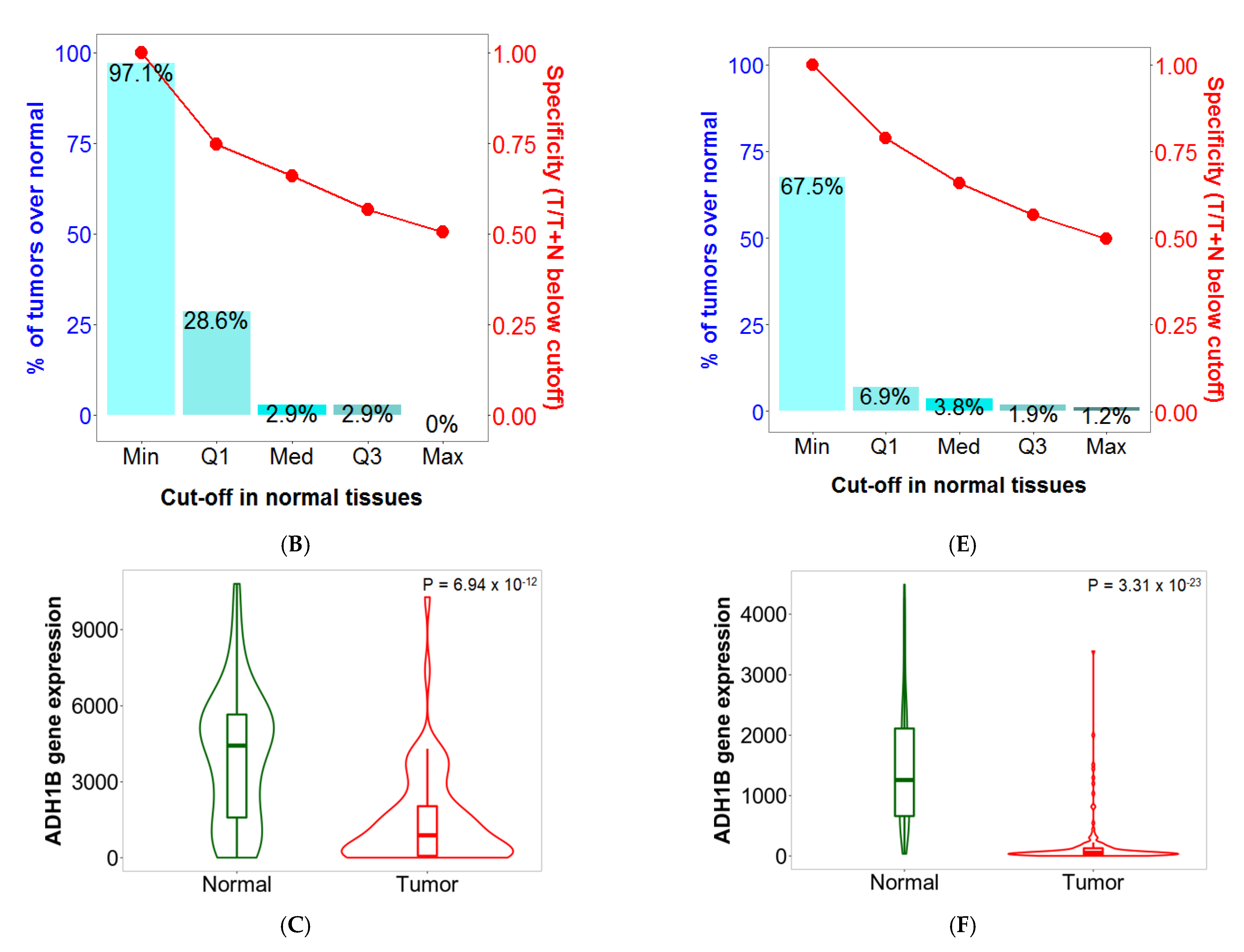
2.5. Sensitivity and Specificity
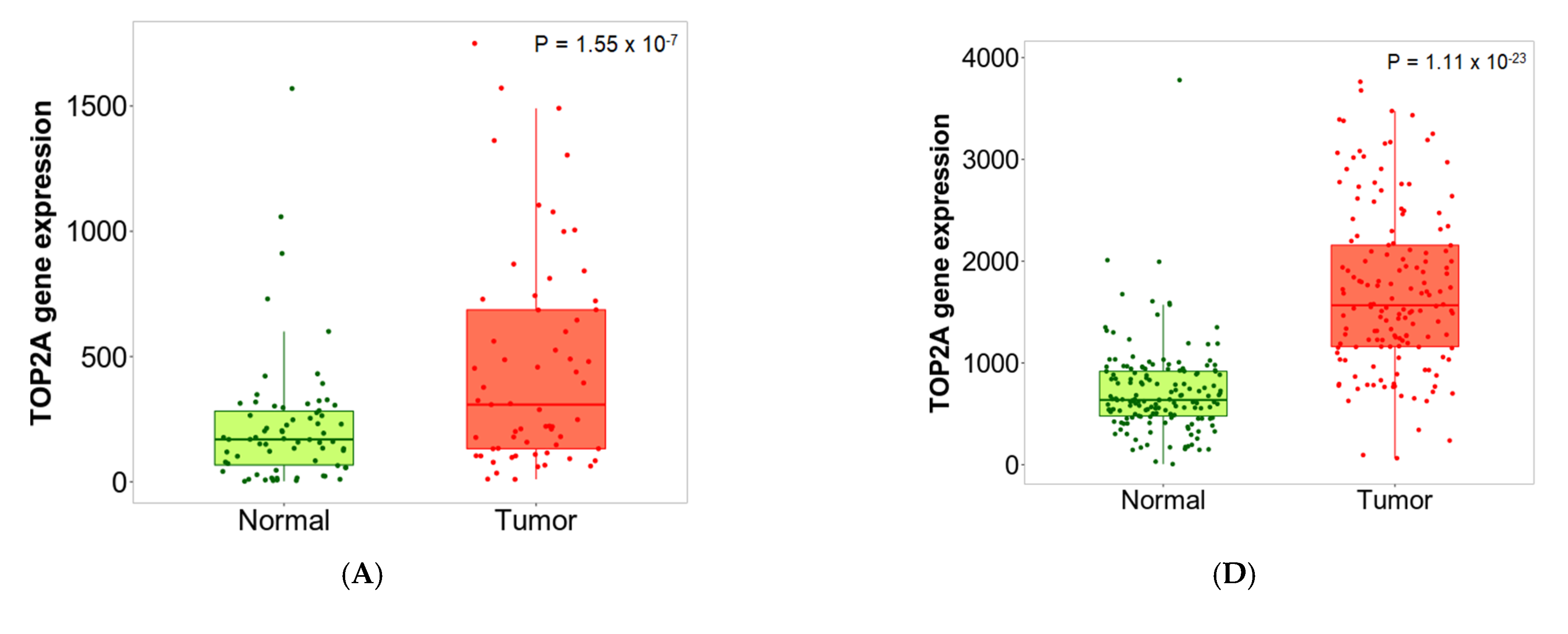
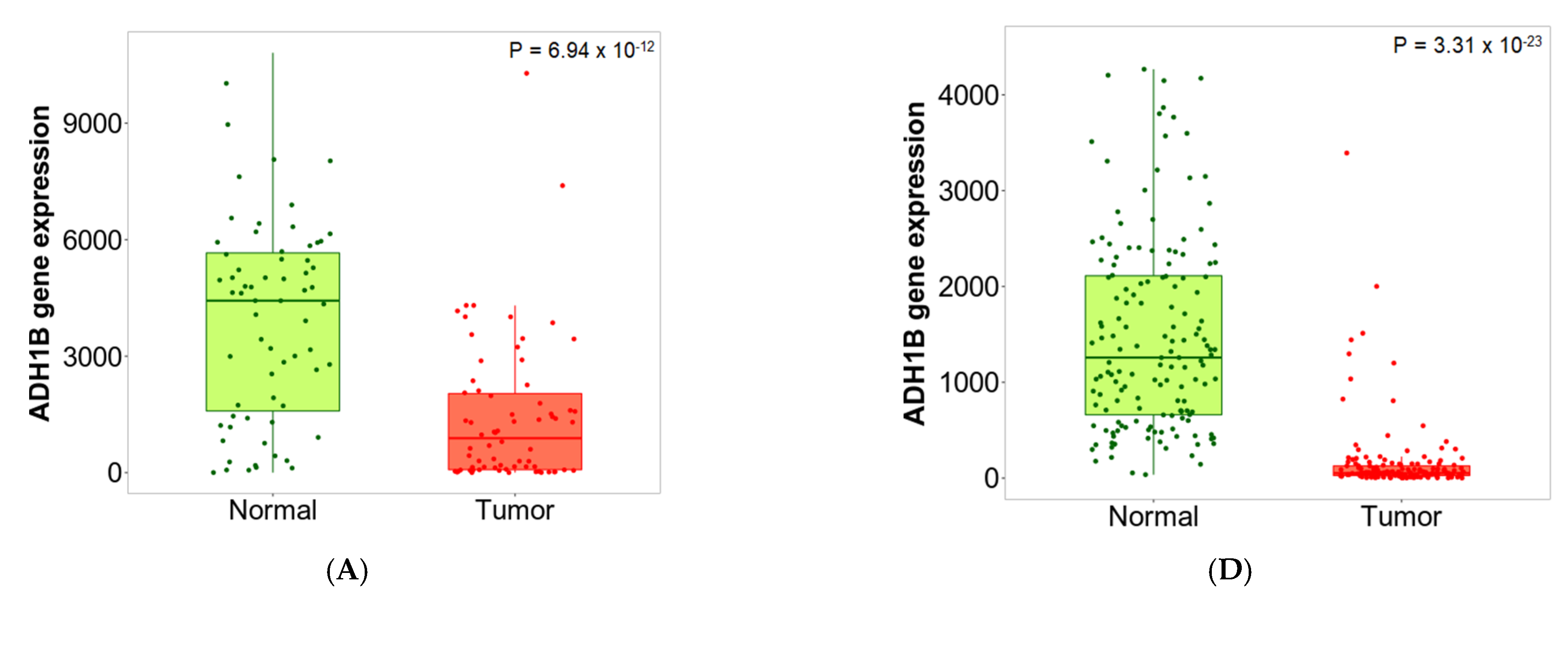
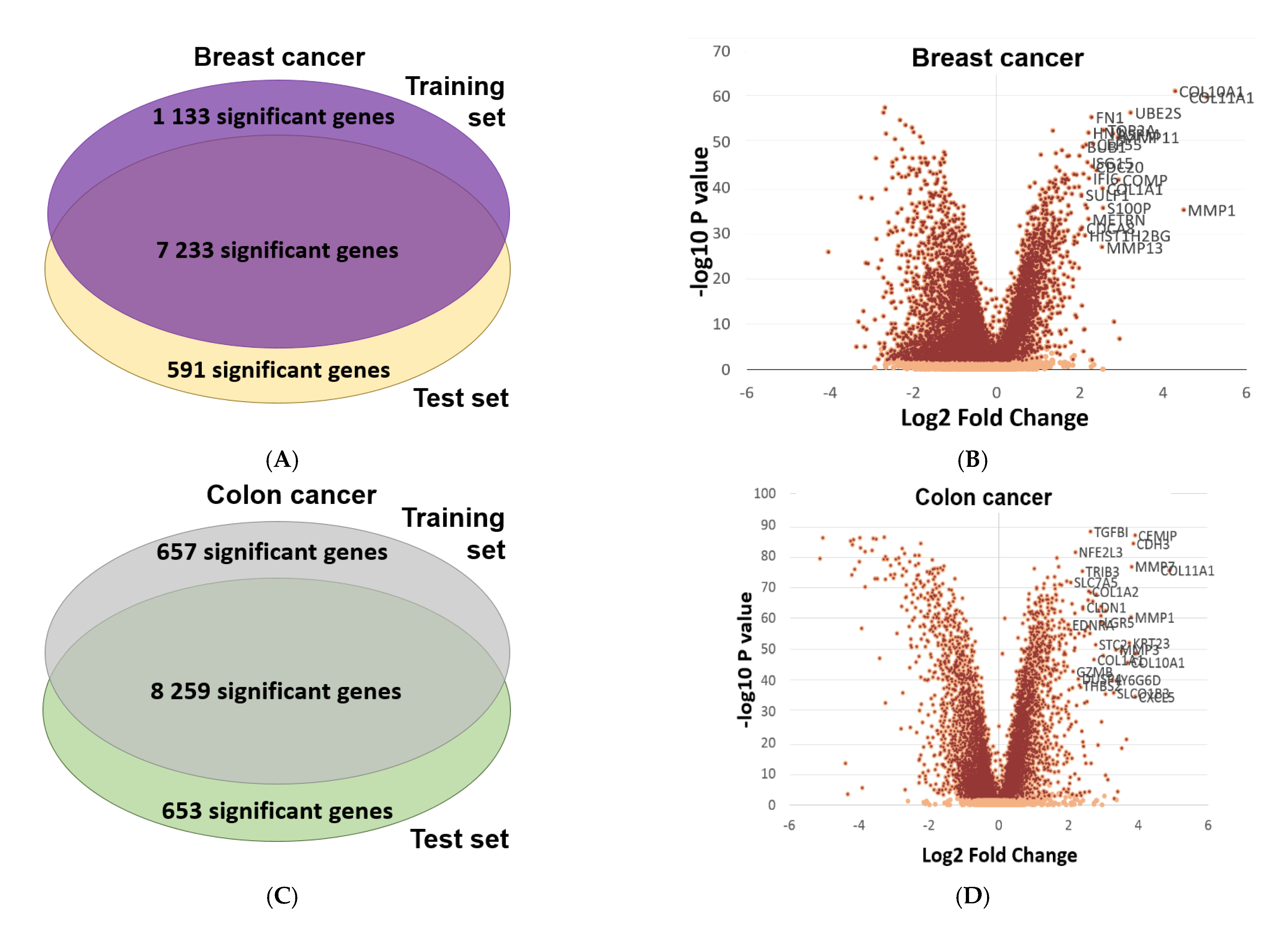
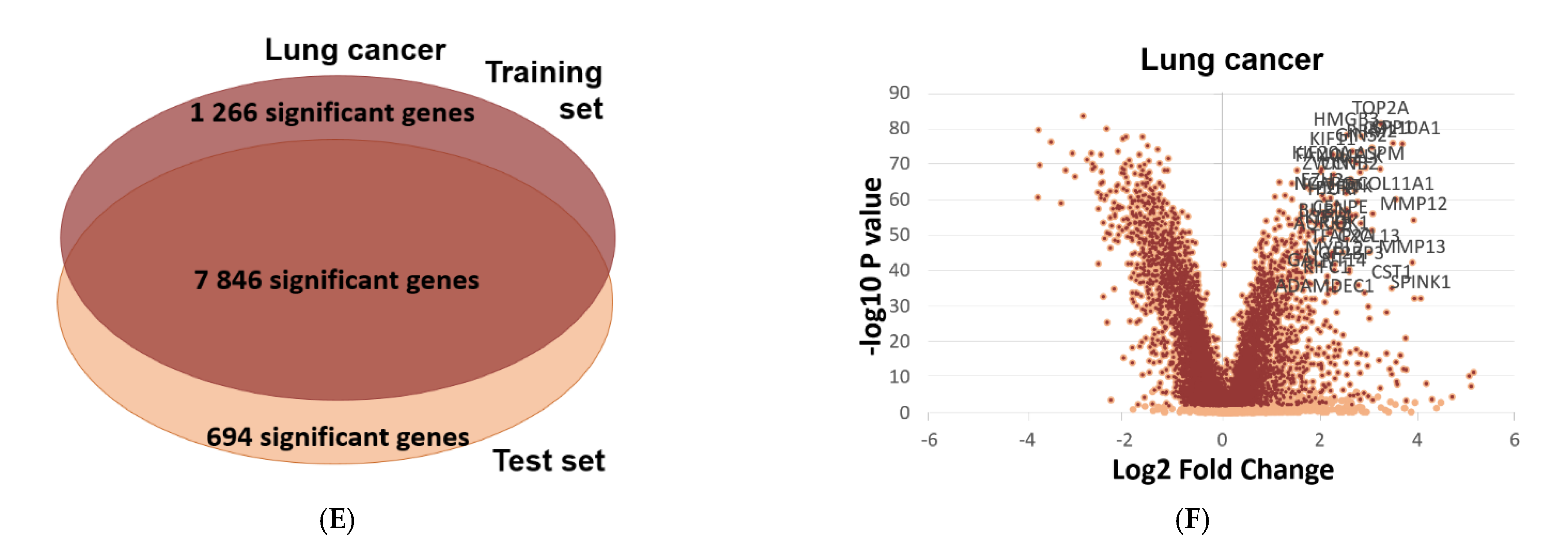
2.6. Validation of Differential Expression between Normal and Tumor Samples
3. Discussion
4. Materials and Methods
4.1. Database Setup—Gene Arrays
4.2. Database Setup—RNA-seq
4.3. Gene Annotation
4.4. Statistical Analysis
4.5. Shiny User Interface
4.6. Validation of Differential Expression
4.7. Cancer Biomarker Genes
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, L.; Zhou, W.; Velculescu, V.E.; Kern, S.E.; Hruban, R.H.; Hamilton, S.R.; Vogelstein, B.; Kinzler, K.W. Gene expression profiles in normal and cancer cells. Science 1997, 276, 1268–1272. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Tamura, S.; Buchdunger, E.; Ohno, S.; Segal, G.M.; Fanning, S.; Zimmermann, J.; Lydon, N.B. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med. 1996, 2, 561–566. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menyhart, O.; Harami-Papp, H.; Sukumar, S.; Schafer, R.; Magnani, L.; de Barrios, O.; Gyorffy, B. Guidelines for the selection of functional assays to evaluate the hallmarks of cancer. Biochim. Biophys. Acta 2016, 1866, 300–319. [Google Scholar] [CrossRef] [Green Version]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research, N.; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.T. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Birkbak, N.J.; Gyorffy, B.; Szallasi, Z.; Eklund, A.C. Jetset: Selecting the optimal microarray probe set to represent a gene. BMC Bioinform. 2011, 12, 474. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagès, H.; Carlson, M.; Falcon, S.; Li, N. AnnotationDbi: Manipulation of SQLite-Based Annotations in Bioconductor. Available online: http://www.bioconductor.org/packages/release/bioc/html/AnnotationDbi.html (accessed on 4 March 2021).
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fekete, J.T.; Gyorffy, B. ROCplot.org: Validating predictive biomarkers of chemotherapy/hormonal therapy/anti-HER2 therapy using transcriptomic data of 3,104 breast cancer patients. Int. J. Cancer 2019, 145, 3140–3151. [Google Scholar] [CrossRef] [PubMed]
- Kou, F.; Sun, H.; Wu, L.; Li, B.; Zhang, B.; Wang, X.; Yang, L. TOP2A Promotes Lung Adenocarcinoma Cells’ Malignant Progression and Predicts Poor Prognosis in Lung Adenocarcinoma. J. Cancer 2020, 11, 2496–2508. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Xu, J.; Zhao, J.; Bai, J.H. Proliferation and invasion of colon cancer cells are suppressed by knockdown of TOP2A. J. Cell Biochem. 2018, 119, 7256–7263. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Xu, F.; Luo, R.; Zheng, Q.; Lu, J.; Yang, Y.; Qin, T.; Yuan, Z.; Shi, Y.; Jiang, W.; et al. The prognostic significance of topoisomerase II alpha protein in early stage luminal breast cancer. BMC Cancer 2018, 18, 331. [Google Scholar] [CrossRef] [Green Version]
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef] [PubMed]
- Jasra, S.; Anampa, J. Anthracycline Use for Early Stage Breast Cancer in the Modern Era: A Review. Curr. Treat. Options Oncol. 2018, 19, 30. [Google Scholar] [CrossRef]
- Hallek, M. Chronic lymphocytic leukemia: 2020 update on diagnosis, risk stratification and treatment. Am. J. Hematol. 2019, 94, 1266–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cederleuf, H.; Bjerregard Pedersen, M.; Jerkeman, M.; Relander, T.; d’Amore, F.; Ellin, F. The addition of etoposide to CHOP is associated with improved outcome in ALK+ adult anaplastic large cell lymphoma: A Nordic Lymphoma Group study. Br. J. Haematol. 2017, 178, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Kropotova, E.S.; Zinovieva, O.L.; Zyryanova, A.F.; Dybovaya, V.I.; Prasolov, V.S.; Beresten, S.F.; Oparina, N.Y.; Mashkova, T.D. Altered expression of multiple genes involved in retinoic acid biosynthesis in human colorectal cancer. Pathol. Oncol. Res. 2014, 20, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhang, L.; Huang, C.; Huang, P.; Zhang, J. Distinct Prognostic Values of Alcohol Dehydrogenase Family Members for Non-Small Cell Lung Cancer. Med. Sci. Monit. 2018, 24, 3578–3590. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Huang, H.Y.; Lee, S.W.; Chen, T.J.; Tai, H.C.; Hsu, H.P.; Chang, K.Y.; Li, C.F. TOP2A overexpression as a poor prognostic factor in patients with nasopharyngeal carcinoma. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Sulakhe, D.; D’Souza, M.; Wang, S.; Balasubramanian, S.; Athri, P.; Xie, B.; Canzar, S.; Agam, G.; Gilliam, T.C.; Maltsev, N. Exploring the functional impact of alternative splicing on human protein isoforms using available annotation sources. Brief. Bioinform. 2019, 20, 1754–1768. [Google Scholar] [CrossRef]
- Wu, C.; Ma, S. A selective review of robust variable selection with applications in bioinformatics. Brief. Bioinform. 2015, 16, 873–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Chang, W.; Cheng, J.; Allaire, J.J.; Xie, Y.; McPherson, J. Shiny: Web Application Framework for R. Available online: https://CRAN.R-project.org/package=shiny (accessed on 4 March 2021).
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Manual Screening | Computational Screening | Result | T | N | M | ||||
|---|---|---|---|---|---|---|---|---|---|
| NCBI GEO | GSE screened: 3180 datasets | Primary tissue series n = 554 (38,897 Samples) | Data cleaning | MAS5 [10] normalization and scaling | JetSet [11] Annotation | 38,431 Samples 38 tumor types | 29,376 | 3691 | 453 |
| TARGET | 1193 samples | - | Data cleaning | DESeq2 [12] normalization and scaling | AnnotationDBI [13] annotation | 1193 samples 7 tumor types | 1180 | 12 | 1 |
| TCGA | 11,050 samples | Removal of non-primary tissues | Data cleaning | DESeq2 normalization and scaling | AnnotationDBI annotation | 11,010 samples 33 tumor types | 9886 | 730 | 394 |
| GTEx | 11,688 samples | Removal of non-primary tissues | Data cleaning | DESeq2 normalization and scaling | biomaRt [14] andAnnotationDBI annotation | 11,215 samples 51 tumor types | - | 11,215 | - |
| Gene | Mean Fold Change | Gene | Mean Fold Change |
|---|---|---|---|
| TOP2A | 7.80 | RUVBL2 | 1.77 |
| SPP1 | 7.00 | TMSB10 | 1.76 |
| CENPA | 6.03 | RPN1 | 1.75 |
| NEK2 | 5.63 | CHPF2 | 1.67 |
| MELK | 5.46 | CERS2 | 1.63 |
| HMMR | 5.29 | SH3BGRL3 | 1.61 |
| KIF20A | 4.96 | APRT | 1.60 |
| NEIL3 | 4.89 | IRAK1 | 1.56 |
| TTK | 4.85 | SEC61A1 | 1.54 |
| ASPM | 4.82 | PSME2 | 1.52 |
| CCNB2 | 4.76 | SPAST | 1.49 |
| DTL | 4.44 | DNASE1L1 | 1.42 |
| NCAPG | 4.44 | PGLS | 1.40 |
| ZWINT | 4.15 | DIRAS3 | 0.60 |
| CCNB1 | 4.14 | ECHDC3 | 0.59 |
| BUB1B | 3.79 | PDE8B | 0.56 |
| TK1 | 3.76 | PCDH9 | 0.52 |
| PRC1 | 3.72 | PEG3 | 0.46 |
| CENPU | 3.58 | PKNOX2 | 0.44 |
| KPNA2 | 3.23 | CXCL12 | 0.42 |
| CENPN | 3.03 | PHYHIP | 0.33 |
| CKAP2 | 2.62 | GPM6A | 0.32 |
| KNOP1 | 2.26 | FHL1 | 0.27 |
| SNRPB | 2.00 | DPT | 0.25 |
| MAGOHB | 1.90 | C7 | 0.24 |
| RPN2 | 1.83 | AOX1 | 0.22 |
| SNRPF | 1.82 | ADH1B | 0.15 |
| ENO1 | 1.79 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartha, Á.; Győrffy, B. TNMplot.com: A Web Tool for the Comparison of Gene Expression in Normal, Tumor and Metastatic Tissues. Int. J. Mol. Sci. 2021, 22, 2622. https://doi.org/10.3390/ijms22052622
Bartha Á, Győrffy B. TNMplot.com: A Web Tool for the Comparison of Gene Expression in Normal, Tumor and Metastatic Tissues. International Journal of Molecular Sciences. 2021; 22(5):2622. https://doi.org/10.3390/ijms22052622
Chicago/Turabian StyleBartha, Áron, and Balázs Győrffy. 2021. "TNMplot.com: A Web Tool for the Comparison of Gene Expression in Normal, Tumor and Metastatic Tissues" International Journal of Molecular Sciences 22, no. 5: 2622. https://doi.org/10.3390/ijms22052622