
Overexpression of miR-124 in Motor Neurons Plays a Key Role in ALS Pathological Processes

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
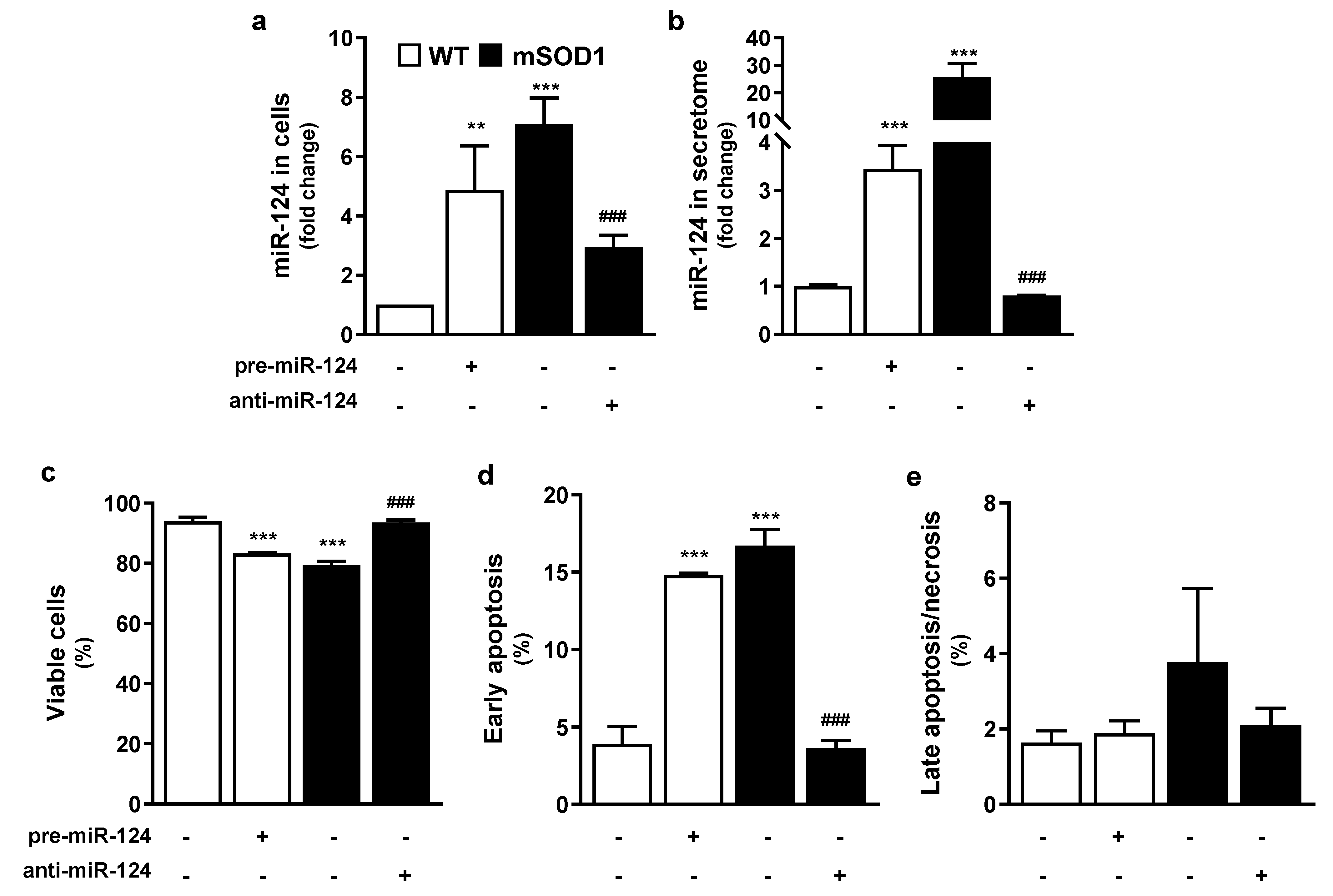
2.1. Overexpression of miR-124 in mSOD1 MNs and in Pre-miR-124-Treated WT MNs Compromise Cell Viability, Which Is Prevented by Restoring Its Normal Values
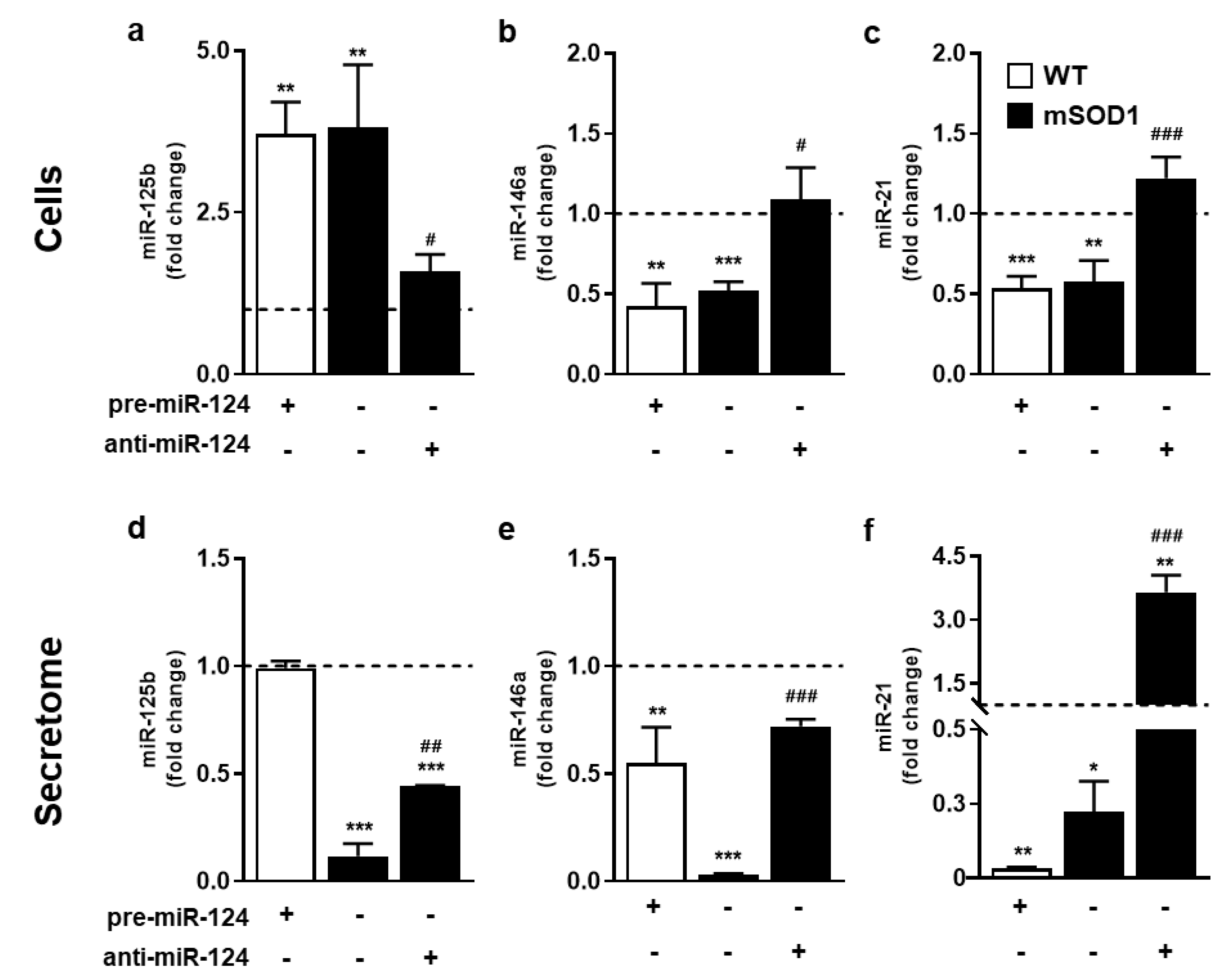
2.2. Modulation of miR-124 toward Normal Values in mSOD1 MNs Reverts Their Aberrant Inflamma-miRNA Profile and Partialy Recovers WT Secretome Homeostatic Signature
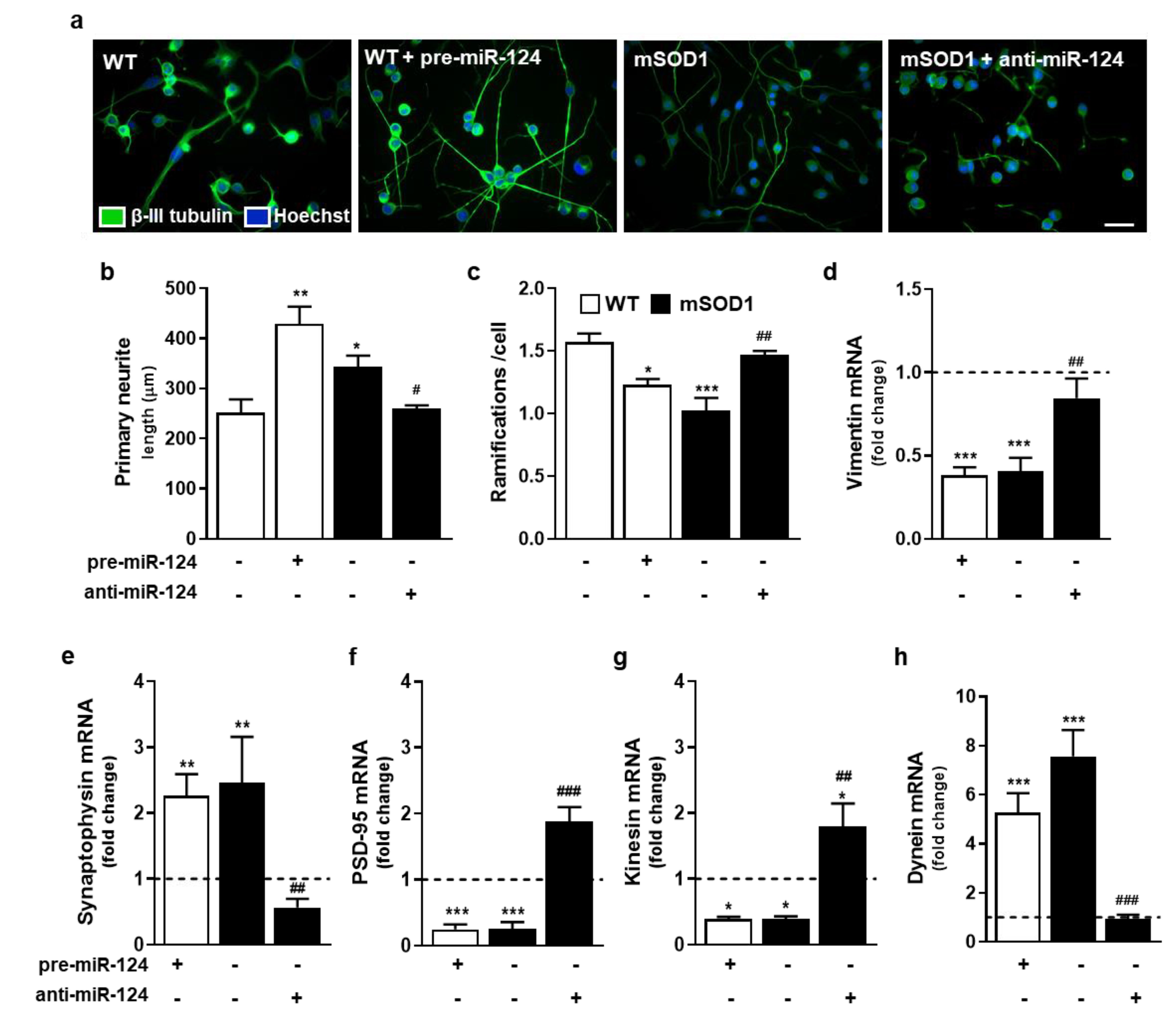
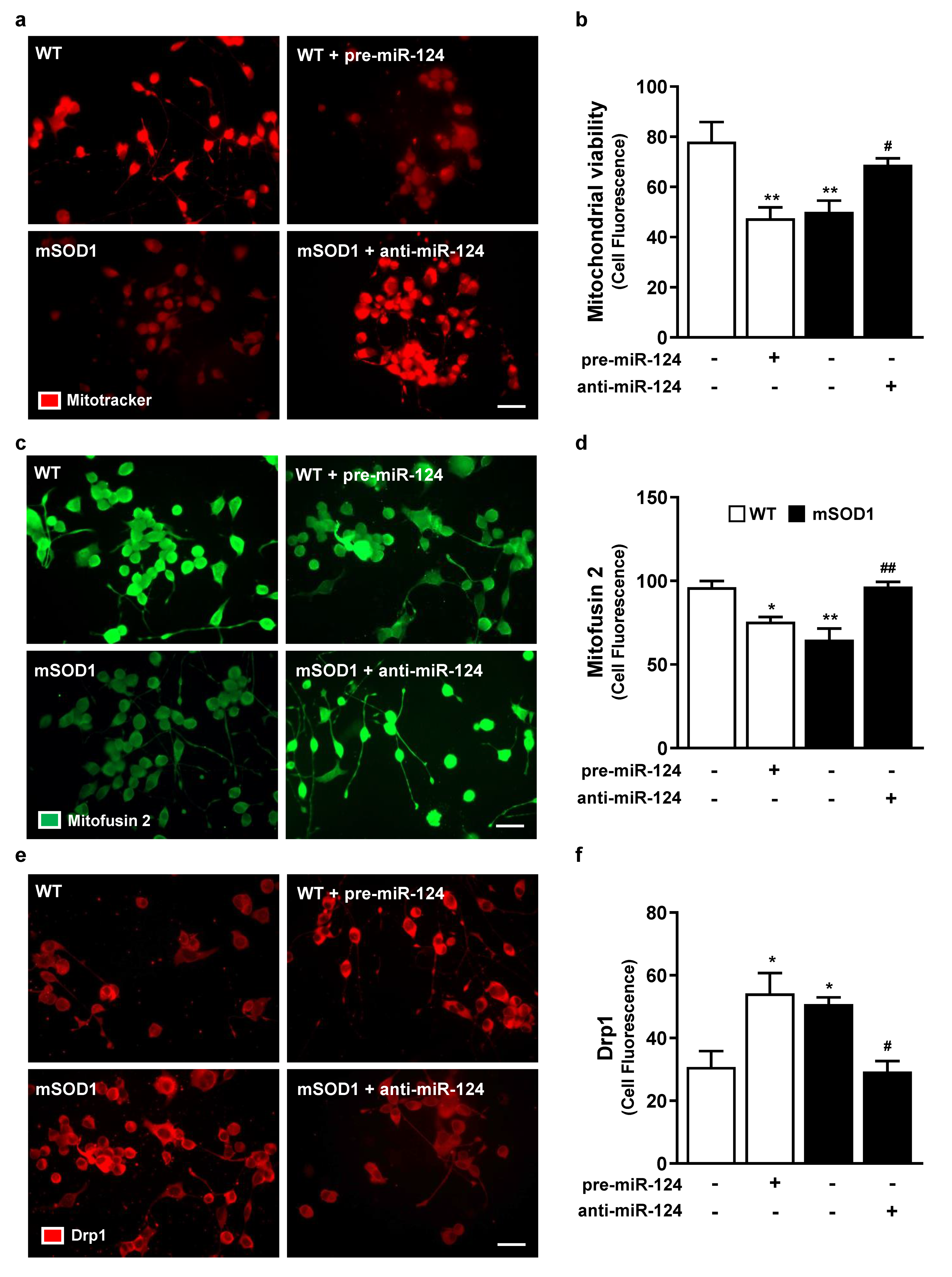
2.3. Impairment of Neurite Outgrowth, Axonal Transport, Synaptic Signaling, and Mitochondria Dynamics in miR-124-Enriched MNs Is Prevented when miR-124 Normal Levels Are Reestablished in mSOD1 MNs
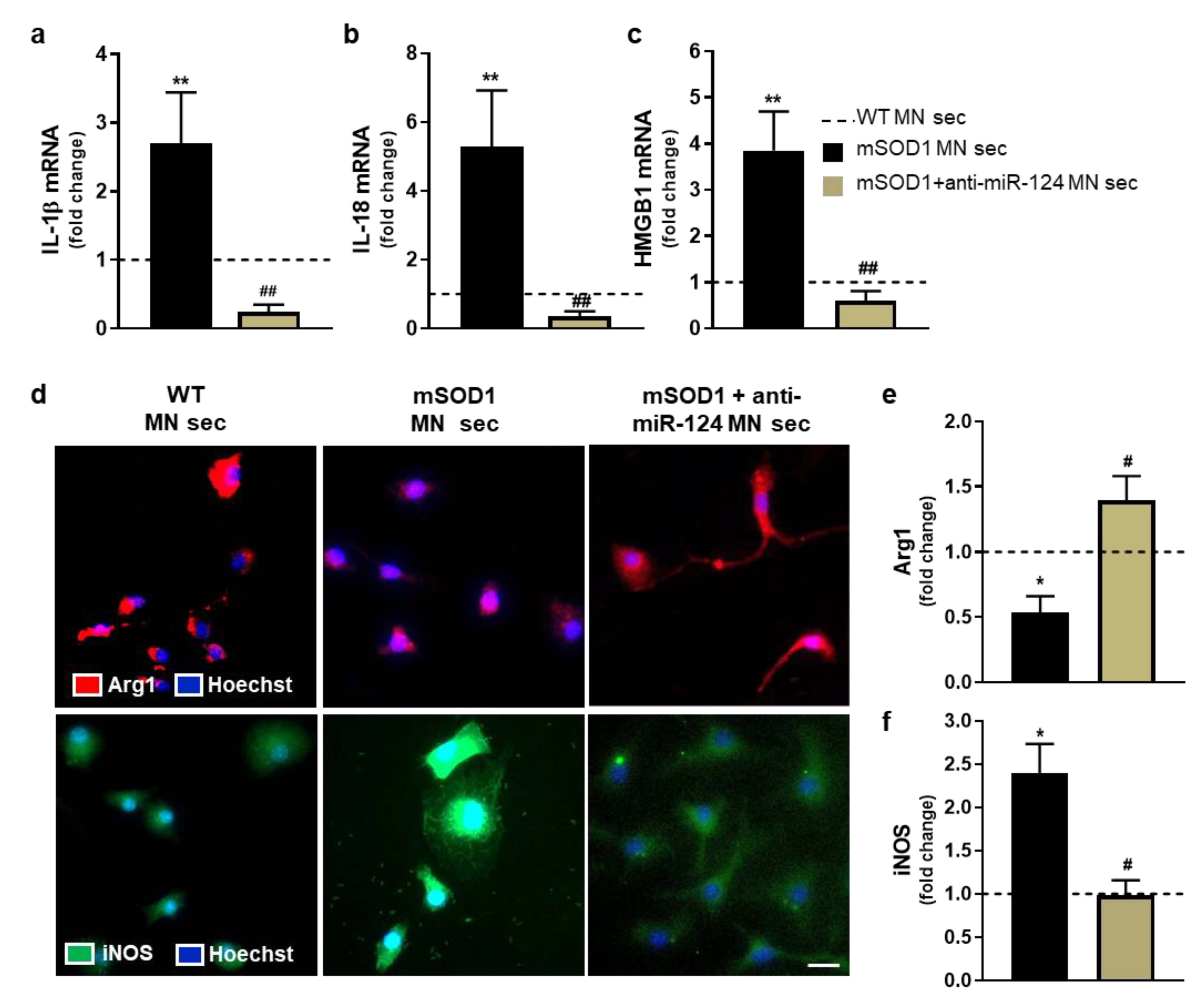
2.4. Microglia Polarization into a Pro-Inflammatory Phenotype by the Secretome from miR-124-Enriched mSOD1 MNs Is Prevented if the Mutated Cells Are Treated with Anti-miR-124
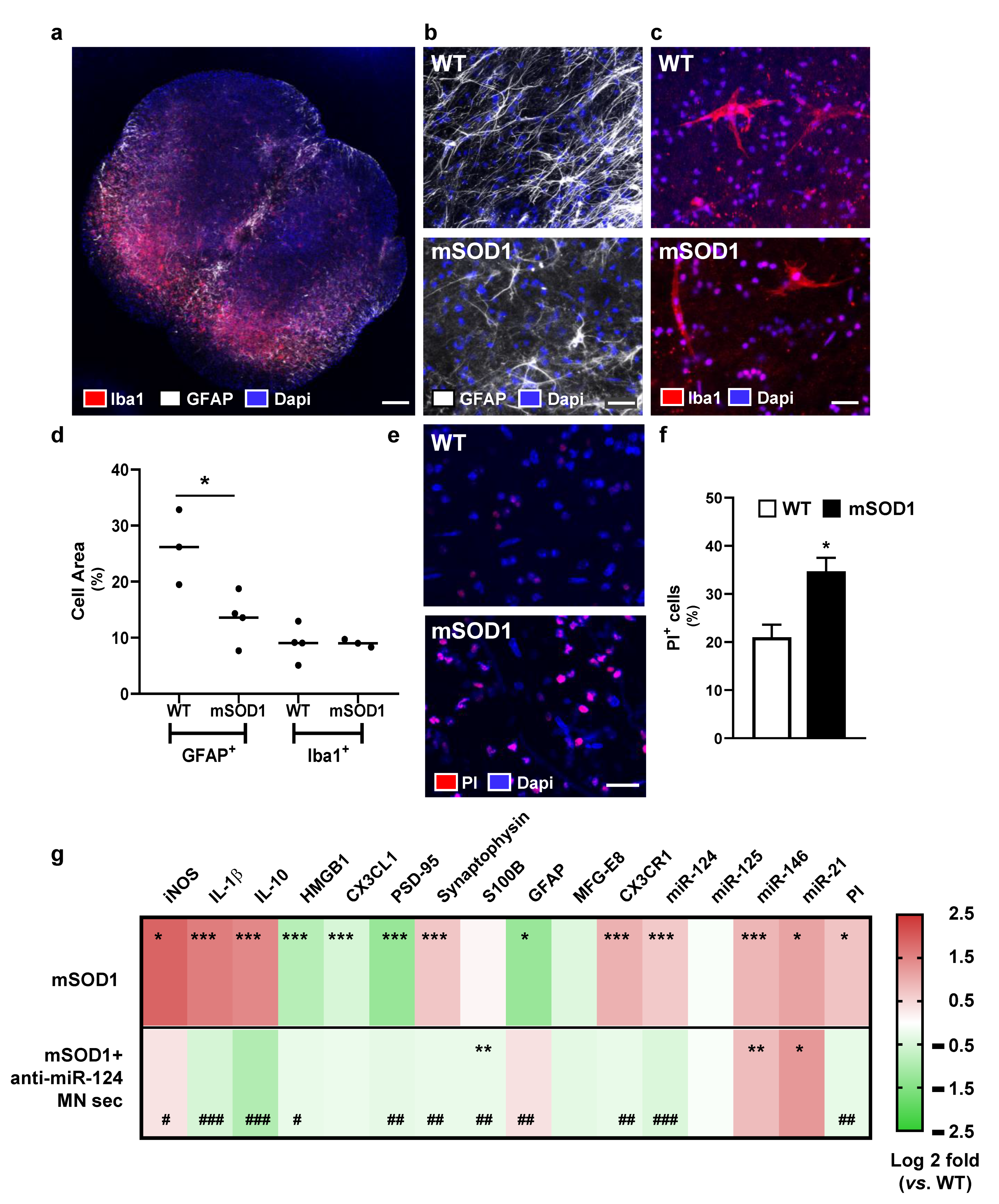
2.5. Pathogenicity of Spinal Organotypic Cultures from Early Symptomatic mSOD1 Mice Is Minimized upon Addition of Secretome from Anti-miR-124-Treated mSOD1 MNs
3. Discussion
4. Materials and Methods
4.1. Culture and Transfection of NSC-34 MN-Like Cell Line
4.2. Mice Models
4.2.1. Primary Spinal Microglia Cultures and Treatments
4.2.2. Spinal Cord Organotypic Cultures and Treatments
4.3. RT-qPCR
4.4. Cell Death by Nexin Assay
4.5. Immunocytochemistry
4.6. Immunohistochemistry
4.7. Mitochondrial Viability
4.8. Cell Death Determination by Propidium Iodide Uptake
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Khalid, S.I.; Ampie, L.; Kelly, R.; Ladha, S.S.; Dardis, C. Immune Modulation in the Treatment of Amyotrophic Lateral Sclerosis: A Review of Clinical Trials. Front. Neurol. 2017, 8, 486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brites, D.; Vaz, A.R. Microglia centered pathogenesis in ALS: Insights in cell interconnectivity. Front. Cell Neurosci. 2014, 8, 117. [Google Scholar] [CrossRef]
- Cunha, C.; Santos, C.; Gomes, C.; Fernandes, A.; Correia, A.M.; Sebastião, A.M.; Vaz, A.R.; Brites, D. Downregulated Glia Interplay and Increased miRNA-155 as Promising Markers to Track ALS at an Early Stage. Mol. Neurobiol. 2018, 55, 4207–4224. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.; Cunha, C.; Nascimento, F.; Ribeiro, J.A.; Vaz, A.R.; Brites, D. Cortical Neurotoxic Astrocytes with Early ALS Pathology and miR-146a Deficit Replicate Gliosis Markers of Symptomatic SOD1G93A Mouse Model. Mol. Neurobiol. 2019, 56, 2137–2158. [Google Scholar] [CrossRef] [PubMed]
- Alrafiah, A.R. From Mouse Models to Human Disease: An Approach for Amyotrophic Lateral Sclerosis. In Vivo 2018, 32, 983–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Bento-Abreu, A.; Van Damme, P.; Van Den Bosch, L.; Robberecht, W. The neurobiology of amyotrophic lateral sclerosis. Eur. J. Neurosci. 2010, 31, 2247–2265. [Google Scholar] [CrossRef]
- Van Damme, P.; Robberecht, W.; Van Den Bosch, L. Modelling amyotrophic lateral sclerosis: Progress and possibilities. Dis. Model Mech. 2017, 10, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Brites, D. Regulatory function of microRNAs in microglia. Glia 2020, 68, 1631–1642. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Cialic, R.; Krasemann, S.; Murugaiyan, G.; Fanek, Z.; Greco, D.J.; Wu, P.M.; Doykan, C.E.; Kiner, O.; et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann. Neurol. 2015, 77, 75–99. [Google Scholar] [CrossRef]
- Campos-Melo, D.; Droppelmann, C.A.; He, Z.; Volkening, K.; Strong, M.J. Altered microRNA expression profile in Amyotrophic Lateral Sclerosis: A role in the regulation of NFL mRNA levels. Mol. Brain 2013, 6, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foggin, S.; Mesquita-Ribeiro, R.; Dajas-Bailador, F.; Layfield, R. Biological Significance of microRNA Biomarkers in ALS-Innocent Bystanders or Disease Culprits? Front. Neurol. 2019, 10, 578. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Romero, C.; Hur, J.; Lunn, J.S.; Paez-Colasante, X.; Bender, D.E.; Yung, R.; Sakowski, S.A.; Feldman, E.L. Expression of microRNAs in human post-mortem amyotrophic lateral sclerosis spinal cords provides insight into disease mechanisms. Mol. Cell Neurosci. 2016, 71, 34–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Luo, Z.M.; Guo, X.M.; Su, D.F.; Liu, X. An updated role of microRNA-124 in central nervous system disorders: A review. Front. Cell Neurosci. 2015, 9, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neo, W.H.; Yap, K.; Lee, S.H.; Looi, L.S.; Khandelia, P.; Neo, S.X.; Makeyev, E.V.; Su, I.H. MicroRNA miR-124 controls the choice between neuronal and astrocyte differentiation by fine-tuning Ezh2 expression. J. Biol. Chem. 2014, 289, 20788–20801. [Google Scholar] [CrossRef] [Green Version]
- Ponomarev, E.D.; Veremeyko, T.; Barteneva, N.; Krichevsky, A.M.; Weiner, H.L. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-alpha-PU.1 pathway. Nat. Med. 2011, 17, 64–70. [Google Scholar] [CrossRef]
- Ponomarev, E.D.; Veremeyko, T.; Weiner, H.L. MicroRNAs are universal regulators of differentiation, activation, and polarization of microglia and macrophages in normal and diseased CNS. Glia 2013, 61, 91–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morel, L.; Regan, M.; Higashimori, H.; Ng, S.K.; Esau, C.; Vidensky, S.; Rothstein, J.; Yang, Y. Neuronal exosomal miRNA-dependent translational regulation of astroglial glutamate transporter GLT1. J. Biol. Chem. 2013, 288, 7105–7116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Zhang, C.; Guan, Y.; Chen, Y.; Lu, Q.; Jie, L.; Gao, H.; Du, H.; Zhang, H.; Liu, Y.; et al. Screening the expression characteristics of several miRNAs in G93A-SOD1 transgenic mouse: Altered expression of miRNA-124 is associated with astrocyte differentiation by targeting Sox2 and Sox9. J. Neurochem. 2018, 145, 51–67. [Google Scholar] [CrossRef] [Green Version]
- Yardeni, T.; Fine, R.; Joshi, Y.; Gradus-Pery, T.; Kozer, N.; Reichenstein, I.; Yanowski, E.; Nevo, S.; Weiss-Tishler, H.; Eisenberg-Bord, M.; et al. High content image analysis reveals function of miR-124 upstream of Vimentin in regulating motor neuron mitochondria. Sci. Rep. 2018, 8, 59. [Google Scholar] [CrossRef] [Green Version]
- Marcuzzo, S.; Bonanno, S.; Kapetis, D.; Barzago, C.; Cavalcante, P.; D’Alessandro, S.; Mantegazza, R.; Bernasconi, P. Up-regulation of neural and cell cycle-related microRNAs in brain of amyotrophic lateral sclerosis mice at late disease stage. Mol. Brain 2015, 8, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parisi, C.; Arisi, I.; D’Ambrosi, N.; Storti, A.E.; Brandi, R.; D’Onofrio, M.; Volonte, C. Dysregulated microRNAs in amyotrophic lateral sclerosis microglia modulate genes linked to neuroinflammation. Cell Death Dis. 2013, 4, e959. [Google Scholar] [CrossRef]
- Waller, R.; Wyles, M.; Heath, P.R.; Kazoka, M.; Wollff, H.; Shaw, P.J.; Kirby, J. Small RNA Sequencing of Sporadic Amyotrophic Lateral Sclerosis Cerebrospinal Fluid Reveals Differentially Expressed miRNAs Related to Neural and Glial Activity. Front. Neurosci. 2017, 11, 731. [Google Scholar] [CrossRef] [PubMed]
- Yelick, J.; Men, Y.; Jin, S.; Seo, S.; Espejo-Porras, F.; Yang, Y. Elevated exosomal secretion of miR-124–3p from spinal neurons positively associates with disease severity in ALS. Exp. Neurol. 2020, 333, 113414. [Google Scholar] [CrossRef] [PubMed]
- Pinto, S.; Cunha, C.; Barbosa, M.; Vaz, A.R.; Brites, D. Exosomes from NSC-34 Cells Transfected with hSOD1-G93A Are Enriched in miR-124 and Drive Alterations in Microglia Phenotype. Front. Neurosci. 2017, 11, 273. [Google Scholar] [CrossRef] [Green Version]
- Vaz, A.R.; Cunha, C.; Gomes, C.; Schmucki, N.; Barbosa, M.; Brites, D. Glycoursodeoxycholic acid reduces matrix metalloproteinase-9 and caspase-9 activation in a cellular model of superoxide dismutase-1 neurodegeneration. Mol. Neurobiol. 2015, 51, 864–877. [Google Scholar] [CrossRef]
- Caldeira, C.; Oliveira, A.F.; Cunha, C.; Vaz, A.R.; Falcão, A.S.; Fernandes, A.; Brites, D. Microglia change from a reactive to an age-like phenotype with the time in culture. Front. Cell Neurosci. 2014, 8, 152. [Google Scholar] [CrossRef]
- Berthod, F.; Gros-Louis, F. In Vivo and In Vitro Models to Study Amyotrophic Lateral Sclerosis. In Amyotrophic Lateral Sclerosis (ALS); Maurer, M.H., Ed.; IntechOpen: London, UK, 2012; Chapter 4. [Google Scholar]
- Hayden, P.J.; Harbell, J.W. Special review series on 3D organotypic culture models: Introduction and historical perspective. Vitro Cell Dev. Biol. Anim. 2021, 57, 95–103. [Google Scholar] [CrossRef]
- Yu, J.Y.; Chung, K.H.; Deo, M.; Thompson, R.C.; Turner, D.L. MicroRNA miR-124 regulates neurite outgrowth during neuronal differentiation. Exp. Cell Res. 2008, 314, 2618–2633. [Google Scholar] [CrossRef] [Green Version]
- Han, D.; Dong, X.; Zheng, D.; Nao, J. MiR-124 and the Underlying Therapeutic Promise of Neurodegenerative Disorders. Front. Pharmacol. 2019, 10, 1555. [Google Scholar] [CrossRef]
- Kim, K.Y.; Kim, Y.R.; Choi, K.W.; Lee, M.; Lee, S.; Im, W.; Shin, J.Y.; Kim, J.Y.; Hong, Y.H.; Kim, M.; et al. Downregulated miR-18b-5p triggers apoptosis by inhibition of calcium signaling and neuronal cell differentiation in transgenic SOD1 (G93A) mice and SOD1 (G17S and G86S) ALS patients. Transl. Neurodegener. 2020, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Cho, G.W.; Kim, G.Y.; Baek, S.; Kim, H.; Kim, T.; Kim, H.J.; Kim, S.H. Recombinant human erythropoietin reduces aggregation of mutant Cu/Zn-binding superoxide dismutase (SOD1) in NSC-34 cells. Neurosci. Lett. 2011, 504, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.; Ruan, H.; Gilbert, J.; Wang, G.; Ma, Q.; Yao, W.D.; Man, H.Y. MicroRNA miR124 is required for the expression of homeostatic synaptic plasticity. Nat. Commun. 2015, 6, 10045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Z.; Wang, P.Y.; Su, D.F.; Liu, X. miRNA-124 in Immune System and Immune Disorders. Front. Immunol. 2016, 7, 406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, X.T.; Lei, P.; Wang, H.C.; Zhang, A.L.; Han, Z.L.; Chen, X.; Li, S.H.; Jiang, R.C.; Kang, C.S.; Zhang, J.N. miR-21 improves the neurological outcome after traumatic brain injury in rats. Sci. Rep. 2014, 4, 6718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz-Lasso, D.C.; Roma-Mateo, C.; Pallardo, F.V.; Gonzalez-Cabo, P. Much More Than a Scaffold: Cytoskeletal Proteins in Neurological Disorders. Cells 2020, 9, 358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruangjaroon, T.; Chokchaichamnankit, D.; Srisomsap, C.; Svasti, J.; Paricharttanakul, N.M. Involvement of vimentin in neurite outgrowth damage induced by fipronil in SH-SY5Y cells. Biochem. Biophys. Res. Commun. 2017, 486, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [PubMed] [Green Version]
- Liu, W.; Yamashita, T.; Tian, F.; Morimoto, N.; Ikeda, Y.; Deguchi, K.; Abe, K. Mitochondrial fusion and fission proteins expression dynamically change in a murine model of amyotrophic lateral sclerosis. Curr. Neurovasc. Res. 2013, 10, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Liu, Y.J.; McIntyre, R.L.; Janssens, G.E.; Houtkooper, R.H. Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech. Ageing Dev. 2020, 186, 111212. [Google Scholar] [CrossRef]
- Barbosa, M.; Gomes, C.; Sequeira, C.; Goncalves-Ribeiro, J.; Pina, C.C.; Carvalho, L.A.; Moreira, R.; Vaz, S.H.; Vaz, A.R.; Brites, D. Recovery of Depleted miR-146a in ALS Cortical Astrocytes Reverts Cell Aberrancies and Prevents Paracrine Pathogenicity on Microglia and Motor Neurons. Front. Cell Dev. Biol. 2021, 9, 634355. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.J.; Talbot, K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog. Neurobiol. 2008, 85, 94–134. [Google Scholar] [CrossRef]
- Cavaliere, F.; Benito-Munoz, M.; Matute, C. Organotypic Cultures as a Model to Study Adult Neurogenesis in CNS Disorders. Stem. Cells Int. 2016, 2016, 3540568. [Google Scholar] [CrossRef] [Green Version]
- Lossi, L.; Merighi, A. The Use of ex Vivo Rodent Platforms in Neuroscience Translational Research With Attention to the 3Rs Philosophy. Front. Vet. Sci. 2018, 5, 164. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol. 2020, 41, 758–770. [Google Scholar] [CrossRef]
- Hopperton, K.E.; Mohammad, D.; Trepanier, M.O.; Giuliano, V.; Bazinet, R.P. Markers of microglia in post-mortem brain samples from patients with Alzheimer’s disease: A systematic review. Mol. Psychiatry 2018, 23, 177–198. [Google Scholar] [CrossRef]
- Díaz-Amarilla, P.; Olivera-Bravo, S.; Trias, E.; Cragnolini, A.; Martinez-Palma, L.; Cassina, P.; Beckman, J.; Barbeito, L. Phenotypically aberrant astrocytes that promote motoneuron damage in a model of inherited amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2011, 108, 18126–18131. [Google Scholar] [CrossRef] [Green Version]
- Staal, J.A.; Alexander, S.R.; Liu, Y.; Dickson, T.D.; Vickers, J.C. Characterization of cortical neuronal and glial alterations during culture of organotypic whole brain slices from neonatal and mature mice. PLoS ONE 2011, 6, e22040. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Prieto, M.; Gutierrez, I.L.; Garcia-Bueno, B.; Caso, J.R.; Leza, J.C.; Ortega-Hernandez, A.; Gomez-Garre, D.; Madrigal, J.L.M. MicroglialCX3CR1production increases in Alzheimer’s disease and is regulated by noradrenaline. Glia 2021, 69, 73–90. [Google Scholar] [CrossRef]
- Gomes, C.; Sequeira, C.; Barbosa, M.; Cunha, C.; Vaz, A.R.; Brites, D. Astrocyte regional diversity in ALS includes distinct aberrant phenotypes with common and causal pathological processes. Exp. Cell Res. 2020, 395, 112209. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 2002, 12, 735–739. [Google Scholar] [CrossRef] [Green Version]
- Krichevsky, A.M.; King, K.S.; Donahue, C.P.; Khrapko, K.; Kosik, K.S. A microRNA array reveals extensive regulation of microRNAs during brain development. RNA 2003, 9, 1274–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghafouri-Fard, S.; Shoorei, H.; Bahroudi, Z.; Abak, A.; Majidpoor, J.; Taheri, M. An update on the role of miR-124 in the pathogenesis of human disorders. Biomed. Pharmacother. 2021, 135, 111198. [Google Scholar] [CrossRef]
- Zhao, Y.; Yan, M.; Chen, C.; Gong, W.; Yin, Z.; Li, H.; Fan, J.; Zhang, X.A.; Wang, D.W.; Zuo, H. MiR-124 aggravates failing hearts by suppressing CD151-facilitated angiogenesis in heart. Oncotarget 2018, 9, 14382–14396. [Google Scholar] [CrossRef] [PubMed]
- Vuokila, N.; Lukasiuk, K.; Bot, A.M.; van Vliet, E.A.; Aronica, E.; Pitkanen, A.; Puhakka, N. miR-124–3p is a chronic regulator of gene expression after brain injury. Cell Mol. Life Sci. 2018, 75, 4557–4581. [Google Scholar] [CrossRef]
- An, F.M.; Gong, G.H.; Wang, Y.; Bian, M.; Yu, L.J.; Wei, C.X. MiR-124 acts as a target for Alzheimer’s disease by regulating BACE1. Oncotarget 2017, 8, 114065–114071. [Google Scholar] [CrossRef] [Green Version]
- Bell, E.; Taylor, M.A. Functional Roles for Exosomal MicroRNAs in the Tumour Microenvironment. Comput. Struct. Biotechnol. J. 2017, 15, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Diaz Quiroz, J.F.; Tsai, E.; Coyle, M.; Sehm, T.; Echeverri, K. Precise control of miR-125b levels is required to create a regeneration-permissive environment after spinal cord injury: A cross-species comparison between salamander and rat. Dis. Model Mech. 2014, 7, 601–611. [Google Scholar] [CrossRef] [Green Version]
- Edbauer, D.; Neilson, J.R.; Foster, K.A.; Wang, C.F.; Seeburg, D.P.; Batterton, M.N.; Tada, T.; Dolan, B.M.; Sharp, P.A.; Sheng, M. Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron 2010, 65, 373–384. [Google Scholar] [CrossRef] [Green Version]
- Harrison, E.B.; Hochfelder, C.G.; Lamberty, B.G.; Meays, B.M.; Morsey, B.M.; Kelso, M.L.; Fox, H.S.; Yelamanchili, S.V. Traumatic brain injury increases levels of miR-21 in extracellular vesicles: Implications for neuroinflammation. FEBS Open Bio 2016, 6, 835–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, A.; Ribeiro, A.R.; Monteiro, M.; Garcia, G.; Vaz, A.R.; Brites, D. Secretome from SH-SY5Y APP Swe cells trigger time-dependent CHME3 microglia activation phenotypes, ultimately leading to miR-21 exosome shuttling. Biochimie 2018, 155, 67–82. [Google Scholar] [CrossRef]
- Strickland, I.T.; Richards, L.; Holmes, F.E.; Wynick, D.; Uney, J.B.; Wong, L.F. Axotomy-induced miR-21 promotes axon growth in adult dorsal root ganglion neurons. PLoS ONE 2011, 6, e23423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Q.; Yu, C.; Wang, Y.; Liu, L.; Zhang, K.; Fang, C.; Liu, F.; Bian, G.; Song, B.; Yang, A.; et al. miR-9 and miR-124 synergistically affect regulation of dendritic branching via the AKT/GSK3beta pathway by targeting Rap2a. Sci. Rep. 2016, 6, 26781. [Google Scholar] [CrossRef] [PubMed]
- Osking, Z.; Ayers, J.I.; Hildebrandt, R.; Skruber, K.; Brown, H.; Ryu, D.; Eukovich, A.R.; Golde, T.E.; Borchelt, D.R.; Read, T.A.; et al. ALS-Linked SOD1 Mutants Enhance Neurite Outgrowth and Branching in Adult Motor Neurons. iScience 2019, 19, 448–449. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Huang, Y.; Wang, L.-L.; Zhang, Y.-F.; Xu, J.; Zhou, Y.; Lourenco, G.F.; Zhang, B.; Wang, Y.; Ren, R.-J.; et al. MicroRNA-146a suppresses ROCK1 allowing hyperphosphorylation of tau in Alzheimer’s disease. Sci. Rep. 2016, 6, 26697. [Google Scholar] [CrossRef] [PubMed]
- Gatto, R.G.; Amin, M.Y.; Deyoung, D.; Hey, M.; Mareci, T.H.; Magin, R.L. Ultra-High Field Diffusion MRI Reveals Early Axonal Pathology in Spinal Cord of ALS mice. Transl. Neurodegener. 2018, 7, 20. [Google Scholar] [CrossRef]
- Zayia, L.C.; Tadi, P. Neuroanatomy, Motor Neuron. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Nagata, K.; Hama, I.; Kiryu-Seo, S.; Kiyama, H. microRNA-124 is down regulated in nerve-injured motor neurons and it potentially targets mRNAs for KLF6 and STAT3. Neuroscience 2014, 256, 426–432. [Google Scholar] [CrossRef] [Green Version]
- Hawley, Z.C.E.; Campos-Melo, D.; Droppelmann, C.A.; Strong, M.J. MotomiRs: miRNAs in Motor Neuron Function and Disease. Front. Mol. Neurosci. 2017, 10, 127. [Google Scholar] [CrossRef]
- Genç, B.; Jara, J.H.; Lagrimas, A.K.; Pytel, P.; Roos, R.P.; Mesulam, M.M.; Geula, C.; Bigio, E.H.; Ozdinler, P.H. Apical dendrite degeneration, a novel cellular pathology for Betz cells in ALS. Sci. Rep. 2017, 7, 41765. [Google Scholar] [CrossRef] [Green Version]
- De Vos, K.J.; Hafezparast, M. Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research? Neurobiol. Dis. 2017, 105, 283–299. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef]
- Warita, H.; Itoyama, Y.; Abe, K. Selective impairment of fast anterograde axonal transport in the peripheral nerves of asymptomatic transgenic mice with a G93A mutant SOD1 gene. Brain Res. 1999, 819, 120–131. [Google Scholar] [CrossRef]
- De Vos, K.J.; Chapman, A.L.; Tennant, M.E.; Manser, C.; Tudor, E.L.; Lau, K.F.; Brownlees, J.; Ackerley, S.; Shaw, P.J.; McLoughlin, D.M.; et al. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 2007, 16, 2720–2728. [Google Scholar] [CrossRef]
- Delic, V.; Kurien, C.; Cruz, J.; Zivkovic, S.; Barretta, J.; Thomson, A.; Hennessey, D.; Joseph, J.; Ehrhart, J.; Willing, A.E.; et al. Discrete mitochondrial aberrations in the spinal cord of sporadic ALS patients. J. Neurosci. Res. 2018, 96, 1353–1366. [Google Scholar] [CrossRef] [PubMed]
- Onesto, E.; Colombrita, C.; Gumina, V.; Borghi, M.O.; Dusi, S.; Doretti, A.; Fagiolari, G.; Invernizzi, F.; Moggio, M.; Tiranti, V.; et al. Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol. Commun. 2016, 4, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magrane, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, A.U.; Saw, N.L.; Vogel, H.; Cunnigham, A.D.; Shamloo, M.; Mochly-Rosen, D. Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol. Med. 2018, 10, e8166. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Yan, L.; Jiang, X.; Yu, Y.; Liu, H.; Gu, T.; Shi, E. Acquired inhibition of microRNA-124 protects against spinal cord ischemia-reperfusion injury partially through a mitophagy-dependent pathway. J. Thorac. Cardiovasc. Surg. 2017, 154, 1498–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veremeyko, T.; Kuznetsova, I.S.; Dukhinova, M.; Yung, A.W.Y.; Kopeikina, E.; Barteneva, N.S.; Ponomarev, E.D. Neuronal extracellular microRNAs miR-124 and miR-9 mediate cell-cell communication between neurons and microglia. J. Neurosci. Res. 2019, 97, 162–184. [Google Scholar] [CrossRef] [Green Version]
- Zullo, J.; Matsumoto, K.; Xavier, S.; Ratliff, B.; Goligorsky, M.S. The cell secretome, a mediator of cell-to-cell communication. Prostaglandins Lipid Mediat. 2015, 120, 17–20. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.; Kaspar, B.K. Glia-neuron interactions in neurological diseases: Testing non-cell autonomy in a dish. Brain Res. 2017, 1656, 27–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaz, A.R.; Pinto, S.; Ezequiel, C.; Cunha, C.; Carvalho, L.A.; Moreira, R.; Brites, D. Phenotypic Effects of Wild-Type and Mutant SOD1 Expression in N9 Murine Microglia at Steady State, Inflammatory and Immunomodulatory Conditions. Front. Cell Neurosci. 2019, 13, 109. [Google Scholar] [CrossRef] [Green Version]
- Gugliandolo, A.; Giacoppo, S.; Bramanti, P.; Mazzon, E. NLRP3 Inflammasome Activation in a Transgenic Amyotrophic Lateral Sclerosis Model. Inflammation 2018, 41, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Franklin, T.C.; Wohleb, E.S.; Zhang, Y.; Fogaca, M.; Hare, B.; Duman, R.S. Persistent Increase in Microglial RAGE Contributes to Chronic Stress-Induced Priming of Depressive-like Behavior. Biol. Psychiatry. 2018, 83, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Guzmán-Lenis, M.S.; Navarro, X.; Casas, C. Drug screening of neuroprotective agents on an organotypic-based model of spinal cord excitotoxic damage. Restor. Neurol. Neurosci. 2009, 27, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, Y.; Liu, X.; Li, S.; Cheng, C.; Chen, S.; Le, W. Dynamic changes of CX3CL1/CX3CR1 axis during microglial activation and motor neuron loss in the spinal cord of ALS mouse model. Transl. Neurodegener. 2018, 7, 35. [Google Scholar] [CrossRef]
- Slota, J.A.; Booth, S.A. MicroRNAs in Neuroinflammation: Implications in Disease Pathogenesis, Biomarker Discovery and Therapeutic Applications. Noncoding RNA 2019, 5, 35. [Google Scholar] [CrossRef] [Green Version]
- Saura, J.; Tusell, J.M.; Serratosa, J. High-yield isolation of murine microglia by mild trypsinization. Glia 2003, 44, 183–189. [Google Scholar] [CrossRef]
- Lossi, L.; Alasia, S.; Salio, C.; Merighi, A. Cell death and proliferation in acute slices and organotypic cultures of mammalian CNS. Prog. Neurobiol. 2009, 88, 221–245. [Google Scholar] [CrossRef] [PubMed]
- Vaz, A.R.; Falcão, A.S.; Scarpa, E.; Semproni, C.; Brites, D. Microglia Susceptibility to Free Bilirubin Is Age-Dependent. Front. Pharmacol. 2020, 11, 1012. [Google Scholar] [CrossRef] [PubMed]
- Popko, J.; Fernandes, A.; Brites, D.; Lanier, L.M. Automated analysis of NeuronJ tracing data. Cytometry A 2009, 75, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.L.; Vaz, A.R.; Diógenes, M.J.; van Rooijen, N.; Sebastião, A.M.; Fernandes, A.; Silva, R.F.; Brites, D. Neuritic growth impairment and cell death by unconjugated bilirubin is mediated by NO and glutamate, modulated by microglia, and prevented by glycoursodeoxycholic acid and interleukin-10. Neuropharmacology 2012, 62, 2398–2408. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaz, A.R.; Vizinha, D.; Morais, H.; Colaço, A.R.; Loch-Neckel, G.; Barbosa, M.; Brites, D. Overexpression of miR-124 in Motor Neurons Plays a Key Role in ALS Pathological Processes. Int. J. Mol. Sci. 2021, 22, 6128. https://doi.org/10.3390/ijms22116128
Vaz AR, Vizinha D, Morais H, Colaço AR, Loch-Neckel G, Barbosa M, Brites D. Overexpression of miR-124 in Motor Neurons Plays a Key Role in ALS Pathological Processes. International Journal of Molecular Sciences. 2021; 22(11):6128. https://doi.org/10.3390/ijms22116128
Chicago/Turabian StyleVaz, Ana Rita, Daniela Vizinha, Hermes Morais, Ana Rita Colaço, Gecioni Loch-Neckel, Marta Barbosa, and Dora Brites. 2021. "Overexpression of miR-124 in Motor Neurons Plays a Key Role in ALS Pathological Processes" International Journal of Molecular Sciences 22, no. 11: 6128. https://doi.org/10.3390/ijms22116128