NF-Y Overexpression in Liver Hepatocellular Carcinoma (HCC)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
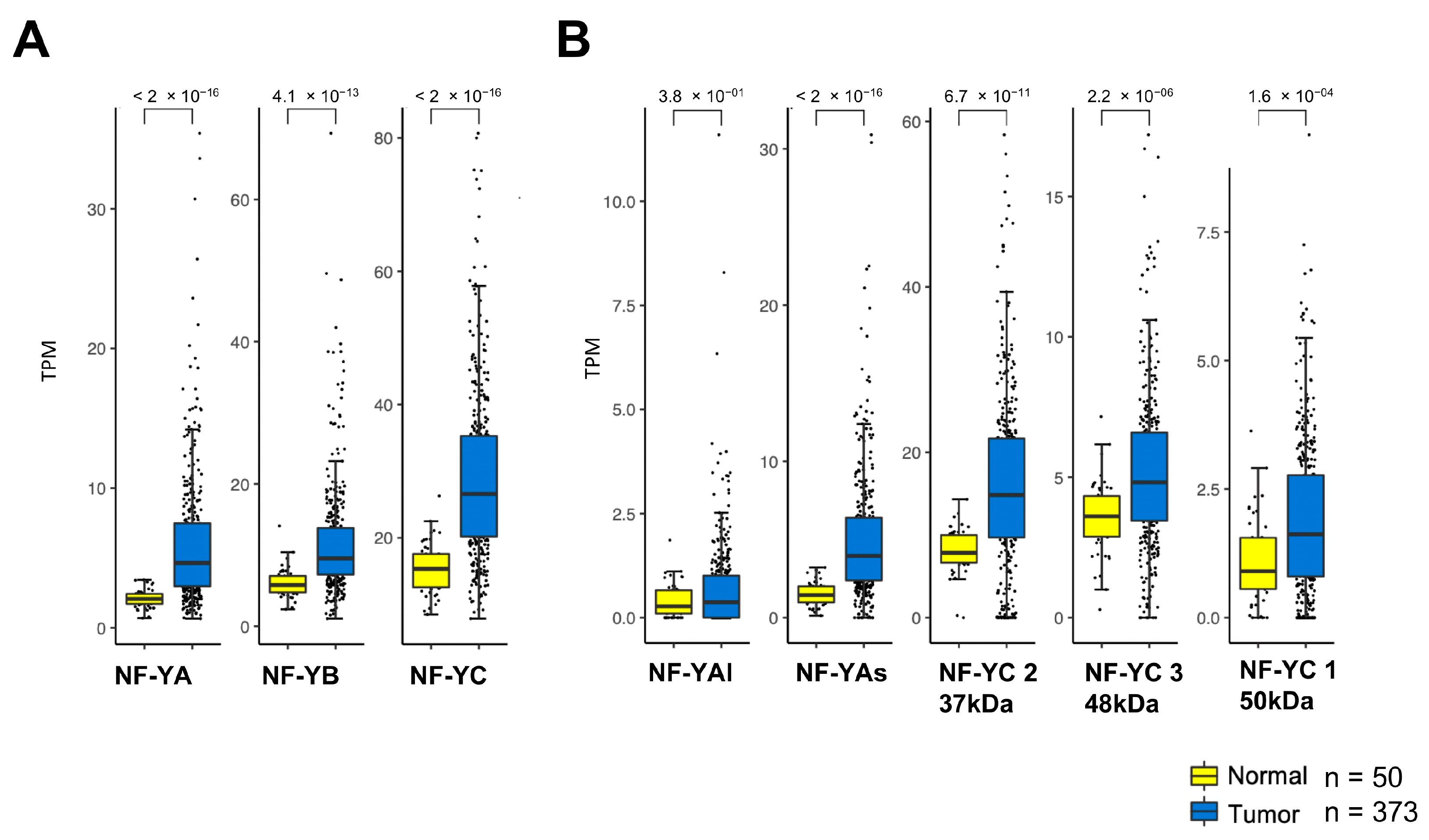
2.1. NF-Y Subunits Are Overexpressed in HCC
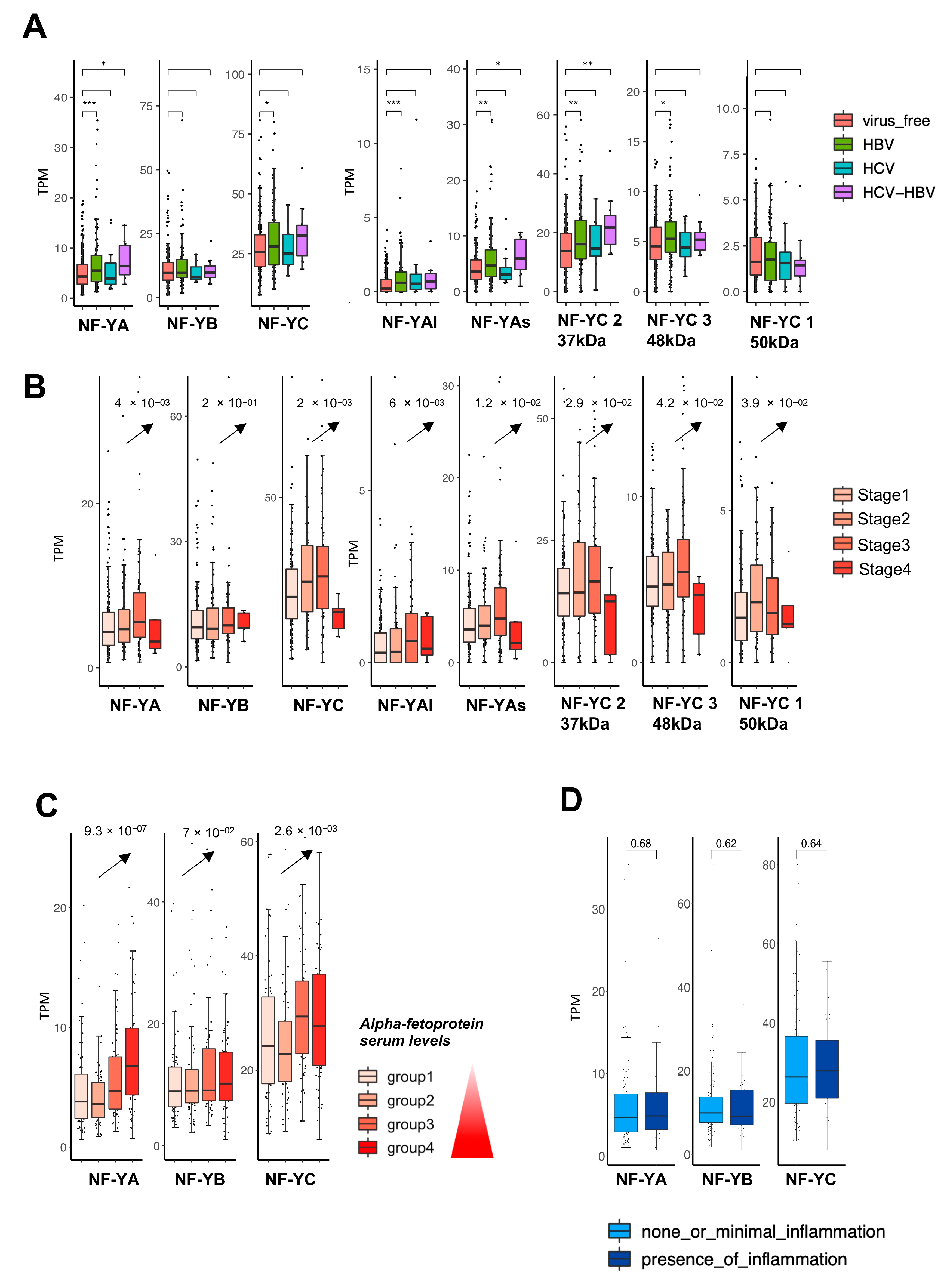
2.2. Expression of NF-Y Isoforms According to Viral Infection, Stage and Inflammation of HCC
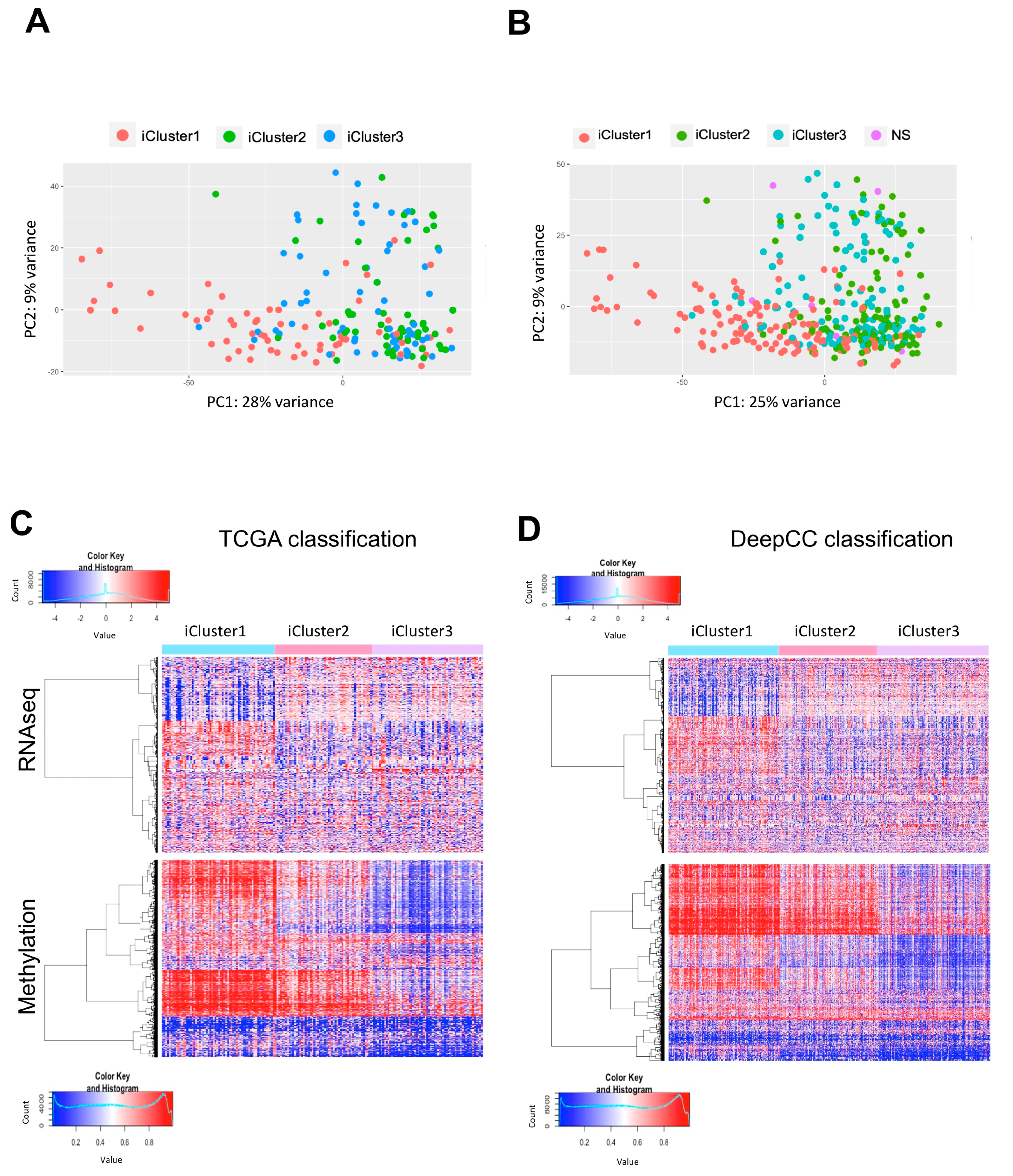
2.3. Partitioning of All TCGA HCC in iClusters According to DeepCC
2.4. Expression of NF-Y Subunits in iClusters
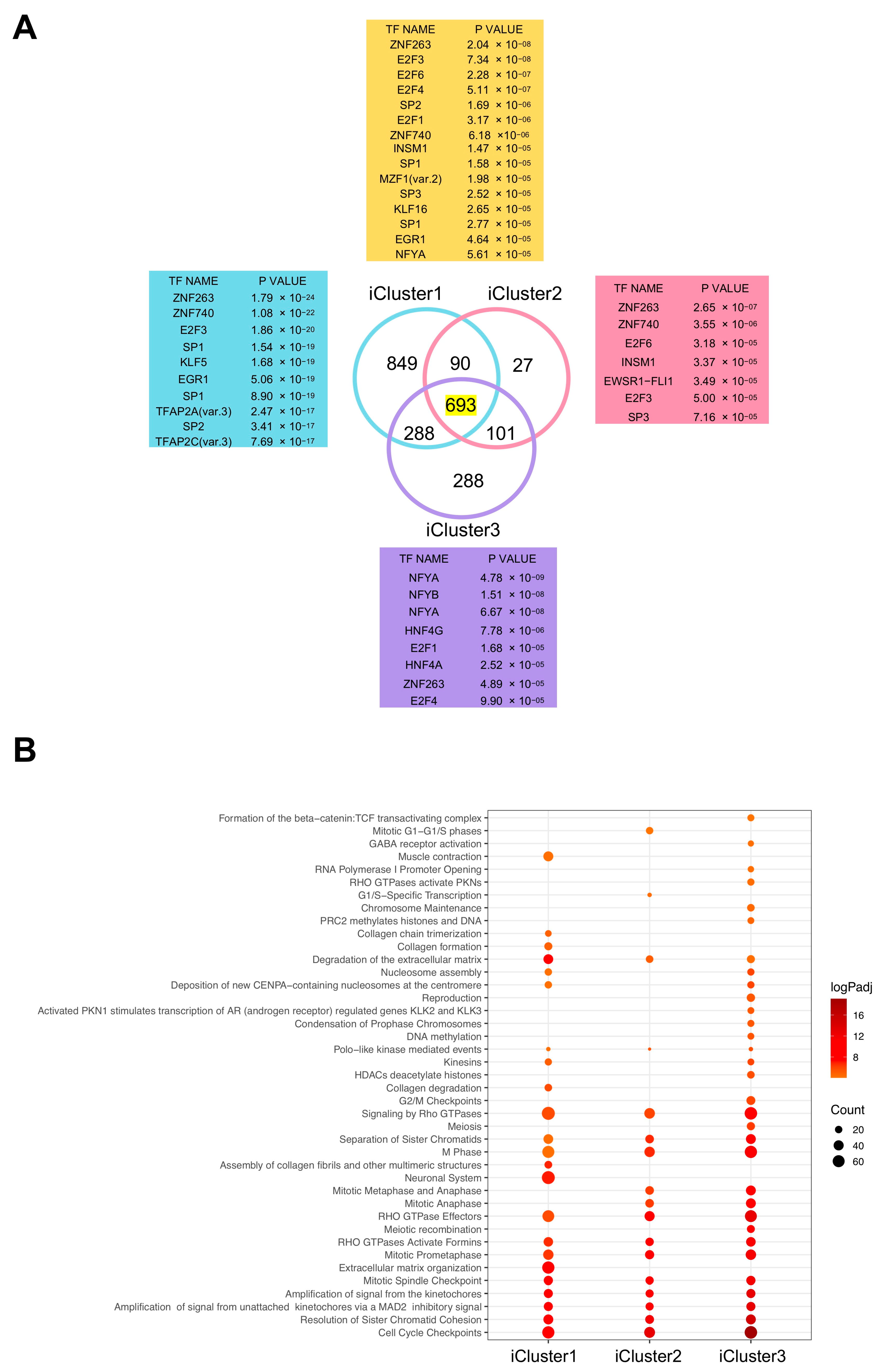
2.5. iCluster3 Differentially Expressed Genes (DEG) Have CCAAT in Promoters
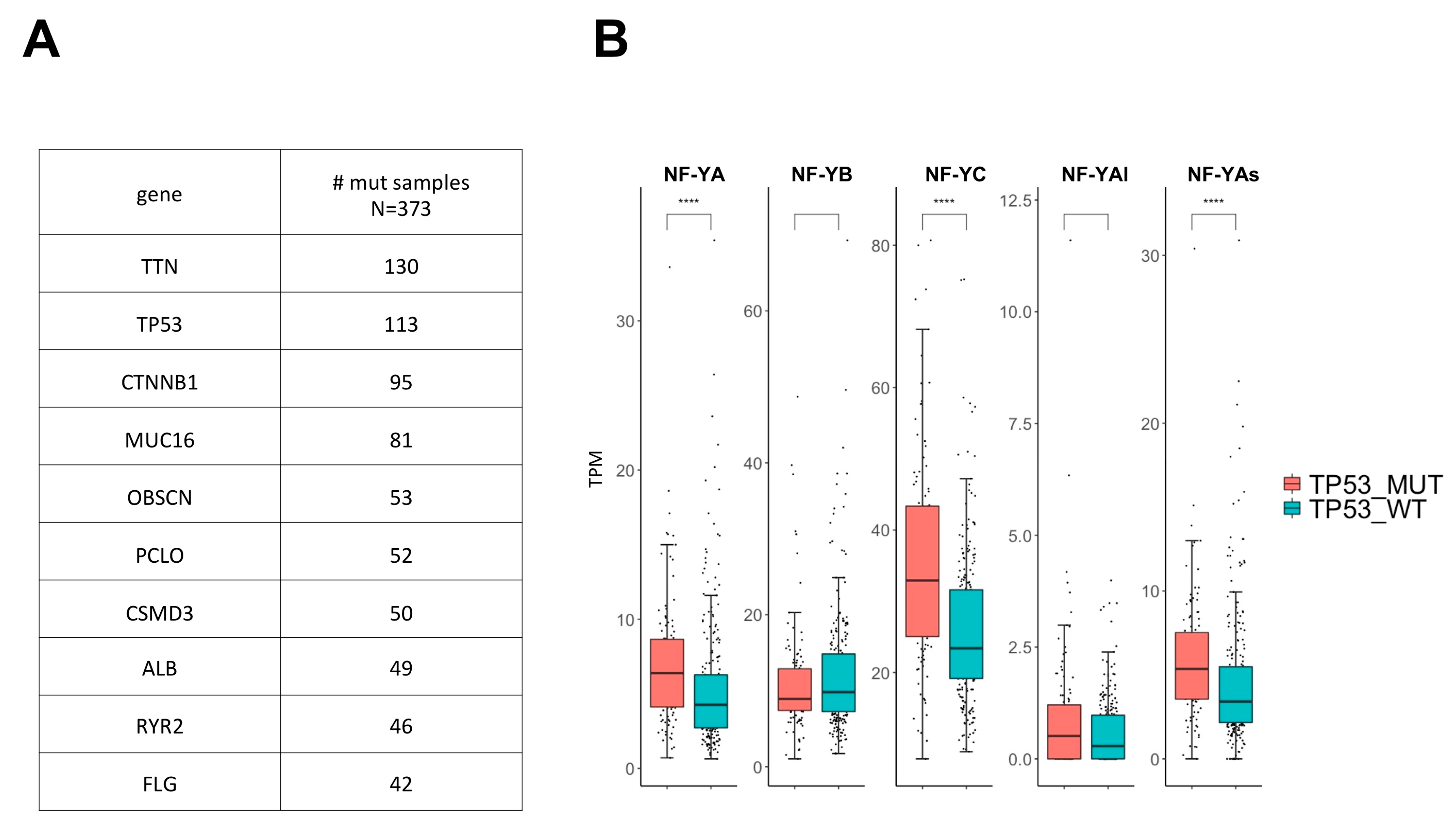
2.6. Analysis of NF-Y Subunit Levels in p53-Mutated HCC
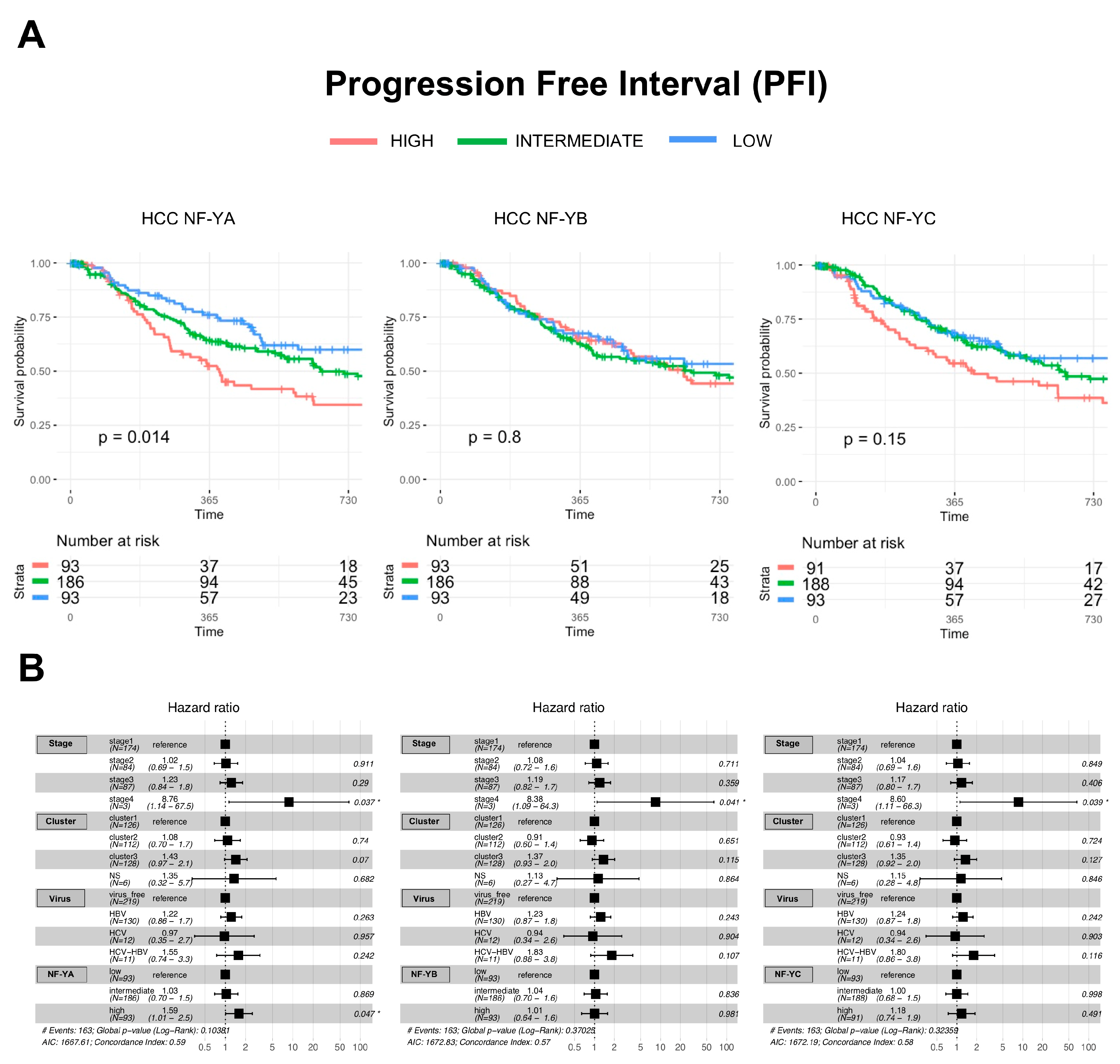
2.7. Clinical Outcomes of NF-Y Overexpression in HCC
3. Discussion
4. Materials and Methods
4.1. TCGA HCC Data
4.2. Classification of All TCGA HCC Tumors
4.3. Global Gene Expression Analysis
4.4. Gene Ontology, Pathway Enrichment and Transcription Factor Binding Site Analysis
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TCGA | The Cancer Genome Atlas |
| NF-YAl | Nuclear Factor Y subunit A isoform long |
| NF-YAs | Nuclear Factor Y subunit A isoform short |
| NF-YB | Nuclear Factor Y subunit B |
| NF-YC | Nuclear factor Y subunit C |
| E2F | E2 Factor |
| KLF | Kruppel-Like Factor |
| TSS | Transcriptional Start Site |
| TF | Transcription Factor |
| TFBS | Transcription Factors Binding Sites |
| FDR | False Discovery Rate |
| EMT | Epithelial to Mesenchymal Transition |
| HFD | Histone Fold Domain |
| BRCA | Breast Carcinoma |
| LUSC | Lung Squamous Cells Carcinoma |
| LUAD | Lung AdenoCarcinoma |
| HCC | HepatoCellular Carcinoma |
| DEG | Differentially Expressed Genes |
| FDR | False Discovery Rate |
| FC | Fold Change |
| PFI | Progression Free Interval |
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer Statistics. Cancer J. Clin. 2012, 62, 10–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.D.; Roberts, L.R. Epidemiology and Management of Hepatocellular Carcinoma. Infect. Dis. Clin. N. Am. 2010, 24, 899–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Bandoh, S.; Roberts, L.R. Molecular pathogenesis of hepatocellular carcinoma and impact of therapeutic advances. F1000Research 2016, 5, 879. [Google Scholar] [CrossRef]
- Ally, A.; Balasundaram, M.; Carlsen, R.; Chuah, E.; Clarke, A.; Dhalla, N.; Holt, R.A.; Jones, S.J.; Lee, D.; Ma, Y.; et al. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341. [Google Scholar] [CrossRef]
- Hoshida, Y.; Nijman, S.M.; Kobayashi, M.; Chan, J.A.; Brunet, J.-P.; Chiang, D.Y.; Villanueva, A.; Newell, P.; Ikeda, K.; Hashimoto, M.; et al. Integrative Transcriptome Analysis Reveals Common Molecular Subclasses of Human Hepatocellular Carcinoma. Cancer Res. 2009, 69, 7385–7392. [Google Scholar] [CrossRef] [Green Version]
- Levine, M.; Cattoglio, C.; Tjian, R. Looping Back to Leap Forward: Transcription Enters a New Era. Cell 2014, 157, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Goodarzi, H.; Elemento, O.; Tavazoie, S. Revealing Global Regulatory Perturbations across Human Cancers. Mol. Cell 2009, 36, 900–911. [Google Scholar] [CrossRef] [Green Version]
- Dolfini, D.; Mantovani, R.; Zambelli, F.; Pavesi, G. A perspective of promoter architecture from the CCAAT box. Cell Cycle 2009, 8, 4127–4137. [Google Scholar] [CrossRef] [Green Version]
- Dolfini, D.; Mantovani, R. Targeting the Y/CCAAT box in cancer: YB-1 (YBX1) or NF-Y? Cell Death Differ. 2013, 20, 676–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldfield, A.J.; Henriques, T.; Kumar, D.; Burkholder, A.B.; Cinghu, S.; Paulet, D.; Bennett, B.D.; Yang, P.; Scruggs, B.S.; Lavender, C.A.; et al. NF-Y controls fidelity of transcription initiation at gene promoters through maintenance of the nucleosome-depleted region. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.Y.; van Huijsduijnen, R.H.; Mantovani, R.; Benoist, C.; Mathis, D. Intron-exon organization of the NF-Y genes. Tissue-specific splicing modifies an activation domain. J. Biol. Chem. 1992, 267, 8984–8990. [Google Scholar]
- Di, A.S.; Imbriano, C.; Mantovani, R. Dissection of the NF-Y transcriptional activation potential. Nucleic Acids Res. 1999, 27, 2578–2584. [Google Scholar] [CrossRef] [Green Version]
- Liberati, C.; di Silvio, A.; Ottolenghi, S.; Mantovani, R. NF-Y binding to twin CCAAT boxes: Role of Q-rich domains and histone fold helices 1 1Edited by M. Yaniv. J. Mol. Biol. 1999, 285, 1441–1455. [Google Scholar] [CrossRef]
- Ceribelli, M.; Benatti, P.; Imbriano, C.; Mantovani, R. NF-YC Complexity Is Generated by Dual Promoters and Alternative Splicing. J. Biol. Chem. 2009, 284, 34189–34200. [Google Scholar] [CrossRef] [Green Version]
- Fleming, J.D.; Pavesi, G.; Benatti, P.; Imbriano, C.; Mantovani, R.; Struhl, K. NF-Y coassociates with FOS at promoters, enhancers, repetitive elements, and inactive chromatin regions, and is stereo-positioned with growth-controlling transcription factors. Genome Res. 2013, 23, 1195–1209. [Google Scholar] [CrossRef] [Green Version]
- Xie, D.; Boyle, A.P.; Wu, L.; Zhai, J.; Kawli, T.; Snyder, M. Dynamic trans-Acting Factor Colocalization in Human Cells. Cell 2013, 155, 713–724. [Google Scholar] [CrossRef] [Green Version]
- Dolfini, D.; Zambelli, F.; Pedrazzoli, M.; Mantovani, R.; Pavesi, G. A high definition look at the NF-Y regulome reveals genome-wide associations with selected transcription factors. Nucleic Acids Res. 2016, 44, 4684–4702. [Google Scholar] [CrossRef] [Green Version]
- Gurtner, A.; Manni, I.; Piaggio, G. NF-Y in cancer: Impact on cell transformation of a gene essential for proliferation. Biochim. Biophys. Acta Bioenerg. 2017, 1860, 604–616. [Google Scholar] [CrossRef]
- Schmidt, E.E.; Schibler, U. Cell size regulation, a mechanism that controls cellular RNA accumulation: Consequences on regulation of the ubiquitous transcription factors Oct1 and NF-Y and the liver-enriched transcription factor DBP. J. Cell Biol. 1995, 128, 467–483. [Google Scholar] [CrossRef]
- Goeman, F.; Manni, I.; Artuso, S.; Ramachandran, B.; Toietta, G.; Bossi, G.; Rando, G.; Cencioni, C.; Germoni, S.; Straino, S.; et al. Molecular imaging of nuclear factor-Y transcriptional activity maps proliferation sites in live animals. Mol. Biol. Cell 2012, 23, 1467–1474. [Google Scholar] [CrossRef]
- Zhang, Y.; Guan, Q.; Liu, Y.; Zhang, Y.; Chen, Y.; Chen, J.; Liu, Y.; Su, Z. Regulation of hepatic gluconeogenesis by nuclear factor Y transcription factor in mice. J. Biol. Chem. 2018, 293, 7894–7904. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; Klumpp, S.A.; Finegold, M.J.; Maity, S.N. Inactivation of CBF/NF-Y in postnatal liver causes hepatocellular degeneration, lipid deposition and endoplasmic reticulum stress. Sci. Rep. 2011, 1, 136. [Google Scholar] [CrossRef] [Green Version]
- Maire, P.; Wuarin, J.; Schibler, U. The role of cis-acting promoter elements in tissue-specific albumin gene expression. Science 1989, 244, 343–346. [Google Scholar] [CrossRef]
- Wuarin, J.; Mueller, C.; Schibler, U. A ubiquitous CCAAT factor is required for efficient in vitro transcription from the mouse albumin promoter. J. Mol. Biol. 1990, 214, 865–874. [Google Scholar] [CrossRef]
- Tronche, F.; Rollier, A.; Herbomel, P.; Bach, I.; Cereghini, S.; Weiss, M.; Yaniv, M. Anatomy of the rat albumin promoter. Mol. Boil. Med. 1990, 7. [Google Scholar]
- Benatti, P.; Chiaramonte, M.L.; Lorenzo, M.; Hartley, J.A.; Hochhauser, D.; Gnesutta, N.; Mantovani, R.; Imbrriano, C.; Dolfini, D. NF-Y activates genes of metabolic pathways altered in cancer cells. Oncotarget 2016, 7, 1633–1650. [Google Scholar] [CrossRef] [Green Version]
- Ishimoto, K.; Tachibana, K.; Hanano, I.; Yamasaki, D.; Nakamura, H.; Kawai, M.; Urano, Y.; Tanaka, T.; Hamakubo, T.; Sakai, J.; et al. Sterol-regulatory-element-binding protein 2 and nuclear factor Y control human farnesyl diphosphate synthase expression and affect cell proliferation in hepatoblastoma cells. Biochem. J. 2010, 429, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.; Chiang, C.-I.; Labhart, P.; Zhao, Y.; Yang, J.; Mistretta, T.-A.; Henning, S.J.; Maity, S.N.; Mori-Akiyama, Y. Context-specific role of SOX9 in NF-Y mediated gene regulation in colorectal cancer cells. Nucleic Acids Res. 2015, 43, 6257–6269. [Google Scholar] [CrossRef]
- Hanley, N.; Oakley, F.; Sugden, S.; Wilson, D.I.; Mann, D.A.; Hanley, N.A. Ectopic SOX9 Mediates Extracellular Matrix Deposition Characteristic of Organ Fibrosis. J. Biol. Chem. 2008, 283, 14063–14071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.G.; Pedersen, P.L. Glucose metabolism in cancer: Importance of transcription factor-DNA interactions within a short segment of the proximal region og the type II hexokinase promoter. J Biol Chem. 2003, 278, 41047–41058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.-C.; Yen, T. Activation of the Hepatitis B Virus S Promoter by Transcription Factor NF-Y via a CCAAT Element. Virology 1996, 225, 387–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Z.; Liu, Y.; Luo, M.; Wang, W.; Liu, J.; Liu, W.; Pan, S.; Xie, Y. Nuclear factor Y regulates ancient budgerigar hepadnavirus core promoter activity. Biochem. Biophys. Res. Commun. 2016, 478, 825–830. [Google Scholar] [CrossRef]
- Li, K.; Ding, S.; Chen, K.; Qin, D.; Qu, J.; Wang, S.; Sheng, Y.; Zou, C.; Chen, L.; Tang, H. Hepatitis B Virus X Protein Up-Regulates AKR1C1 Expression Through Nuclear Factor-Y in Human Hepatocarcinoma Cells. Zahedan J. Res. Med Sci. 2013, 13. [Google Scholar] [CrossRef]
- Mamat, S.; Ikeda, J.-I.; Tian, T.; Wang, Y.; Luo, W.; Aozasa, K.; Morii, E. Transcriptional Regulation of Aldehyde Dehydrogenase 1A1 Gene by Alternative Spliced Forms of Nuclear Factor Y in Tumorigenic Population of Endometrial Adenocarcinoma. Genes Cancer 2011, 2, 979–984. [Google Scholar] [CrossRef]
- Cicchillitti, L.; Corrado, G.; Carosi, M.; Dabrowska, M.E.; Loria, R.; Falcioni, R.; Cutillo, G.; Piaggio, G.; Vizza, E. Prognostic role of NF-YA splicing isoforms and Lamin A status in low grade endometrial cancer. Oncotarget 2016, 8, 7935–7945. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Zhao, X.; Cui, N.; Liang, Y.-L. Cadherins Associate with Distinct Stem Cell-Related Transcription Factors to Coordinate the Maintenance of Stemness in Triple-Negative Breast Cancer. Stem Cells Int. 2017, 2017, 1–13. [Google Scholar] [CrossRef]
- Cao, B.; Zhao, Y.; Zhang, Z.; Li, H.; Xing, J.; Guo, S.; Qiu, X.; Zhang, S.; Min, L.; Zhu, S. Gene regulatory network construction identified NFYA as a diffuse subtype-specific prognostic factor in gastric cancer. Int. J. Oncol. 2018, 53, 1857–1868. [Google Scholar] [CrossRef]
- Bie, L.-Y.; Li, D.; Mu, Y.; Wang, S.; Chen, B.-B.; Lyu, H.-F.; Han, L.-L.; Nie, C.-Y.; Yang, C.-C.; Wang, L.; et al. Analysis of cyclin E co-expression genes reveals nuclear transcription factor Y subunit alpha is an oncogene in gastric cancer. Chronic Dis. Transl. Med. 2019, 5, 44–52. [Google Scholar] [CrossRef]
- Dolfini, D.; Andrioletti, V.; Mantovani, R. Overexpression and alternative splicing of NF-YA in breast cancer. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Bezzecchi, E.; Ronzio, M.; Semeghini, V.; Andrioletti, V.; Mantovani, R.; Dolfini, D. NF-YA Overexpression in Lung Cancer: LUAD. Genes 2020, 11, 198. [Google Scholar] [CrossRef] [Green Version]
- Bezzecchi, E.; Ronzio, M.; Dolfini, D.; Mantovani, R. NF-YA Overexpression in Lung Cancer: LUSC. Genes 2019, 10, 937. [Google Scholar] [CrossRef] [Green Version]
- Danaher, P.; Warren, S.; Lu, R.; Samayoa, J.; Sullivan, A.; Pekker, I.; Wallden, B.; Marincola, F.M.; Cesano, A. Pan-cancer adaptive immune resistance as defined by the Tumor Inflammation Signature (TIS): Results from The Cancer Genome Atlas (TCGA). J. Immunother. Cancer 2018, 6, 63. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Wang, W.; Tan, M.; Zhu, L.; Zhang, Y.; Fessler, E.; Vermeulen, L.; Wang, X. DeepCC: A novel deep learning-based framework for cancer molecular subtype classification. Oncogenesis 2019, 8, 1–12. [Google Scholar] [CrossRef]
- Zambelli, F.; Pesole, G.; Pavesi, G. Pscan: Finding over-represented transcription factor binding site motifs in sequences from co-regulated or co-expressed genes. Nucleic Acids Res. 2009, 37, W247–W252. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; Cao, J.; Li, Q.; Lv, Y.; Jia, W.; Ren, W.; Cheng, Q.; Song, X.; Xu, G. Hepatocellular Carcinoma-propagating Cells are Detectable by Side Population Analysis and Possess an Expression Profile Reflective of a Primitive Origin. Sci. Rep. 2016, 6, 34856. [Google Scholar] [CrossRef] [Green Version]
- Cappabianca, L.; Farina, A.; di Marcotullio, L.; Infante, P.; de Simone, D.; Sebastiano, M.; Mackay, A. Discovery, characterization and potential roles of a novel NF-YAx splice variant in human neuroblastoma. J. Exp. Clin. Cancer Res. 2019, 38, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Frontini, M.; Imbriano, C.; Manni, I.; Mantovani, R. Cell-Cycle Regulation of NF-YC Nuclear Localization. Cell Cycle 2004, 3, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Kahle, J.; Baake, M.; Doenecke, D.; Albig, W. Subunits of the heterotrimeric transcription factor NF-Y are imported into the nucleus by distinct pathways involving importin beta and importin 13. Mol Cell Biol. 2005, 25, 5339–5354. [Google Scholar] [CrossRef] [Green Version]
- Belluti, S.; Semeghini, V.; Basile, V.; Rigillo, G.; Salsi, V.; Genovese, F.; Dolfini, D.; Imbriano, C. An autoregulatory loop controls the expression of the transcription factor NF-Y. Biochim. Biophys. Acta Bioenerg. 2018, 1861, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Di Agostino, S.; Strano, S.; Emiliozzi, V.; Zerbini, V.; Mottolese, M.; Sacchi, A.; Blandino, G.; Piaggio, G. Gain of function of mutant p53: The mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 2006, 10, 191–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurtner, A.; Starace, G.; Norelli, G.; Piaggio, G.; Sacchi, A.; Bossi, G. Mutant p53-induced Up-regulation of Mitogen-activated Protein Kinase Kinase 3 Contributes to Gain of Function. J. Biol. Chem. 2010, 285, 14160–14169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Ling, S.; Lin, W.C. TopBP1 mediates mutant p53 gain of function through NF-Y and p63/p73. Mol. Cell Biol. 2011, 31, 4464–4481. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Zhang, Y.; Tang, Y.; Butler, N.; Kim, J.; Guessous, F.; Schiff, D.; Mandell, J.; Abounader, R. A novel PTEN/mutant p53/c-Myc/Bcl-XL axis mediates context-dependent oncogenic effects of PTEN with implications for cancer prognosis and therapy. Neoplasia 2013, 15, 952–965. [Google Scholar] [CrossRef] [Green Version]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P.; Wagenblast, E.; Davis, C.A.; Moon, S.-H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant p53 Drives Pancreatic Cancer Metastasis through Cell-Autonomous PDGF Receptor β Signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef] [Green Version]
- Alam, S.K.; Yadav, V.K.; Bajaj, S.; Datta, A.; Dutta, S.K.; Bhattacharyya, M.; Bhattacharya, S.; Debnath, S.; Roy, S.; A Boardman, L.; et al. DNA damage-induced ephrin-B2 reverse signaling promotes chemoresistance and drives EMT in colorectal carcinoma harboring mutant p53. Cell Death Differ. 2015, 23, 707–722. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Ma, H.; Wang, Z.; Zhang, S.; Yang, H.; Fang, Z. EZH2 Palmitoylation Mediated by ZDHHC5 in p53-Mutant Glioma Drives Malignant Development and Progression. Cancer Res. 2017, 77, 4998–5010. [Google Scholar] [CrossRef] [Green Version]
- Ferraiuolo, M.; di Agostino, S.; Blandino, G.; Strano, S. Oncogenic Intra-p53 Family Member Interactions in Human Cancers. Front. Oncol. 2016, 6, 77. [Google Scholar] [CrossRef] [Green Version]
- Hussain, S.P.; Schwank, J.; Staib, F.; Wang, X.-W.; Harris, C.C. TP53 mutations and hepatocellular carcinoma: Insights into the etiology and pathogenesis of liver cancer. Oncogene 2007, 26, 2166–2176. [Google Scholar] [CrossRef] [Green Version]
- Guerrieri, F.; Belloni, L.; Pediconi, N.; Levrero, M. Molecular Mechanisms of HBV-Associated Hepatocarcinogenesis. Semin. Liver Dis. 2013, 33, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Gordan, J.D.; Kennedy, E.B.; Abou-Alfa, G.K.; Beg, M.S.; Brower, S.T.; Gade, T.P.; Goff, L.; Gupta, S.; Guy, J.; Harris, W.P.; et al. Systemic Therapy for Advanced Hepatocellular Carcinoma: ASCO Guideline. J. Clin. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Cervello, M.; Emma, M.R.; Augello, G.; Cusimano, A.; Giannitrapani, L.; Soresi, M.; Akula, S.M.; Abrams, S.L.; Steelman, L.S.; Gulino, A.; et al. New landscapes and horizons in hepatocellular carcinoma therapy. Aging 2020, 12, 3053–3094. [Google Scholar] [CrossRef] [PubMed]
- Pelizzaro, F.; Sammarco, A.; Dadduzio, V.; Pastorelli, D.; Giovanis, P.; Soldà, C.; Rizzato, M.D.; Lombardi, G.; Lonardi, S.; Peserico, G.; et al. Capecitabine in advanced hepatocellular carcinoma: A multicenter experience. Dig. Liver Dis. 2019, 51, 1713–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granito, A.; Marinelli, S.; Terzi, E.; Piscaglia, F.; Renzulli, M.; Venerandi, L.; Benevento, F.; Bolondi, L. Metronomic capecitabine as second-line treatment in hepatocellular carcinoma after sorafenib failure. Dig. Liver Dis. 2015, 47, 518–522. [Google Scholar] [CrossRef]
- Nardone, V.; Chaves-Sanjuan, A.; Lapi, M.; Airoldi, C.; Saponaro, A.; Pasqualato, S.; Dolfini, D.; Camilloni, C.; Bernardini, A.; Gnesutta, N.; et al. Structural Basis of Inhibition of the Pioneer Transcription Factor NF-Y by Suramin. Cells 2020, 9, 2370. [Google Scholar] [CrossRef]
- Goldman, M.; Craft, B.; Hastie, M.; Repečka, K.; Kamath, A.; McDade, F.; Rogers, D.; Brooks, A.N.; Zhu, J.; Haussler, D. The UCSC Xena platform for public and private cancer genomics data visualization and interpretation. bioRxiv 2018, 326470. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 002832. [Google Scholar] [CrossRef] [Green Version]
- Therneau, T.M.; Grambsch, P.M. Modeling Survival Data: Extending the Cox Model; Springer: New York, NY, USA, 2020. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bezzecchi, E.; Ronzio, M.; Mantovani, R.; Dolfini, D. NF-Y Overexpression in Liver Hepatocellular Carcinoma (HCC). Int. J. Mol. Sci. 2020, 21, 9157. https://doi.org/10.3390/ijms21239157
Bezzecchi E, Ronzio M, Mantovani R, Dolfini D. NF-Y Overexpression in Liver Hepatocellular Carcinoma (HCC). International Journal of Molecular Sciences. 2020; 21(23):9157. https://doi.org/10.3390/ijms21239157
Chicago/Turabian StyleBezzecchi, Eugenia, Mirko Ronzio, Roberto Mantovani, and Diletta Dolfini. 2020. "NF-Y Overexpression in Liver Hepatocellular Carcinoma (HCC)" International Journal of Molecular Sciences 21, no. 23: 9157. https://doi.org/10.3390/ijms21239157