

Recent Advances in the Synthesis of 3,4-Dihydropyran-2-Ones Organocatalyzed by N-Heterocyclic Carbenes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
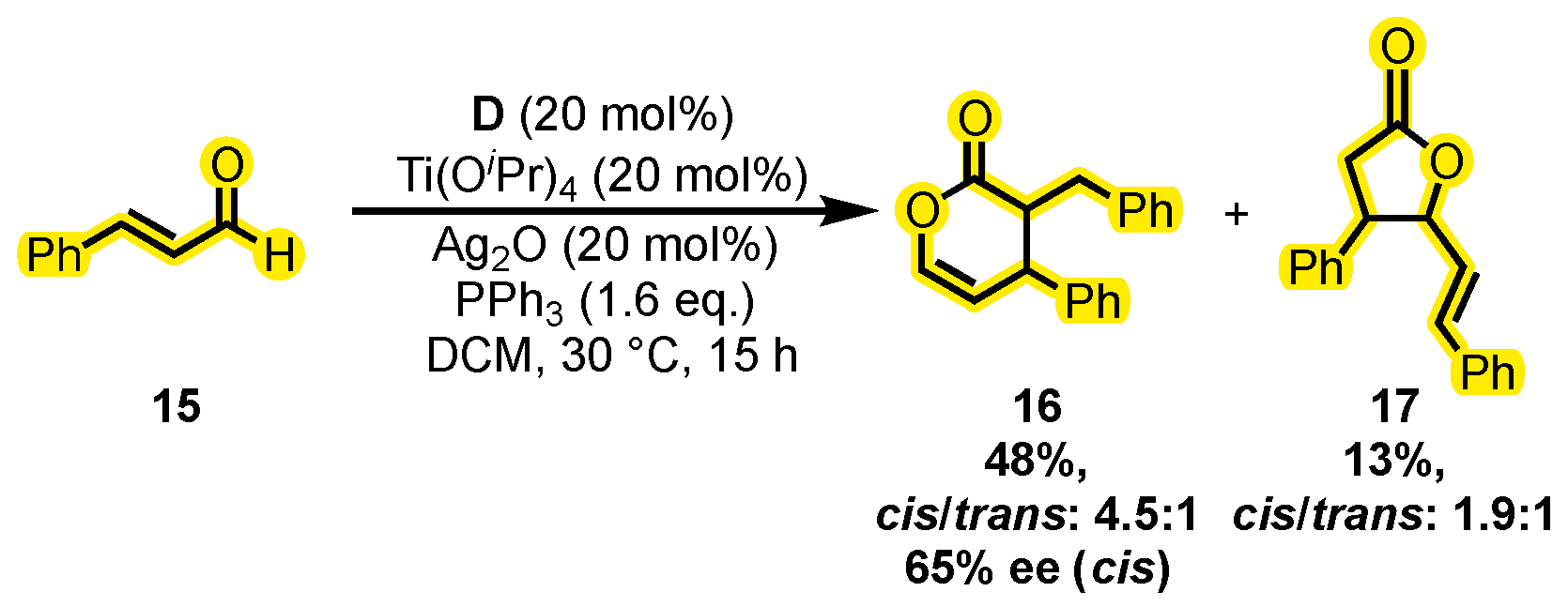
{kind=link}
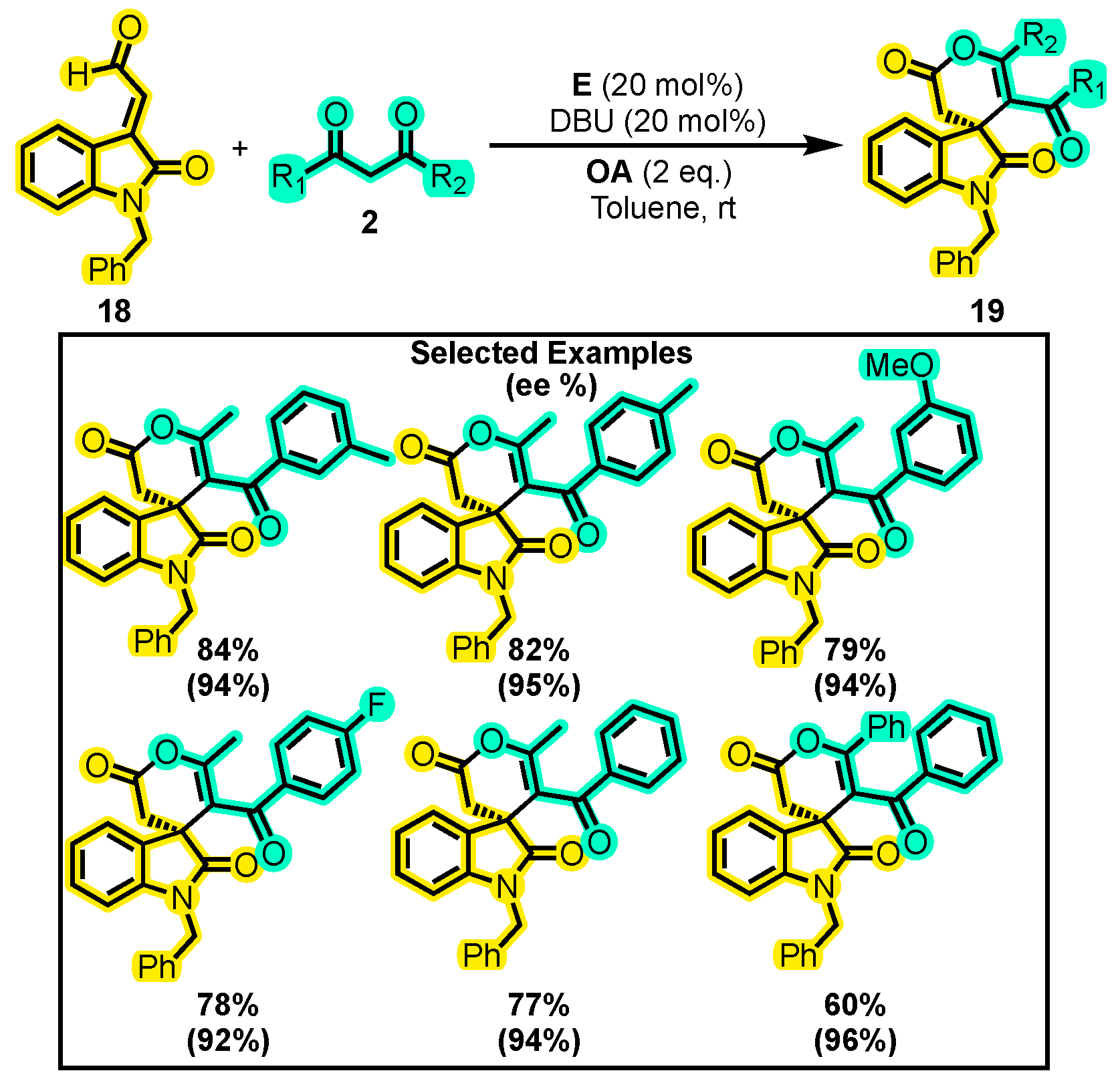
{kind=link}
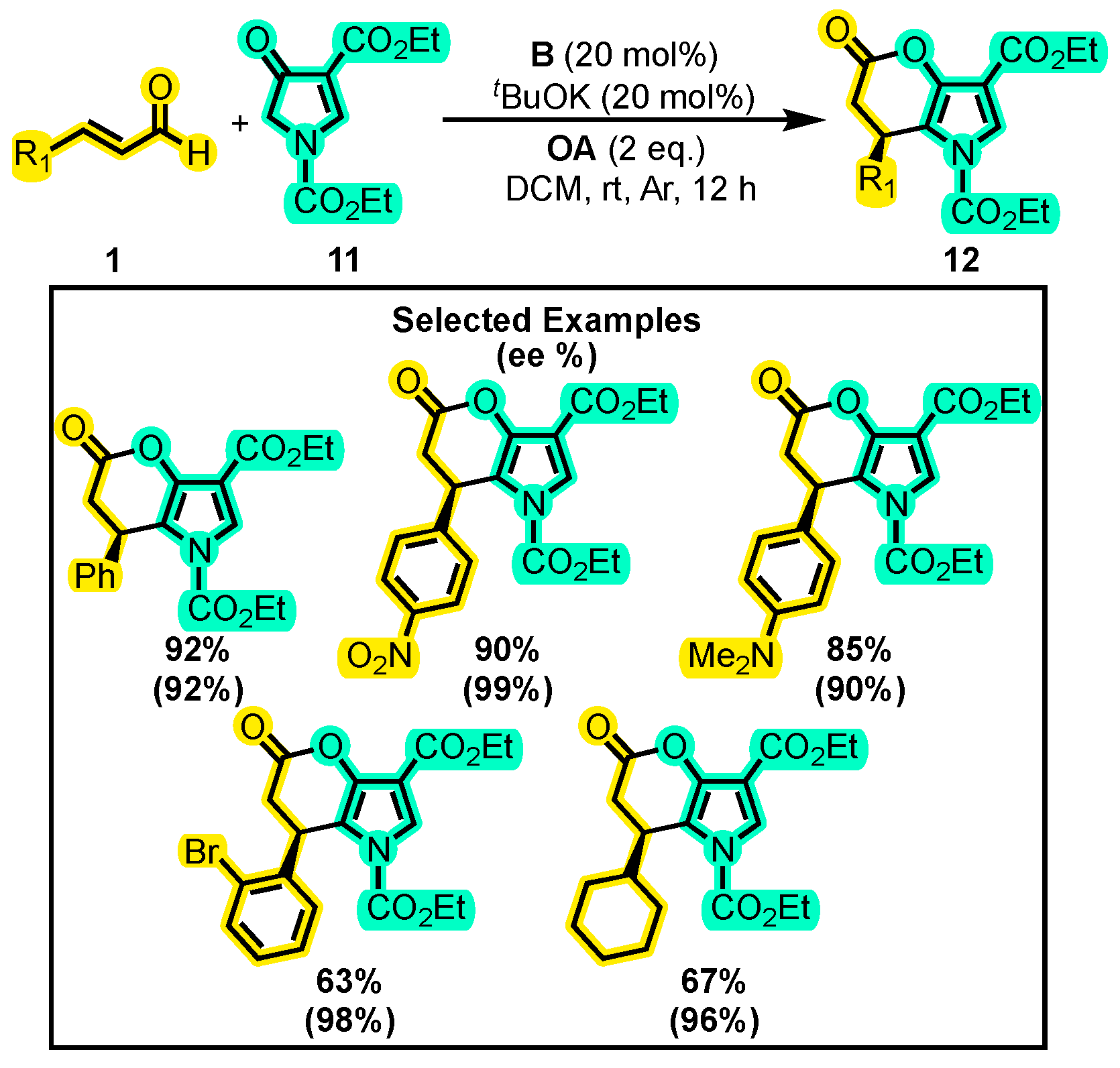
{kind=link}
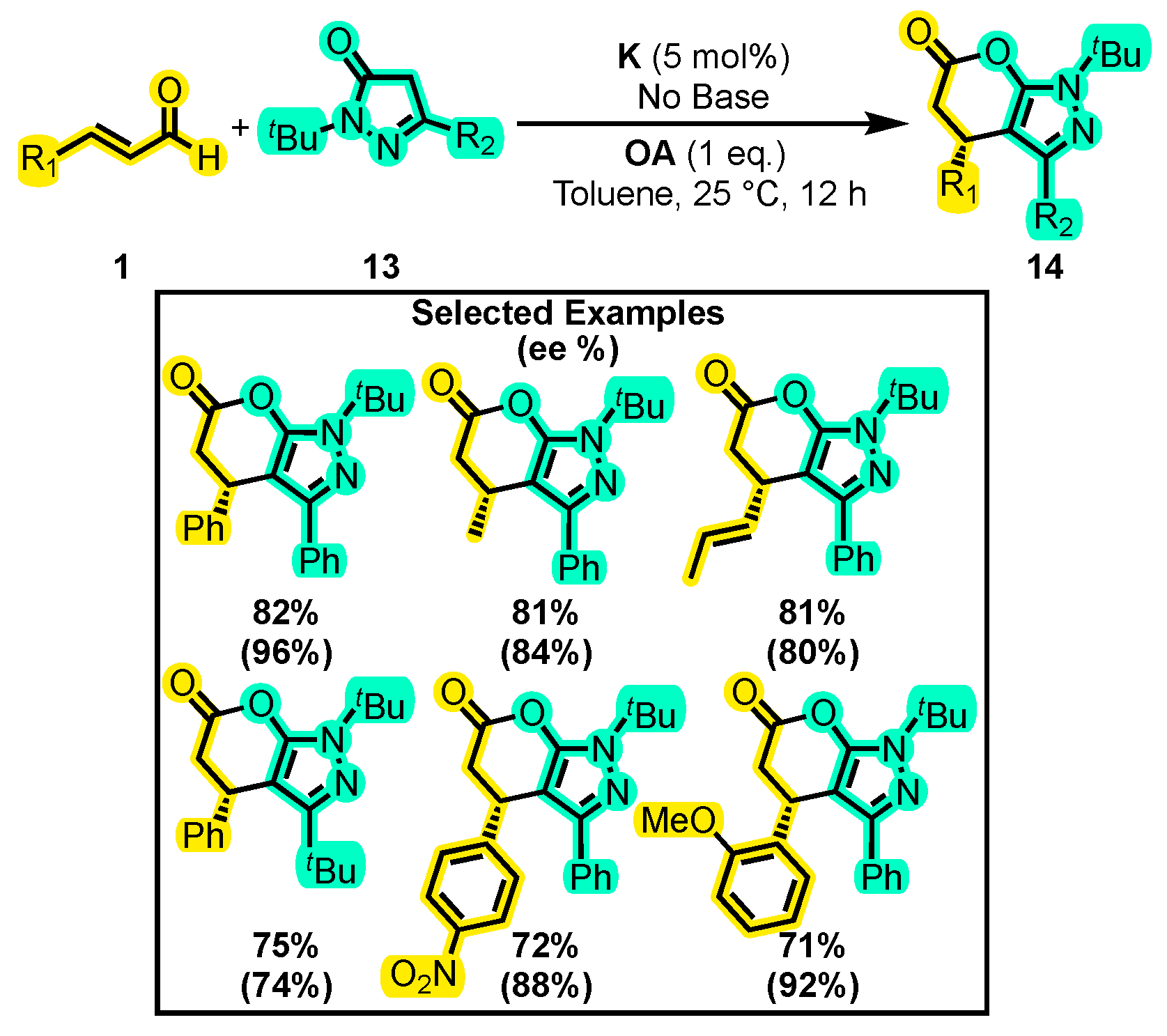
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
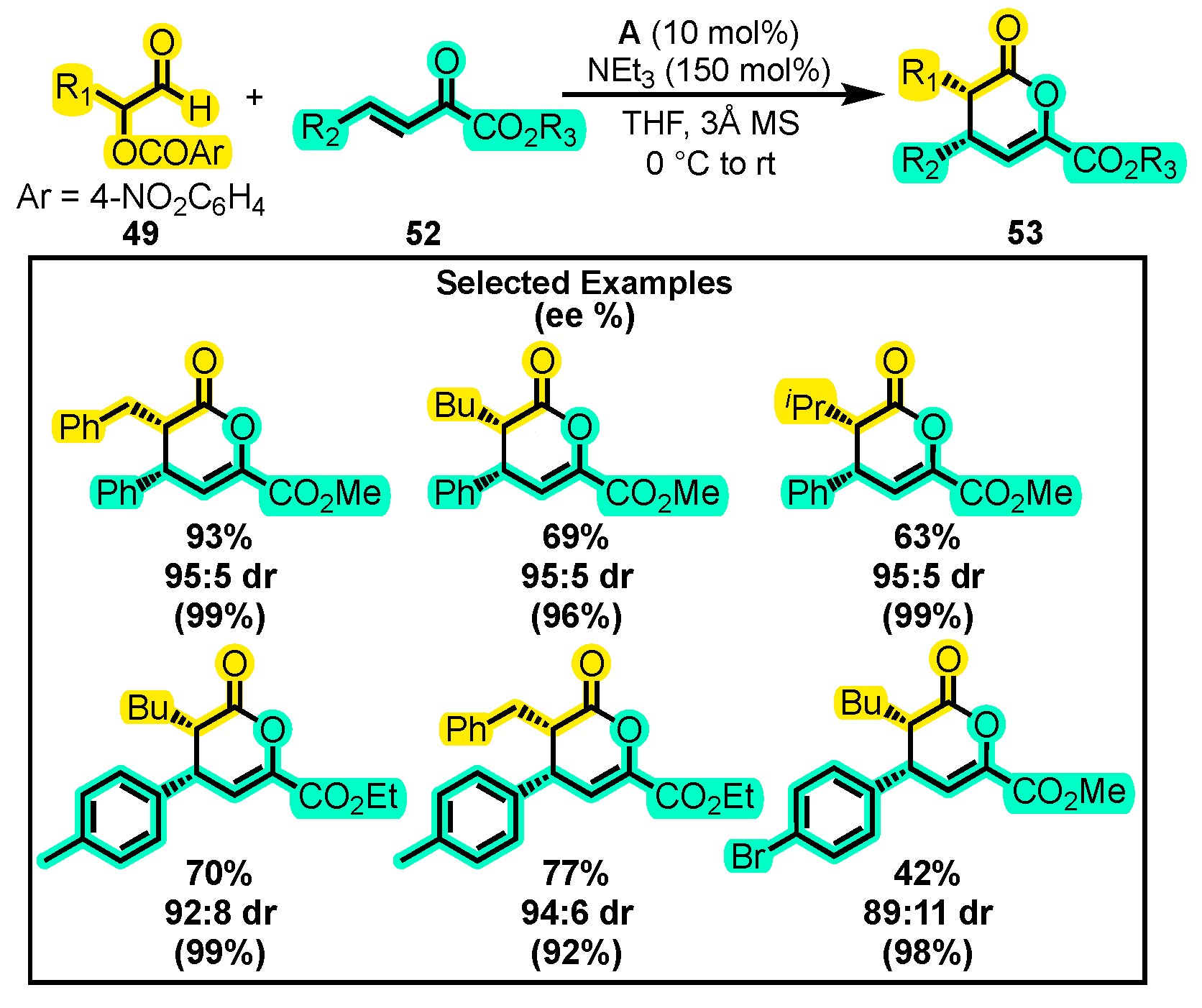{kind=link}
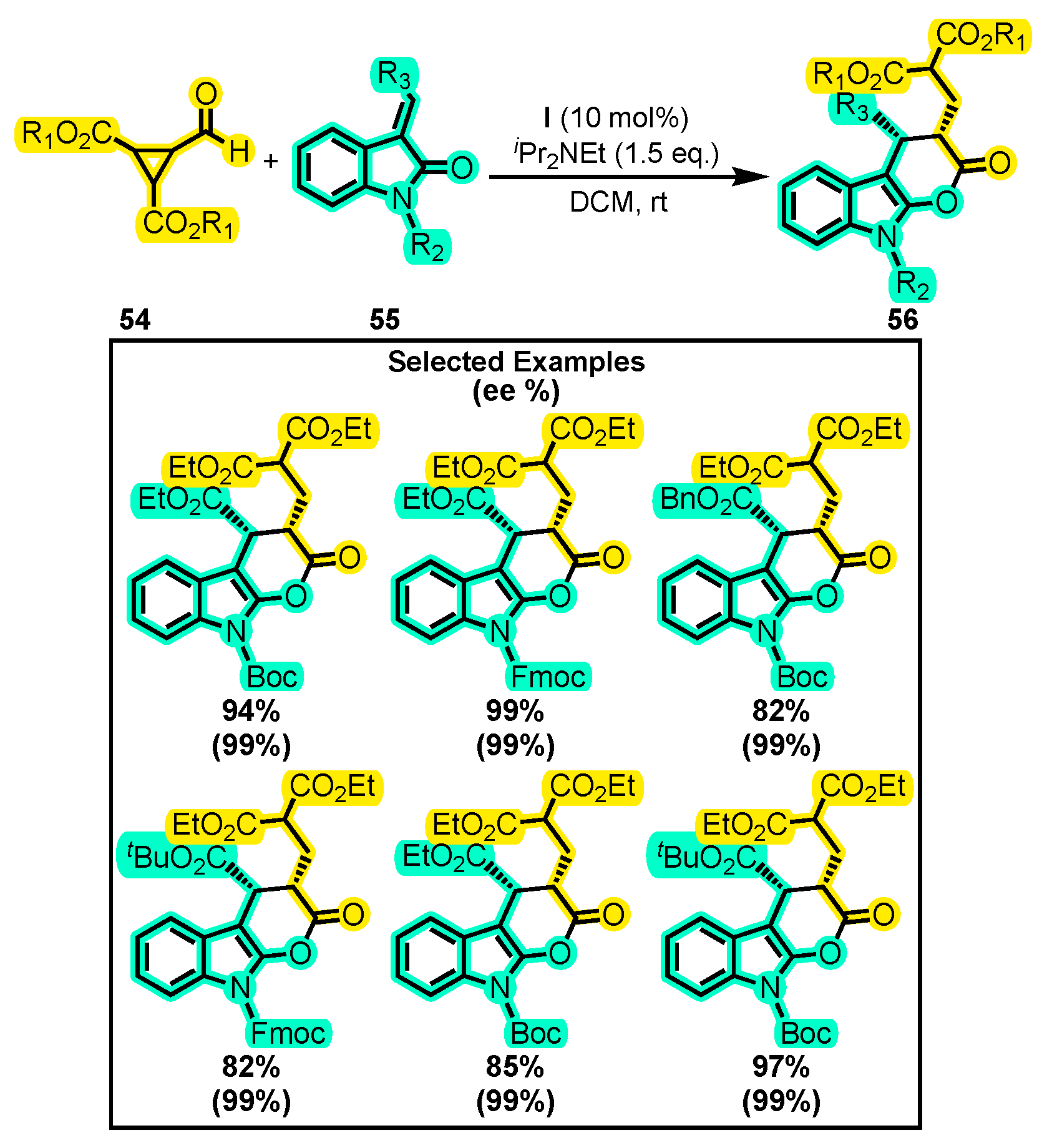{kind=link}
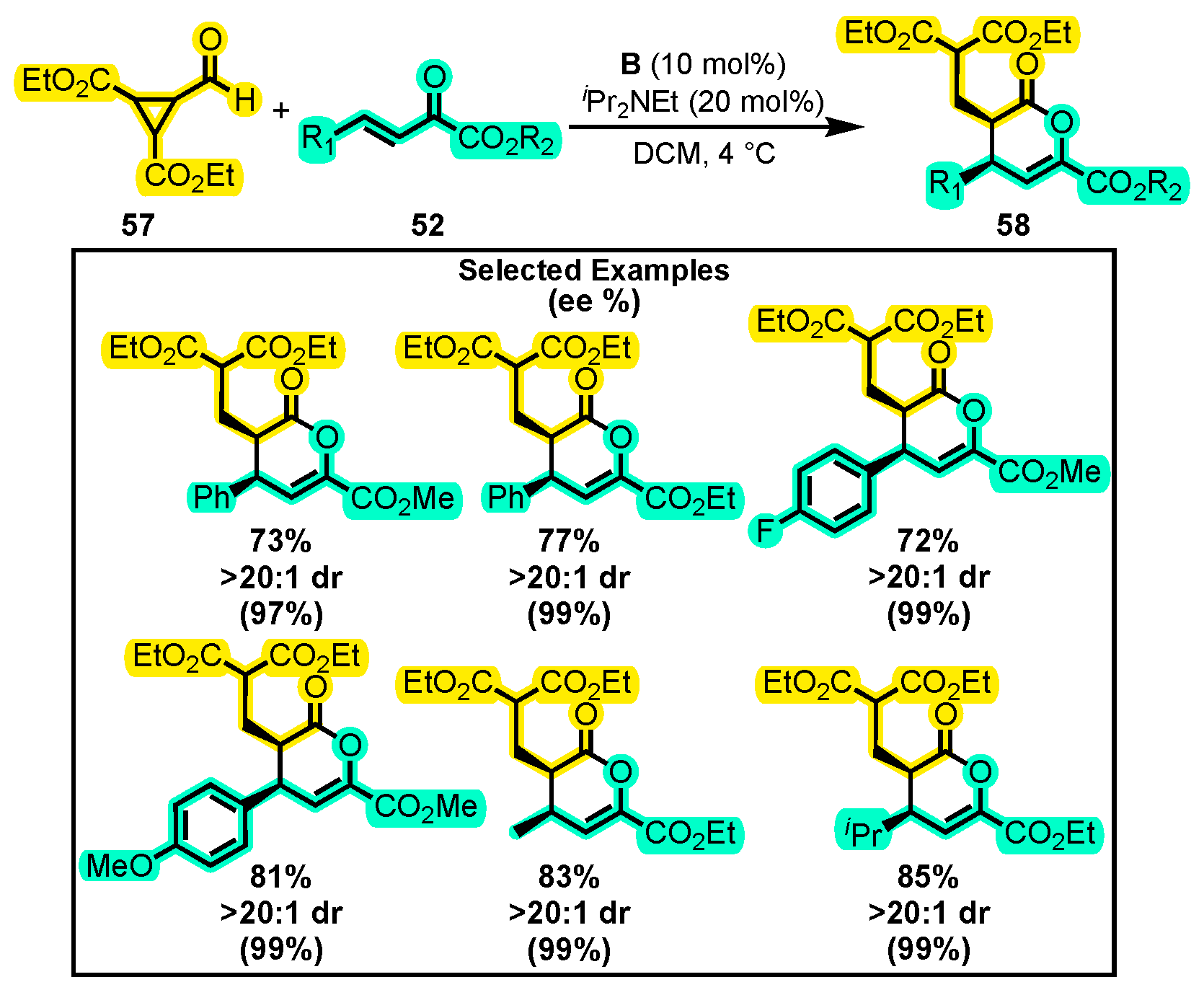{kind=link}
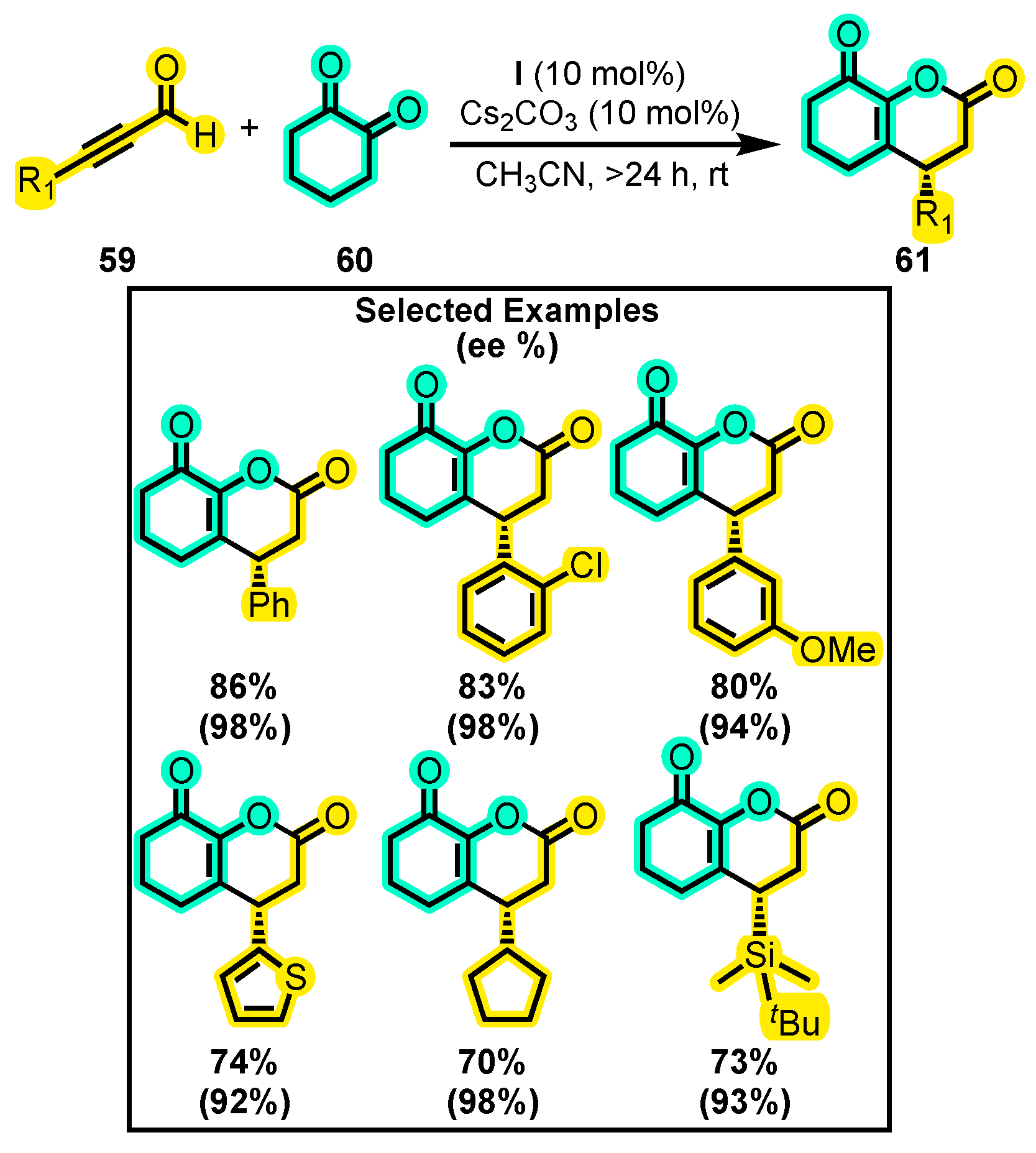{kind=link}
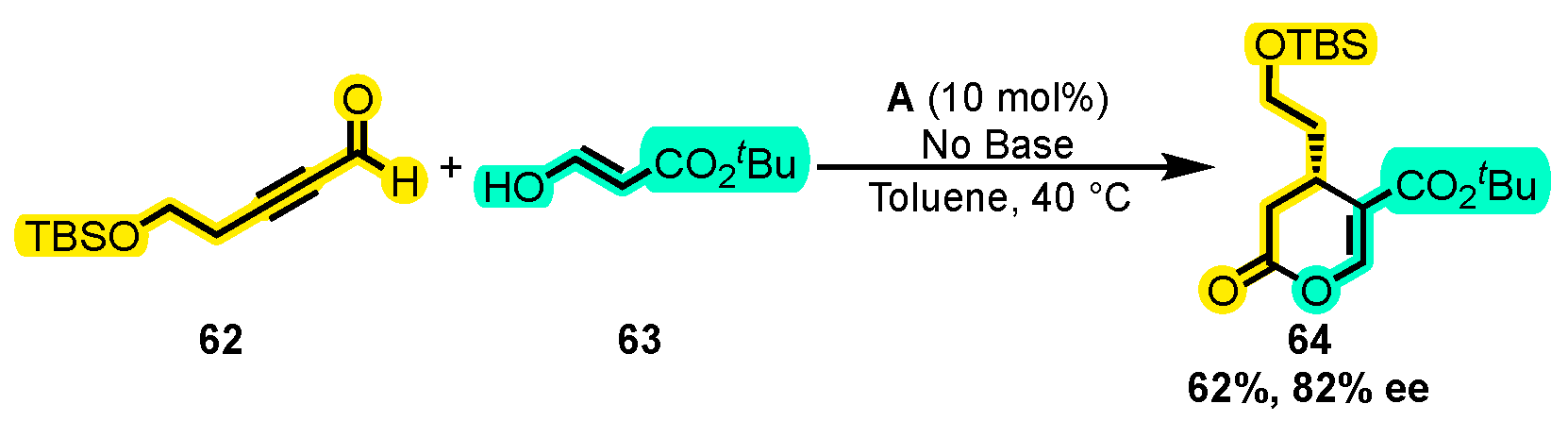{kind=link}
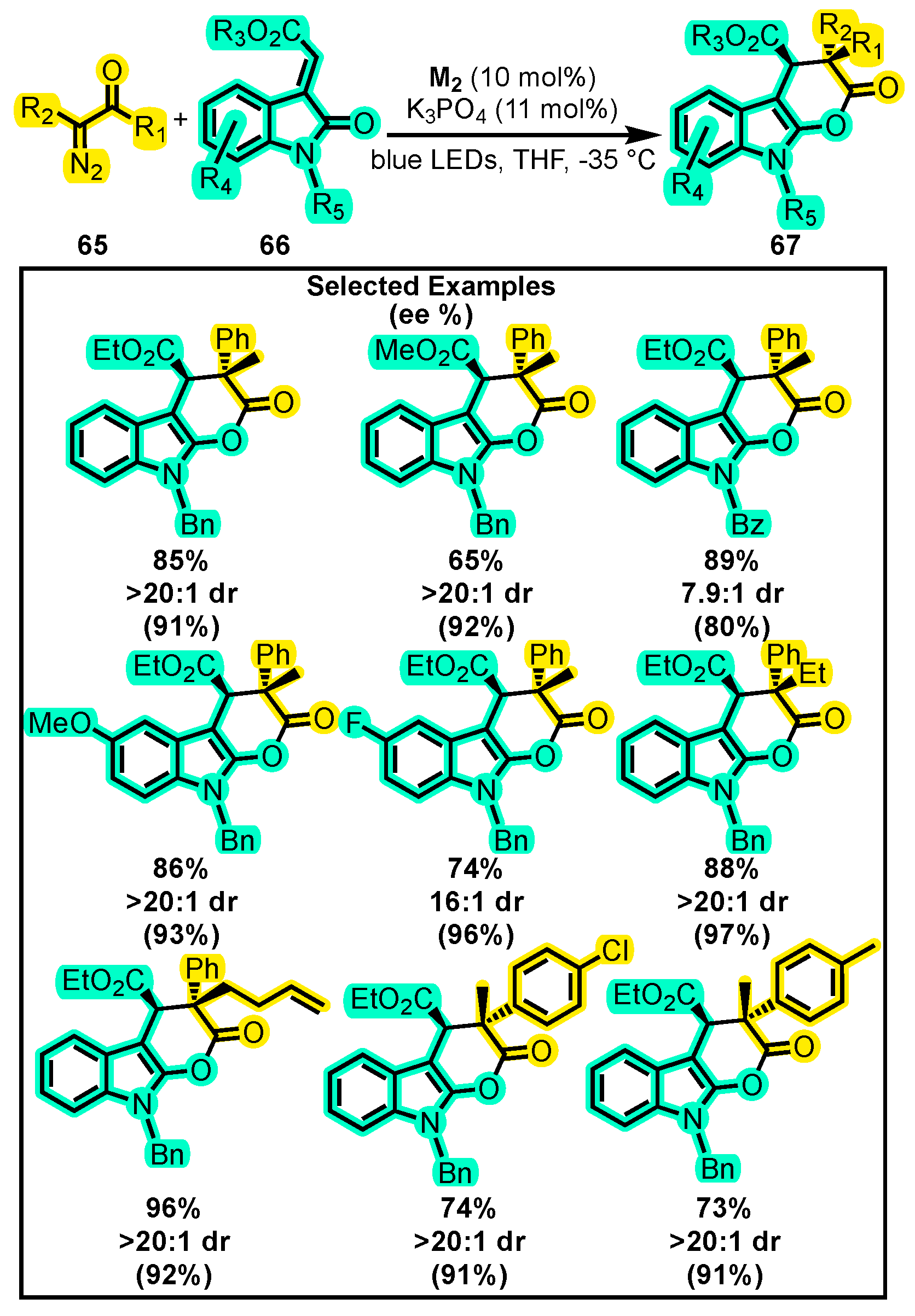{kind=link}
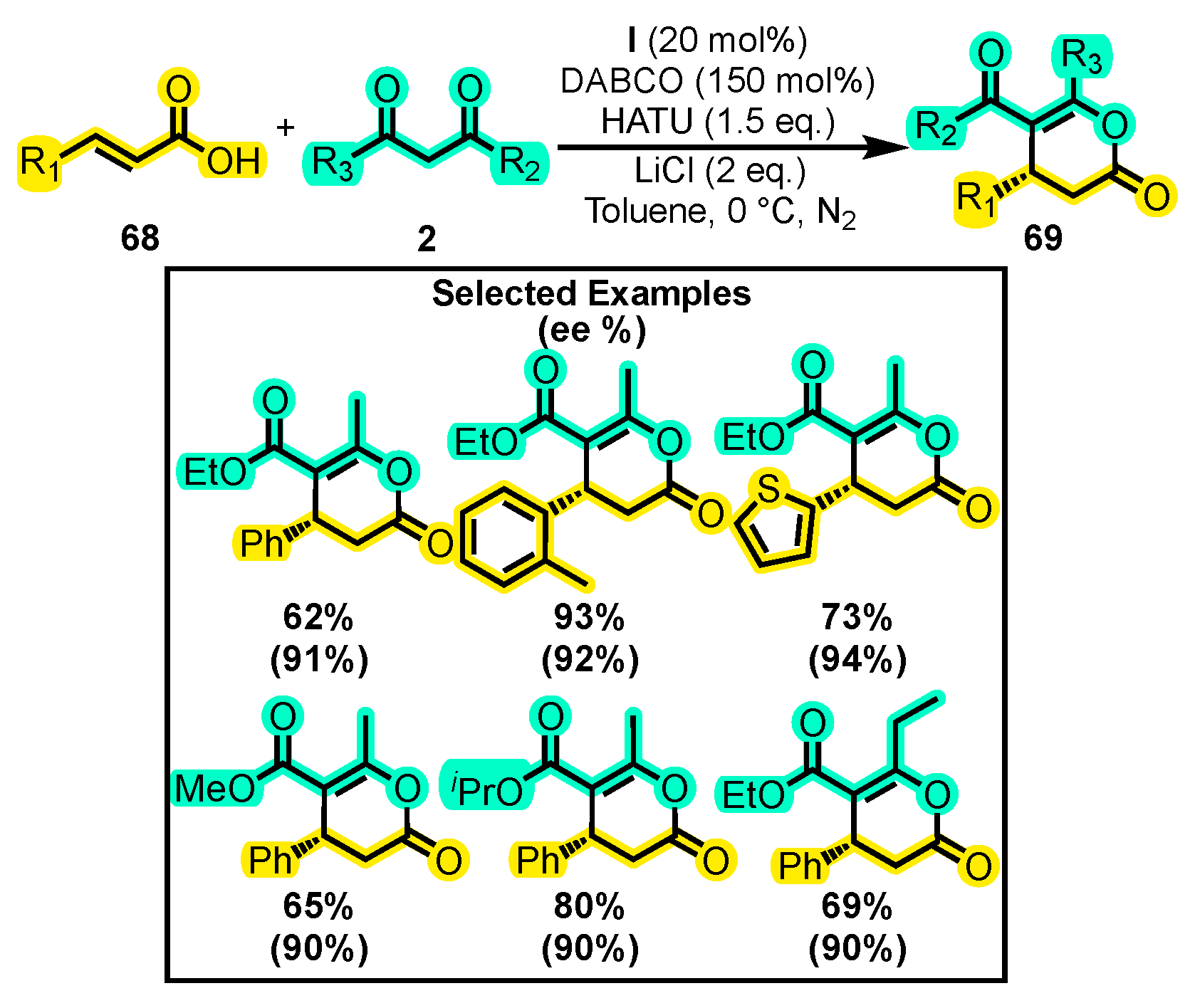{kind=link}
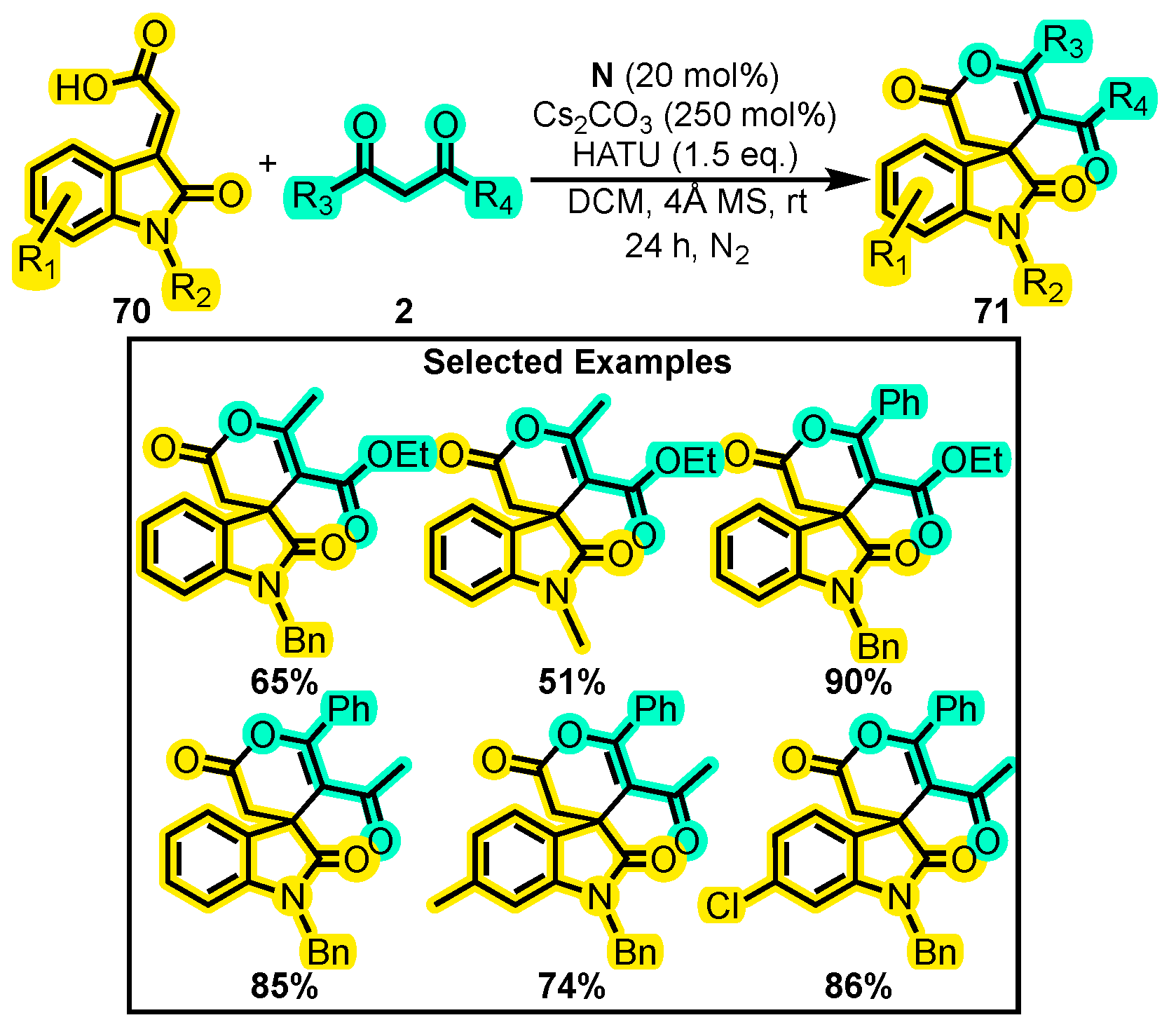{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. α, β-Unsaturated Aldehydes as Reagents for Dihydropyranones
3. Saturated Aldehydes as Reagents for Dihydropyranones
4. Ynals and Ketones as Reagents for Dihydropyranones
5. Carboxylic Acid and Derivatives as Reagents for Dihydropyranones
6. Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Yang, X.; Zhang, Y.C.; Zhu, Q.N.; Tu, M.S.; Shi, F. Diastereo- and Enantioselective Construction of the Hexahydrocoumarin Scaffold via an Organocatalytic Asymmetric [3+3] Cyclization. J. Org. Chem. 2016, 81, 5056–5065. [Google Scholar] [CrossRef] [PubMed]
- Xian, J.; Chen, L.; Ye, L.; Sun, Y.; Shi, Z.; Zhao, Z.; Li, X. Enantioselective Synthesis of Fused Dihydropyranones via Squaramide-Catalyzed Michael Addition/Lactonization Cascade Reaction. Tetrahedron 2019, 75, 2350–2356. [Google Scholar] [CrossRef]
- Destro, D.; Bottinelli, C.; Ferrari, L.; Albanese, D.C.M.; Bencivenni, G.; Gillick-Healy, M.W.; Kelly, B.G.; Adamo, M.F.A. Enantioselective Synthesis of 3,4-Dihydropyran-2-Ones via Phase-Transfer-Catalyzed Addition-Cyclization of Acetylacetone to Cinnamic Thioesters. J. Org. Chem. 2020, 85, 5183–5192. [Google Scholar] [CrossRef] [PubMed]
- Barik, S.; Biju, A.T. N-Heterocyclic Carbene (NHC) Organocatalysis Using Aliphatic Aldehydes. Chem. Commun. 2020, 56, 15484–15495. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Y.; Gao, Z.H.; Ye, S. Bifunctional N-Heterocyclic Carbenes Derived from l-Pyroglutamic Acid and Their Applications in Enantioselective Organocatalysis. Acc. Chem. Res. 2020, 53, 690–702. [Google Scholar] [CrossRef]
- Chen, X.Y.; Liu, Q.; Chauhan, P.; Enders, D. N-Heterocyclic Carbene Catalysis via Azolium Dienolates: An Efficient Strategy for Remote Enantioselective Functionalizations. Angew. Chem. Int. Ed. 2018, 57, 3862–3873. [Google Scholar] [CrossRef]
- Chen, X.; Wang, H.; Jin, Z.; Chi, Y.R. N-Heterocyclic Carbene Organocatalysis: Activation Modes and Typical Reactive Intermediates. Chin. J. Chem. 2020, 38, 1167–1202. [Google Scholar] [CrossRef]
- Das, T.K.; Biju, A.T. Imines as Acceptors and Donors in N-Heterocyclic Carbene (NHC) Organocatalysis. Chem. Commun. 2020, 56, 8537–8552. [Google Scholar] [CrossRef]
- Flanigan, D.M.; Romanov-Michailidis, F.; White, N.A.; Rovis, T. Organocatalytic Reactions Enabled by N-Heterocyclic Carbenes. Chem. Rev. 2015, 115, 9307–9387. [Google Scholar] [CrossRef]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An Overview of N-Heterocyclic Carbenes. Nature 2014, 510, 485–496. [Google Scholar] [CrossRef]
- Ishii, T.; Nagao, K.; Ohmiya, H. Recent Advances in N-Heterocyclic Carbene-Based Radical Catalysis. Chem. Sci. 2020, 11, 5630–5636. [Google Scholar] [CrossRef]
- Mukherjee, S.; Biju, A.T. Recent Advances in the Organocatalytic Enantioselective Synthesis of Functionalized β-Lactones. Chem. Asian J. 2018, 13, 2333–2349. [Google Scholar] [CrossRef] [PubMed]
- Murauski, K.J.R.; Jaworski, A.A.; Scheidt, K.A. A Continuing Challenge: N-Heterocyclic Carbene-Catalyzed Syntheses of γ-Butyrolactones. Chem. Soc. Rev. 2018, 47, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- Ohmiya, H. N-Heterocyclic Carbene-Based Catalysis Enabling Cross-Coupling Reactions. ACS Catal. 2020, 10, 6862–6869. [Google Scholar] [CrossRef]
- Nair, V.; Poonoth, M.; Vellalath, S.; Suresh, E.; Thirumalai, R.; Cheme, S. An N-Heterocyclic Carbene-Catalyzed [8+3] Annulation of Tropone and Enals via Homoenolate. J. Org. Chem. 2006, 5, 8964–8965. [Google Scholar] [CrossRef] [PubMed]
- Axelsson, A.; Ta, L.; Sundén, H. Biomimetic Oxidative Carbene Catalysis: Enabling Aerial Oxygen as a Terminal Oxidant. Synlett 2017, 28, 873–878. [Google Scholar] [CrossRef]
- Nair, V.; Menon, R.S.; Biju, A.T.; Sinu, C.R.; Paul, R.R.; Jose, A.; Sreekumar, V. Employing Homoenolates Generated by NHC Catalysis in Carbon-Carbon Bond-Forming Reactions: State of the Art. Chem. Soc. Rev. 2011, 40, 5336–5346. [Google Scholar] [CrossRef]
- Zhao, M.; Zhang, Y.T.; Chen, J.; Zhou, L. Enantioselective Reactions Catalyzed by N-Heterocyclic Carbenes. Asian J. Org. Chem. 2018, 7, 54–69. [Google Scholar] [CrossRef]
- Mondal, S.; Yetra, S.R.; Mukherjee, S.; Biju, A.T. NHC-Catalyzed Generation of α,β-Unsaturated Acylazoliums for the Enantioselective Synthesis of Heterocycles and Carbocycles. Acc. Chem. Res. 2019, 52, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lupton, D. N-Heterocyclic Carbene Catalysis via the α,β-Unsaturated Acyl Azolium. In N-Heterocyclic Carbenes in Organocatalysis; John Wiley & Sons: Hoboken, NJ, USA, 2019; pp. 157–185. [Google Scholar] [CrossRef]
- Nair, V.; Vellalath, S.; Pattoorpadi Babu, B. Recent Advances in Carbon-Carbon Bond-Forming Reactions Involving Homoenolates Generated by NHC Catalysis. Chem. Soc. Rev. 2008, 37, 2691–2698. [Google Scholar] [CrossRef]
- Dai, L.; Ye, S. Recent Advances in N-Heterocyclic Carbene-Catalyzed Radical Reactions. Chin. Chem. Lett. 2021, 32, 660–667. [Google Scholar] [CrossRef]
- Albanese, D.C.M.; Gaggero, N. N-Heterocyclic Carbene Catalysis as a Tool for Gaining Access to the 3,4-Dihydropyran-2-One Skeleton. Eur. J. Org. Chem. 2014, 2014, 5631–5640. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, B.; Guo, S. Transition Metal Complexes Supported by N-Heterocyclic Carbene-Based Pincer Platforms: Synthesis, Reactivity and Applications. Eur. J. Inorg. Chem. 2021, 2021, 188–204. [Google Scholar] [CrossRef]
- Liang, Q.; Song, D. Iron N-Heterocyclic Carbene Complexes in Homogeneous Catalysis. Chem. Soc. Rev. 2020, 49, 1209–1232. [Google Scholar] [CrossRef]
- Chakraborty, N.; Das, B.; Rajbongshi, K.K.; Patel, B.K. Combined Power of Organo- and Transition Metal Catalysis in Organic Synthesis. Eur. J. Org. Chem. 2022, 2022, e202200273. [Google Scholar] [CrossRef]
- Romain, C.; Bellemin-Laponnaz, S.; Dagorne, S. Recent Progress on NHC-Stabilized Early Transition Metal (Group 3–7) Complexes: Synthesis and Applications. Coord. Chem. Rev. 2020, 422, 213411. [Google Scholar] [CrossRef]
- Jalal, M.; Hammouti, B.; Touzani, R.; Aouniti, A.; Ozdemir, I. Metal-NHC Heterocycle Complexes in Catalysis and Biological Applications: Systematic Review. Mater. Today Proc. 2020, 31, S122–S129. [Google Scholar] [CrossRef]
- Keiko, N.A.; Vchislo, N.V. Synthesis of Diheteroatomic Five-Membered Heterocyclic Compounds from α,β-Unsaturated Aldehydes. Asian J. Org. Chem. 2016, 5, 1169–1197. [Google Scholar] [CrossRef]
- Keiko, N.A.; Vchislo, N.V. α,β-Unsaturated Aldehydes in the Synthesis of Five-Membered Heterocyclic Compounds with One Heteroatom: Recent Advances from Developments in Metal- and Organocatalysis. Asian J. Org. Chem. 2016, 5, 439–461. [Google Scholar] [CrossRef]
- Vchislo, N.V.; Verochkina, E.A. Recent Advances in Total Synthesis of Alkaloids from α,β-Unsaturated Aldehydes. ChemistrySelect 2020, 5, 9579–9589. [Google Scholar] [CrossRef]
- Xie, D.; Shen, D.; Chen, Q.; Zhou, J.; Zeng, X.; Zhong, G. N-Heterocyclic Carbene/Lewis Acid Catalyzed Enantioselective Aerobic Annulation of α,β-Unsaturated Aldehydes with 1,3-Dicarbonyl Compounds. J. Org. Chem. 2016, 81, 6136–6141. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Young, C.M.; Watts, A.A.; Slawin, A.M.Z.; Boyce, G.R.; Bühl, M.; Smith, A.D. Isothiourea-Catalyzed Enantioselective Michael Addition of Malonates to α,β-Unsaturated Aryl Esters. Org. Lett. 2022, 24, 4040–4045. [Google Scholar] [CrossRef] [PubMed]
- Axelsson, A.; Hammarvid, E.; Ta, L.; Sundén, H. Asymmetric Aerobic Oxidative NHC-Catalysed Synthesis of Dihydropyranones Utilising a System of Electron Transfer Mediators. Chem. Commun. 2016, 52, 11571–11574. [Google Scholar] [CrossRef]
- Ta, L.; Axelsson, A.; Sundén, H. Attractive Aerobic Access to the α,β-Unsaturated Acyl Azolium Intermediate: Oxidative NHC Catalysis via Multistep Electron Transfer. Green Chem. 2016, 18, 686–690. [Google Scholar] [CrossRef]
- Wu, Z.; Wang, J. N-Heterocyclic Carbene-Catalyzed Chemoselective S-O Bond Cleavage of Benzenesulfonic Carbamate. Org. Lett. 2018, 20, 7607–7610. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Y.; Wang, Y.; Ke, J.; Jeret, M.; Reddi, R.N.; Yang, S.; Song, B.A.; Chi, Y.R. Polyhalides as Efficient and Mild Oxidants for Oxidative Carbene Organocatalysis by Radical Processes. Angew. Chem. Int. Ed. 2017, 56, 2942–2946. [Google Scholar] [CrossRef]
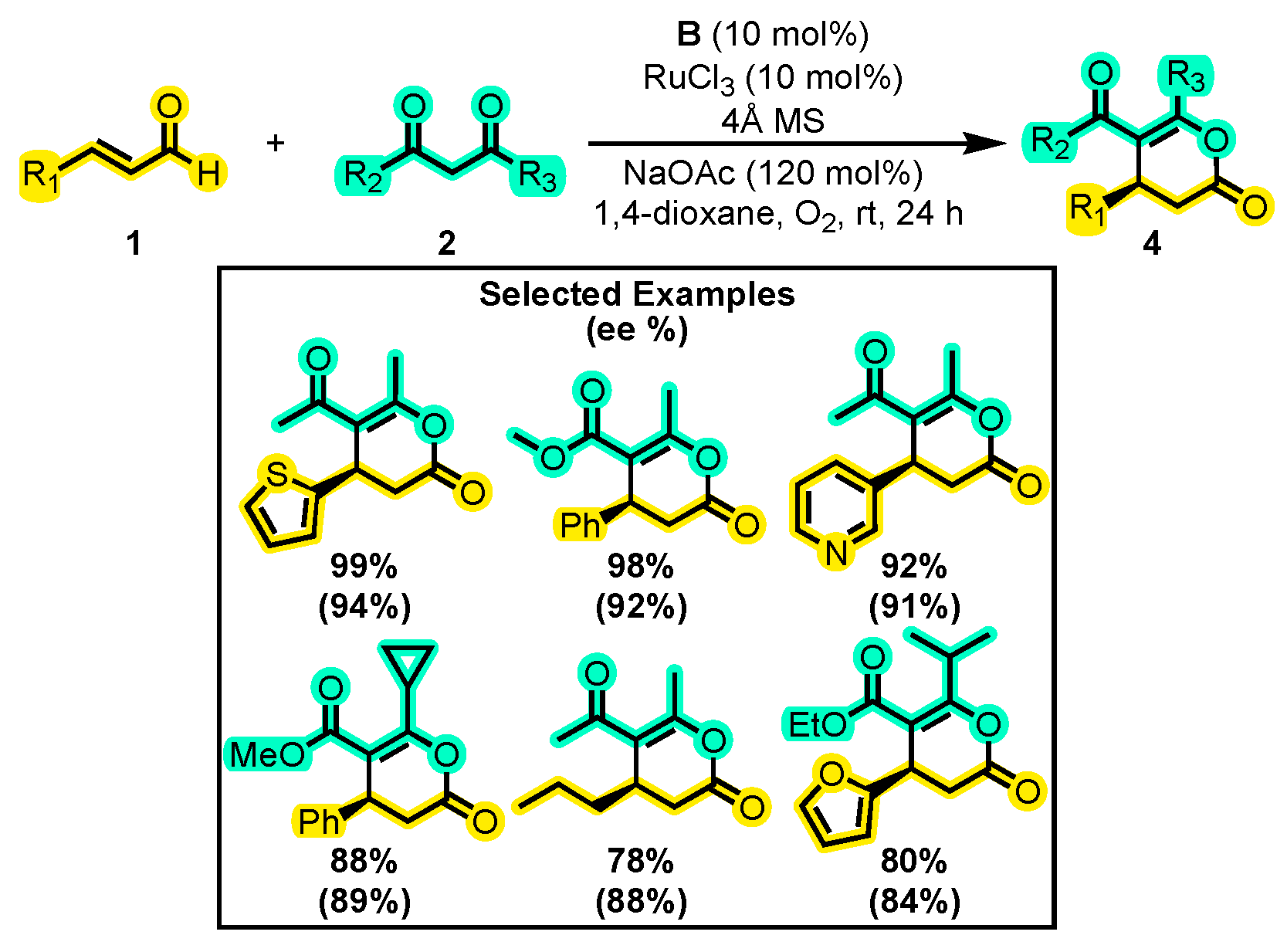
- Wang, Q.; Chen, J.; Huang, Y. Aerobic Oxidation/Annulation Cascades through Synergistic Catalysis of RuCl3 and N-Heterocyclic Carbenes. Chem. Eur. J. 2018, 24, 12806–12810. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.; Shen, L.; Chi, Y.R. Direct β-Activation of Saturated Aldehydes to Formal Michael Acceptors through Oxidative NHC Catalysis. Angew. Chem. Int. Ed. 2013, 52, 8588–8591. [Google Scholar] [CrossRef]
- Evans, D.A.; Thomson, R.J.; Franco, F. Ni(II) Tol-BINAP-Catalyzed Enantioselective Michael Reactions of β-Ketoesters and Unsaturated N-Acylthiazolidinethiones. J. Am. Chem. Soc. 2005, 127, 10816–10817. [Google Scholar] [CrossRef]
- Axelsson, A.; Hammarvid, E.; Rahm, M.; Sundén, H. DBU-Catalyzed Ring-Opening and Retro-Claisen Fragmentation of Dihydropyranones. Eur. J. Org. Chem. 2020, 2020, 5436–5444. [Google Scholar] [CrossRef]
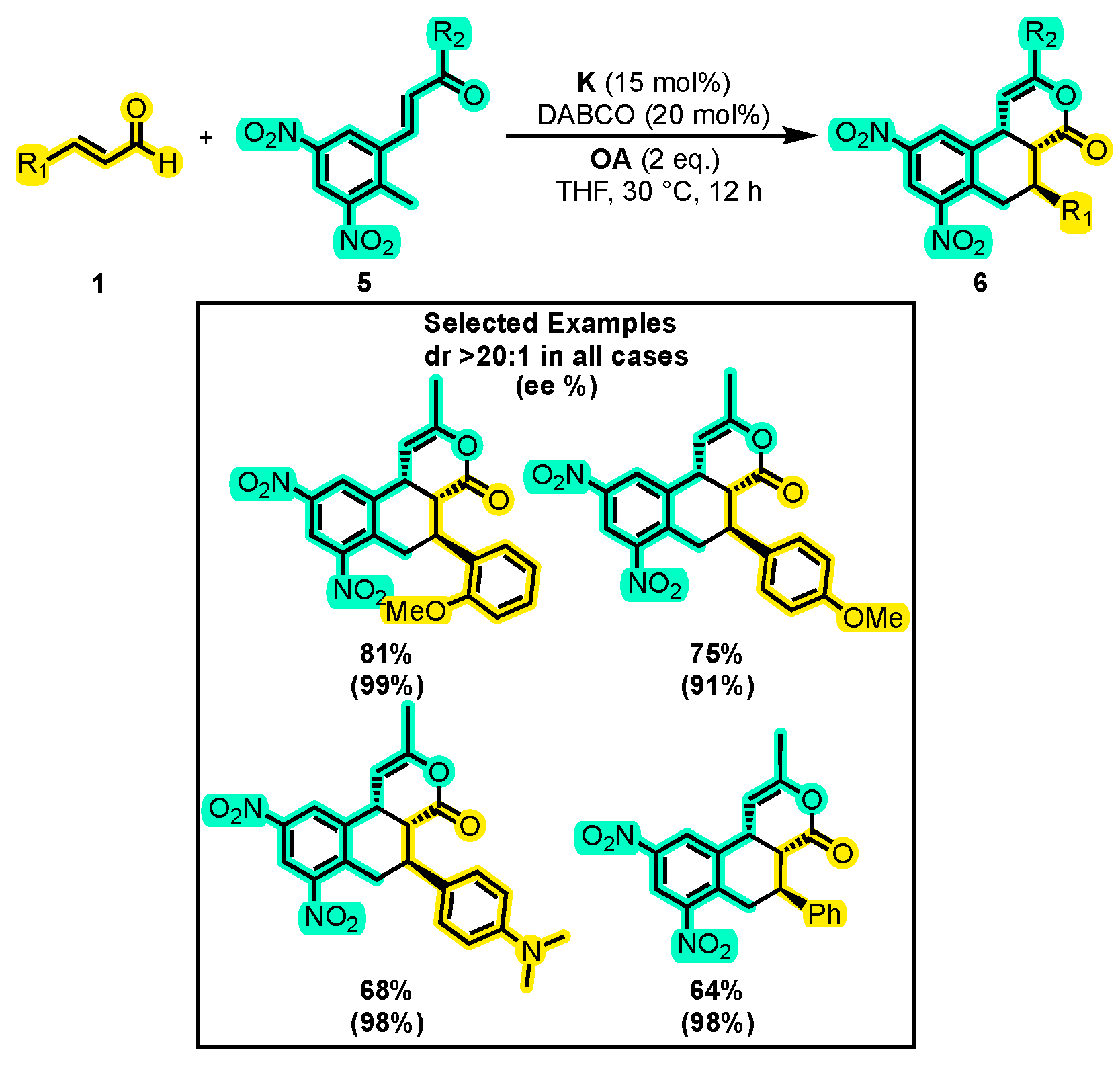
- Mukherjee, S.; Ghosh, A.; Marelli, U.K.; Biju, A.T. N-Heterocyclic Carbene-Catalyzed Michael-Michael-Lactonization Cascade for the Enantioselective Synthesis of Tricyclic δ-Lactones. Org. Lett. 2018, 20, 2952–2955. [Google Scholar] [CrossRef] [PubMed]
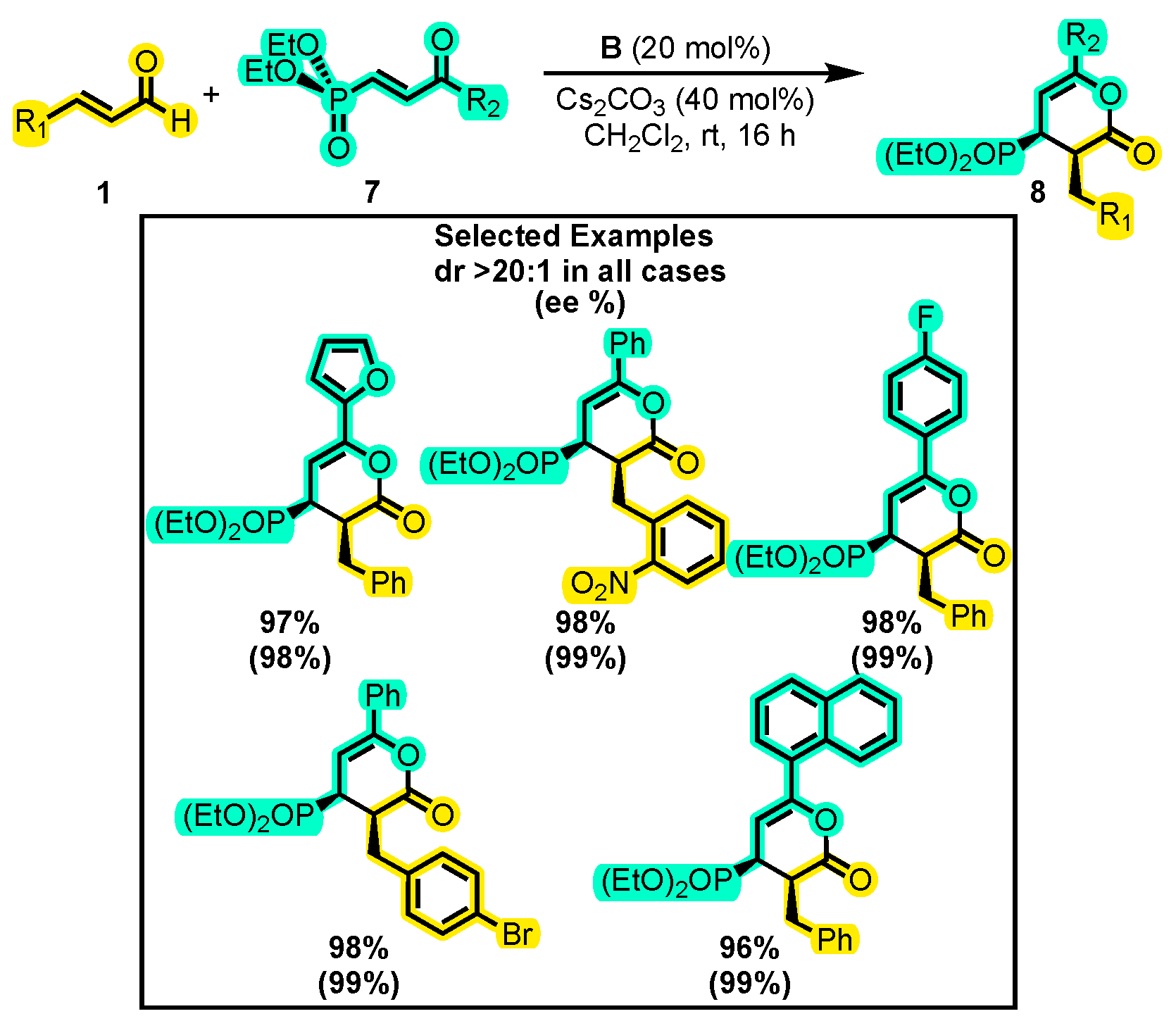
- Verma, R.S.; Khatana, A.K.; Mishra, M.; Kumar, S.; Tiwari, B. Access to Enantioenriched 4-Phosphorylated δ-Lactones from β-Phosphorylenones and Enals: Via Carbene Organocatalysis. Chem. Commun. 2020, 56, 7155–7158. [Google Scholar] [CrossRef]
- Ahn, S.; Ahn, M.; Park, S.; An, S.; Park, I.G.; Hwang, S.Y.; Gong, J.; Oh, S.; Jin, S.H.; Kim, H.J.; et al. Discovery of PPARγ and Glucocorticoid Receptor Dual Agonists to Promote the Adiponectin and Leptin Biosynthesis in Human Bone Marrow Mesenchymal Stem Cells. Eur. J. Med. Chem. 2023, 245, 114927. [Google Scholar] [CrossRef]
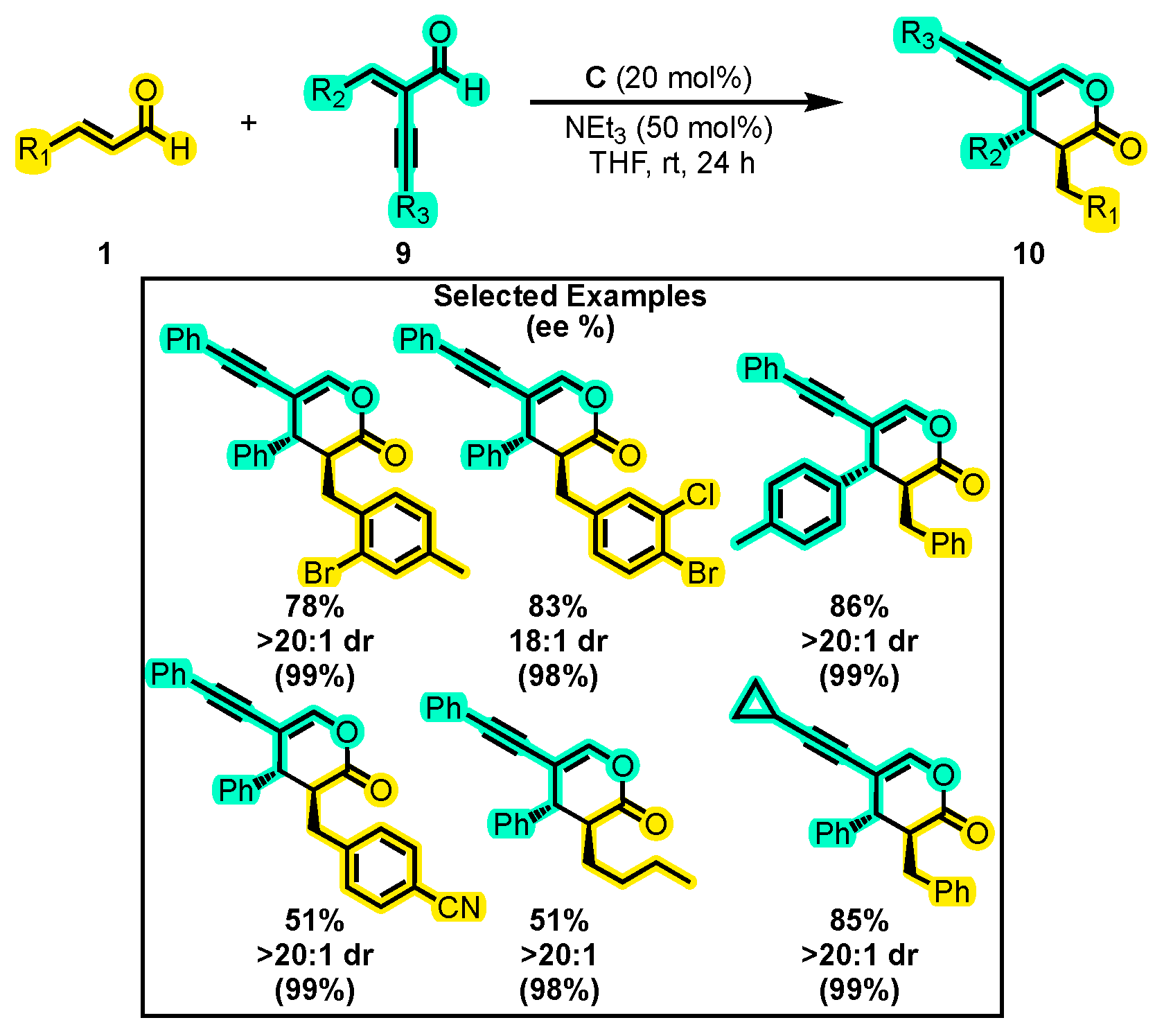
- Peng, X.; Xu, J.; Li, T.; Chi, Y.R.; Jin, Z. Chemo-Selective Cross Reaction of Two Enals: Via Carbene-Catalyzed Dual Activation. Chem. Sci. 2020, 11, 12533–12539. [Google Scholar] [CrossRef]
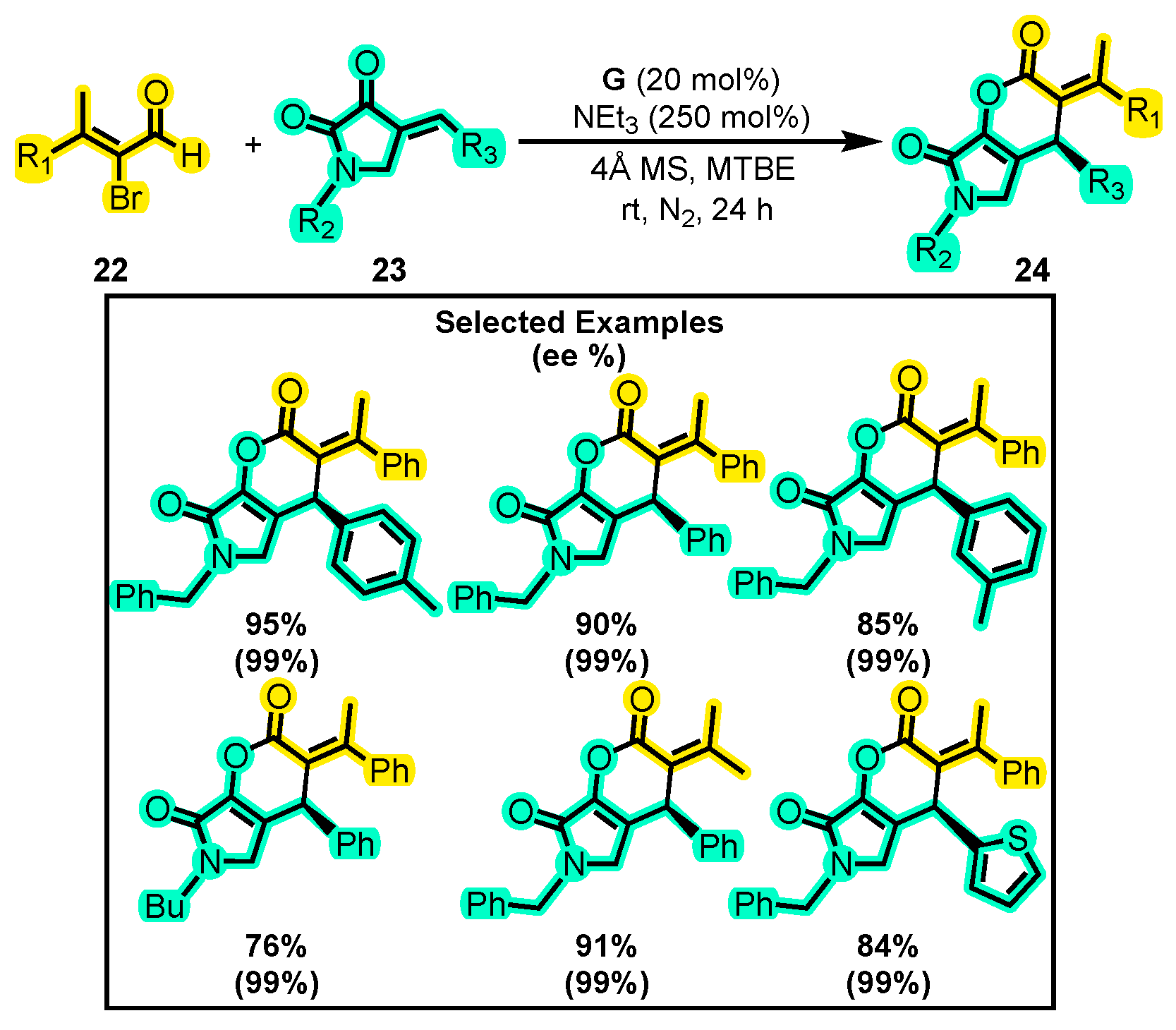
- Wu, Y.T.; Zhang, R.; Duan, X.Y.; Yu, H.F.; Sun, B.Y.; Qi, J. Access to Dihydropyrano [3,2-: B] Pyrrol-5-Ones Skeletons by N-Heterocyclic Carbene-Catalyzed [3+3] Annulations. Chem. Commun. 2020, 56, 9854–9857. [Google Scholar] [CrossRef] [PubMed]
- Yetra, S.R.; Mondal, S.; Suresh, E.; Biju, A.T. Enantioselective Synthesis of Functionalized Pyrazoles by NHC-Catalyzed Reaction of Pyrazolones with α, β-Unsaturated Aldehydes. Org. Lett. 2015, 17, 1417–1420. [Google Scholar] [CrossRef]
- Zhao, Z.; Dai, X.; Li, C.; Wang, X.; Tian, J.; Feng, Y.; Xie, J.; Ma, C.; Nie, Z.; Fan, P.; et al. Pyrazolone Structural Motif in Medicinal Chemistry: Retrospect and Prospect. Eur. J. Med. Chem. 2020, 186, 111893. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; He, Y.; Zhou, J.; Li, X.; Zhu, B.; Chang, J. Organocatalytic Asymmetric Michael Addition of Pyrazol-5-Ones to β-Trifluoromethyl-α,β-Unsaturated Ketones: Stereocontrolled Construction of Vicinal Quaternary and Tertiary Stereocenters. J. Org. Chem. 2020, 85, 574–584. [Google Scholar] [CrossRef]
- Frimurer, T.M.; Mende, F.; Graae, A.S.; Engelstoft, M.S.; Egerod, K.L.; Nygaard, R.; Gerlach, L.O.; Hansen, J.B.; Schwartz, T.W.; Holst, B. Model-Based Discovery of Synthetic Agonists for the Zn2+-Sensing G-Protein-Coupled Receptor 39 (GPR39) Reveals Novel Biological Functions. J. Med. Chem. 2017, 60, 886–898. [Google Scholar] [CrossRef]
- Lee, A.C.H.; Ramanujulu, P.M.; Poulsen, A.; Williams, M.; Blanchard, S.; Ma, D.M.; Bonday, Z.; Goh, K.L.; Goh, K.C.; Goh, M.K.; et al. Thieno [3,2-d]Pyrimidin-4(3H)-One Derivatives as PDK1 Inhibitors Discovered by Fragment-Based Screening. Bioorganic Med. Chem. Lett. 2012, 22, 4023–4027. [Google Scholar] [CrossRef]
- Latendorf, K.; Mechler, M.; Schamne, I.; Mack, D.; Frey, W.; Peters, R. Titanium Salen Complexes with Appended Silver NHC Groups as Nucleophilic Carbene Reservoir for Cooperative Asymmetric Lewis Acid/NHC Catalysis. Eur. J. Org. Chem. 2017, 2017, 4140–4167. [Google Scholar] [CrossRef]
- Miller, E.R.; Hovey, M.T.; Scheidt, K.A. A Concise, Enantioselective Approach for the Synthesis of Yohimbine Alkaloids. J. Am. Chem. Soc. 2020, 142, 2187–2192. [Google Scholar] [CrossRef]
- Lin, J.B.; Cheng, X.N.; Tian, X.D.; Xu, G.Q.; Luo, Y.C.; Xu, P.F. A C1-Symmetric: N-Heterocyclic Carbene Catalysed Oxidative Spiroannulation of Isatin-Derived Enals: Highly Enantioselective Synthesis of Spirooxindole δ-Lactones. RSC Adv. 2018, 8, 15444–15447. [Google Scholar] [CrossRef] [PubMed]
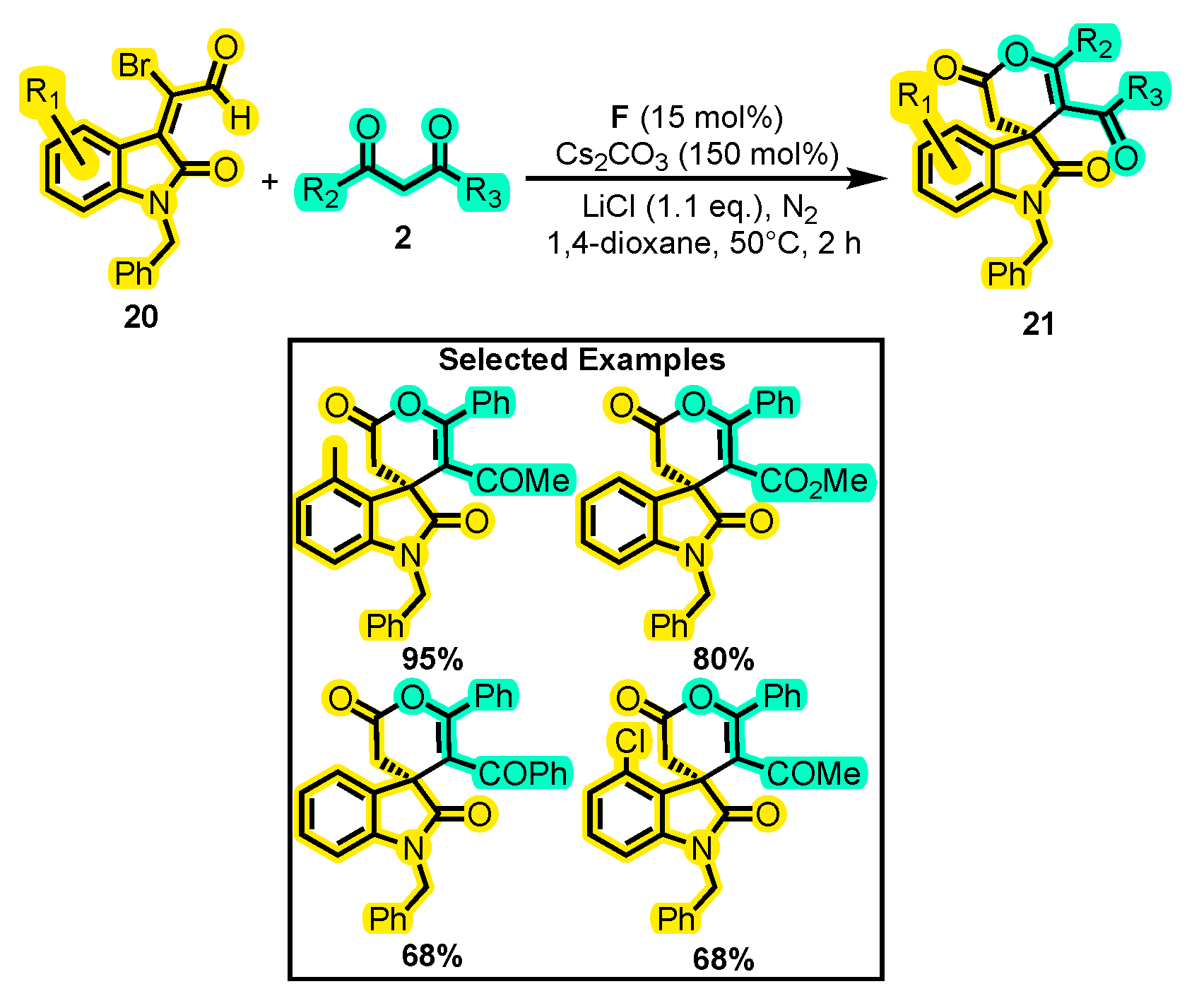
- Xu, J.; Zhang, W.; Liu, Y.; Zhu, S.; Liu, M.; Hua, X.; Chen, S.; Lu, T.; Du, D. Formal [3+3] Annulation of Isatin-Derived 2-Bromoenals with 1,3-Dicarbonyl Compounds Enabled by Lewis Acid/N-Heterocyclic Carbene Cooperative Catalysis. RSC Adv. 2016, 6, 18601–18606. [Google Scholar] [CrossRef]
- Liu, L.; Guo, D.; Wang, J. NHC-Catalyzed Asymmetric α-Regioselective [4+2] Annulation to Construct α-Alkylidene-δ-Lactones. Org. Lett. 2020, 22, 7025–7029. [Google Scholar] [CrossRef]
- Li, J.L.; Fu, L.; Wu, J.; Yang, K.C.; Li, Q.Z.; Gou, X.J.; Peng, C.; Han, B.; Shen, X.D. Highly Enantioselective Synthesis of Fused Bicyclic Dihydropyranones via Low-Loading N-Heterocyclic Carbene Organocatalysis. Chem. Commun. 2017, 53, 6875–6878. [Google Scholar] [CrossRef]
- El-Ablak, F.Z.; Abu-Elenein, N.S.; Sofan, M.A. Synthesis of New Pyrrolo Heterocycles (I): Novel Synthesis of Pyrano [2,3-c]Pyrrole, Isoindoline, Pyrrolo [3,4-b]Pyridine, and Pyrrolo [3,4-d]Pyrimidine Derivatives. J. Heterocycl. Chem. 2016, 53, 1999–2006. [Google Scholar] [CrossRef]
- Li, S.; Chen, X.Y.; Sheng, H.; Von Essen, C.; Rissanen, K.; Enders, D. N-Heterocyclic Carbene Catalyzed Asymmetric Synthesis of Dihydropyranothiazoles via Azolium Enolate Intermediates. Synthesis 2018, 50, 1047–1052. [Google Scholar] [CrossRef]
- Wardkhan, W.W.; Youssef, M.A.; Hamed, F.I.; Ouf, S.A. New Approaches for the Synthesis of Thiazoles and Their Fused Derivatives with Antimicrobial Activities. J. Chin. Chem. Soc. 2008, 55, 1133–1144. [Google Scholar] [CrossRef]
- Petrou, A.; Fesatidou, M.; Geronikaki, A. Thiazole Ring—A Biologically Active Scaffold. Molecules 2021, 26, 3166. [Google Scholar] [CrossRef]
- Ali, S.H.; Sayed, A.R. Review of the Synthesis and Biological Activity of Thiazoles. Synth. Commun. 2021, 51, 670–700. [Google Scholar] [CrossRef]
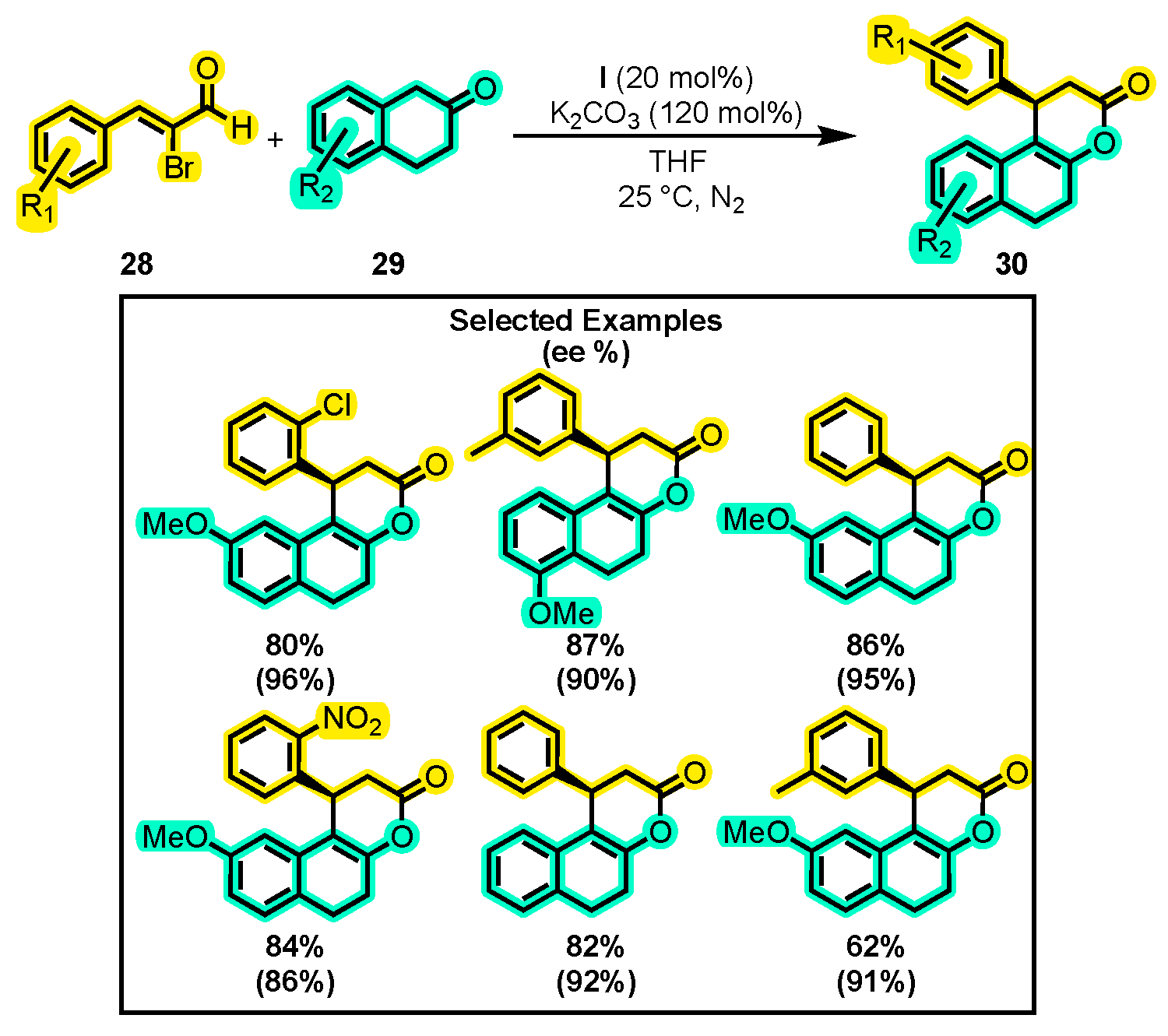
- Li, S.; Yao, Y.; Tang, Z.; Sun, B.; Yu, C.; Li, T.; Yao, C. An Enantioselective Assembly of Naphthopyran: Via NHC-Catalyzed [3+3] Annulation of Bromoenal with β-Tetralone. Org. Biomol. Chem. 2019, 17, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Chang, C.; Chang, H.J.; Chen, K. Synthesis of 2,3,5,6-Tetrahydro-1-Alkyl/Aryl-1H-Benzo[f]Chromen-3-Ol Derivatives from β-Tetralones and α,β-Unsaturated Aldehydes. Org. Biomol. Chem. 2011, 9, 7510–7516. [Google Scholar] [CrossRef]
- Fuchs, P.J.W.; Zeitler, K. An N-Heterocyclic Carbene-Mediated, Enantioselective and Multicatalytic Strategy to Access Dihydropyranones in a Sequential Three-Component One-Pot Reaction. Org. Lett. 2017, 19, 6076–6079. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhang, Y.; Ma, D. Organocatalytic Approach to 3,5,6-Trisubstituted and 4,6-Disubstituted Tetrahydropyran-2-Ones. Tet. Lett. 2010, 51, 3827–3829. [Google Scholar] [CrossRef]
- Breuning, M.; Häuser, T. Unprecedented Formation of Stable Ketene-N,O-Acetals and Their Rearrangement under Basic Conditions. Tetrahedron 2007, 63, 934–940. [Google Scholar] [CrossRef]
- Oh, K.K.; Adnan, M.; Cho, D.H. Network Pharmacology of Bioactives from Sorghum bicolor with Targets Related to Diabetes Mellitus. PLoS ONE 2020, 15, e0240873. [Google Scholar] [CrossRef]
- Gilani, S.A.; Fujii, Y.; Kikuchi, A.; Shinwari, Z.K.; Watanabe, K.N. Ecological Consequences, Genetic and Chemical Variations in Fragmented Populations of a Medicinal Plant, Justicia Adhatoda and Implications for Its Conservation. Pak. J. Bot. 2011, 43, 29–37. [Google Scholar]
- David, O.R.P. A Chemical History of Polycyclic Musks. Chem. Eur. J. 2020, 26, 7537–7555. [Google Scholar] [CrossRef]
- Young, I.S.; Thornton, P.D.; Thompson, A. Synthesis of Natural Products Containing the Pyrrolic Ring. Nat. Prod. Rep. 2010, 27, 1801–1839. [Google Scholar] [CrossRef]
- Xuan, J.; Haelsig, K.T.; Sheremet, M.; Machicao, P.A.; Maimone, T.J. Evolution of a Synthetic Strategy for Complex Polypyrrole Alkaloids: Total Syntheses of Curvulamine and Curindolizine. J. Am. Chem. Soc. 2021, 143, 2970–2983. [Google Scholar] [CrossRef]
- Schmid, M.; Grossmann, A.S.; Wurst, K.; Magauer, T. Total Synthesis of Salimabromide: A Tetracyclic Polyketide from a Marine Myxobacterium. J. Am. Chem. Soc. 2018, 140, 8444–8447. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Barik, S.; Shee, S.; Biju, A.T. Enantioselective Synthesis of Tetra-Substituted Tetralines and Tetrahydro-Indolizines by NHC-Catalyzed Azolium-Enolate Cascade. Chem. Commun. 2021, 57, 7794–7797. [Google Scholar] [CrossRef]
- Satyanarayana, T.; Abraham, S.; Kagan, H.B. Nonlinear Effects in Asymmetric Catalysis. Angew. Chem. Int. Ed. 2009, 48, 456–494. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Xu, J.; Nie, G.; Jin, Z.; Chi, Y.R. NHC-Catalyzed Cascade Reaction between β-Methyl Enals and Dienones for Quick Construction of Complex Multicyclic Lactones. Org. Lett. 2020, 22, 2595–2599. [Google Scholar] [CrossRef]
- Wu, C.; Xiao, H.J.; Wang, S.W.; Tang, M.S.; Tang, Z.L.; Xia, W.; Li, W.F.; Cao, Z.; He, W.M. Natural Deep Eutectic Solvent-Catalyzed Selenocyanation of Activated Alkynes via an Intermolecular H-Bonding Activation Process. ACS Sustain. Chem. Eng. 2019, 7, 2169–2175. [Google Scholar] [CrossRef]
- Lu, L.H.; Zhou, S.J.; He, W.B.; Xia, W.; Chen, P.; Yu, X.; Xu, X.; He, W.M. Metal-Free Difunctionalization of Alkynes Leading to Alkenyl Dithiocyanates and Alkenyl Diselenocyanates at Room Temperature. Org. Biomol. Chem. 2018, 16, 9064–9068. [Google Scholar] [CrossRef] [PubMed]
- Monleón, A.; Blay, G.; Domingo, L.R.; Muñoz, M.C.; Pedro, J.R. Efficient Synthesis of 5-Chalcogenyl-1,3-Oxazin-2-Ones by Chalcogen-Mediated Yne–Carbamate Cyclisation: An Experimental and Theoretical Study. Eur. J. Org. Chem. 2015, 2015, 1020–1027. [Google Scholar] [CrossRef]
- Verma, R.S.; Talukdar, R.; Azaz, T.; Tiwari, B. Carbene Catalyzed Asymmetric Synthesis of Selenylated δ-Lactones via [4+2] Annulation of Selenyl Vinyl Ketones and Enals. Adv. Synth. Catal. 2022, 364, 4031–4035. [Google Scholar] [CrossRef]
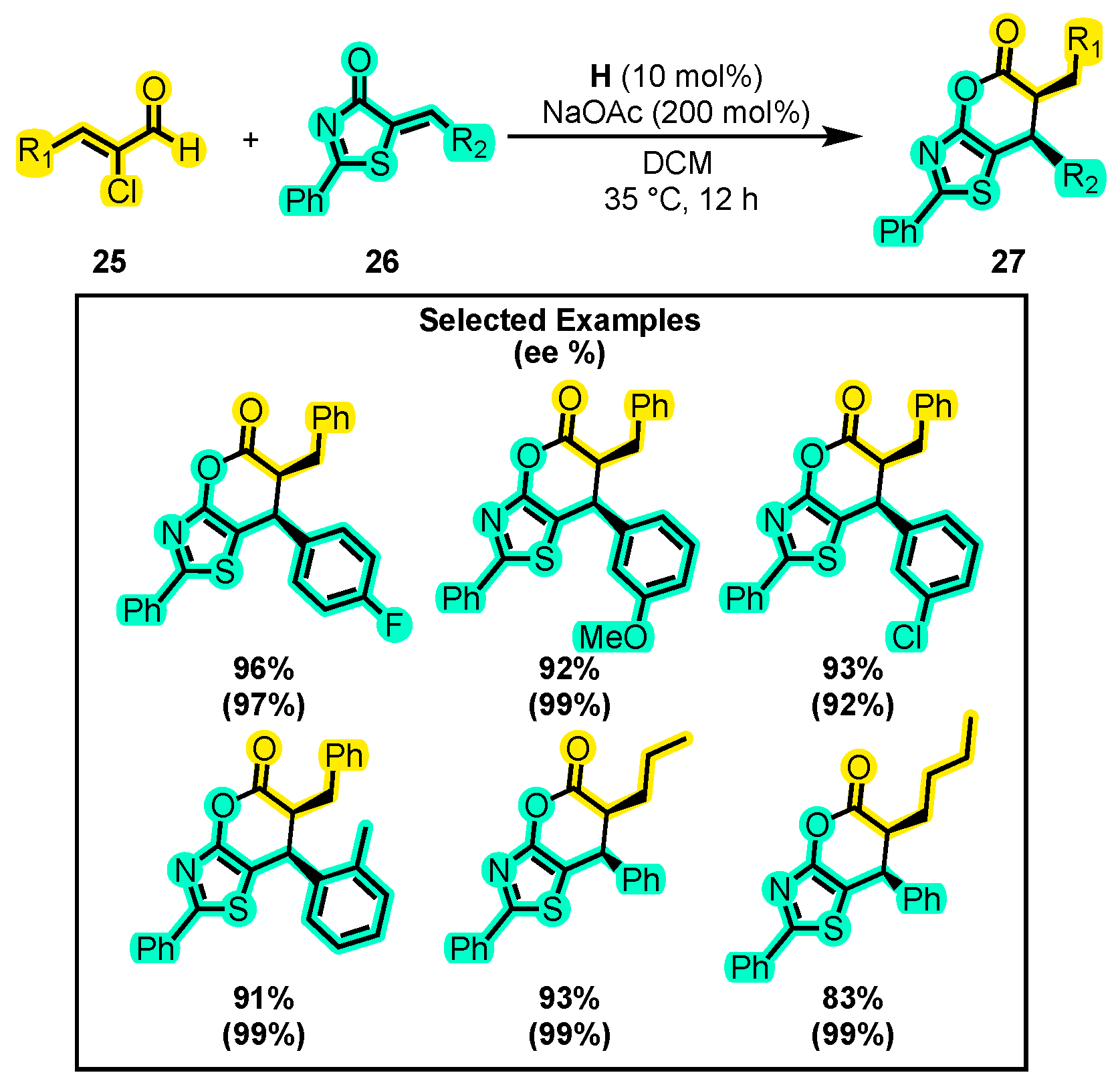
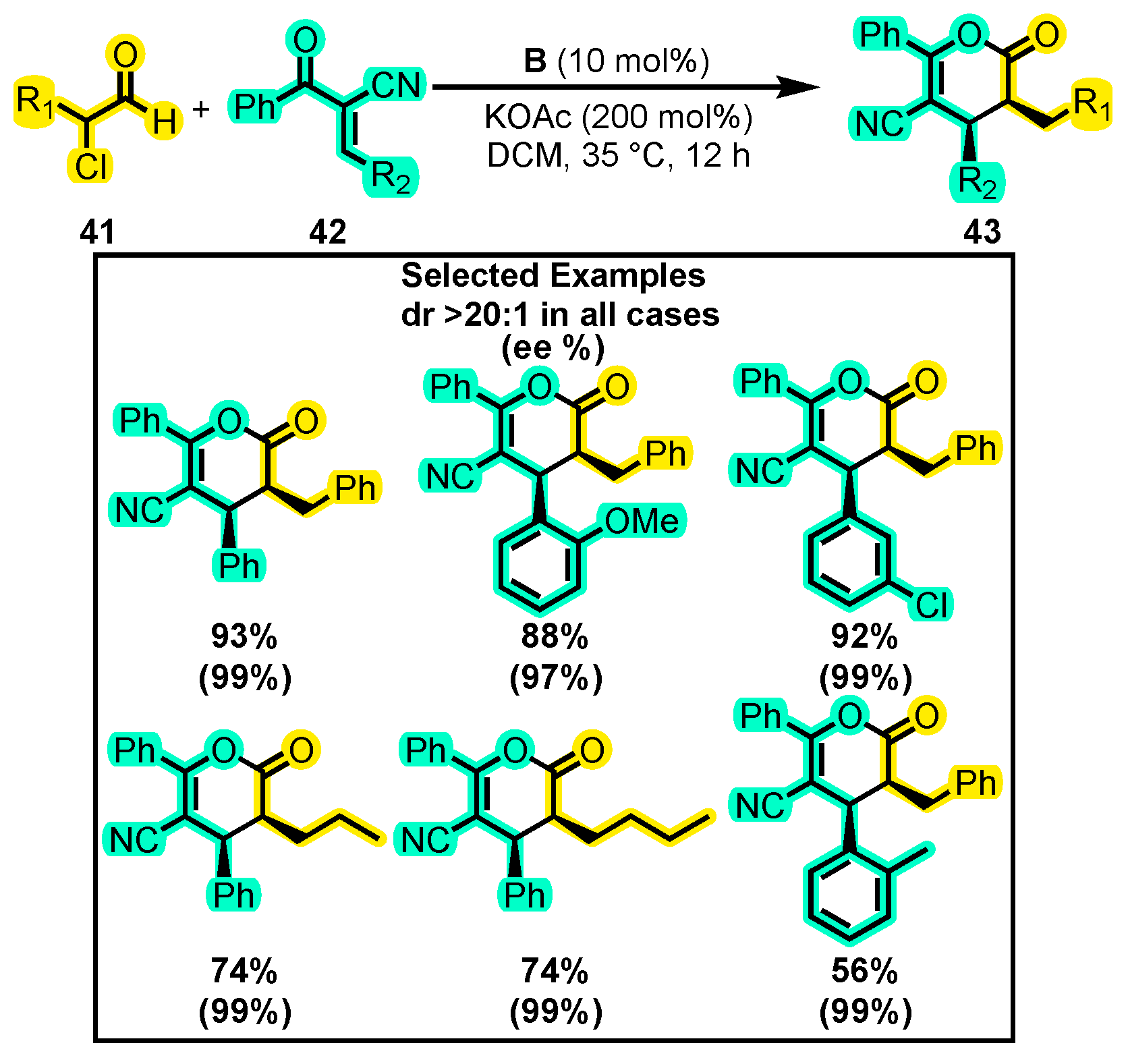
- Li, S.; Chen, X.Y.; Liu, Q.; Peuronen, A.; Rissanen, K.; Enders, D. N-Heterocyclic Carbene-Catalyzed Activation of α-Chloroaldehydes: Asymmetric Synthesis of 5-Cyano-Substituted Dihydropyranones. Synthesis 2017, 49, 4861–4868. [Google Scholar] [CrossRef]
- Saddiqa, A.; Usman, M.; Çakmak, O. Isocoumarins and 3,4-Dihydroisocoumarins, Amazing Natural Products: A Review. Turk. J. Chem. 2017, 41, 153–178. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Y.; Li, X.; Mao, Y.; Chen, W.; Zhan, R.; Huang, H. Enantioselective Synthesis of Pyrano [2,3-: C] Pyrrole via an Organocatalytic [4+2] Cyclization Reaction of Dioxopyrrolidines and Azlactones. Org. Biomol. Chem. 2019, 17, 3945–3950. [Google Scholar] [CrossRef]
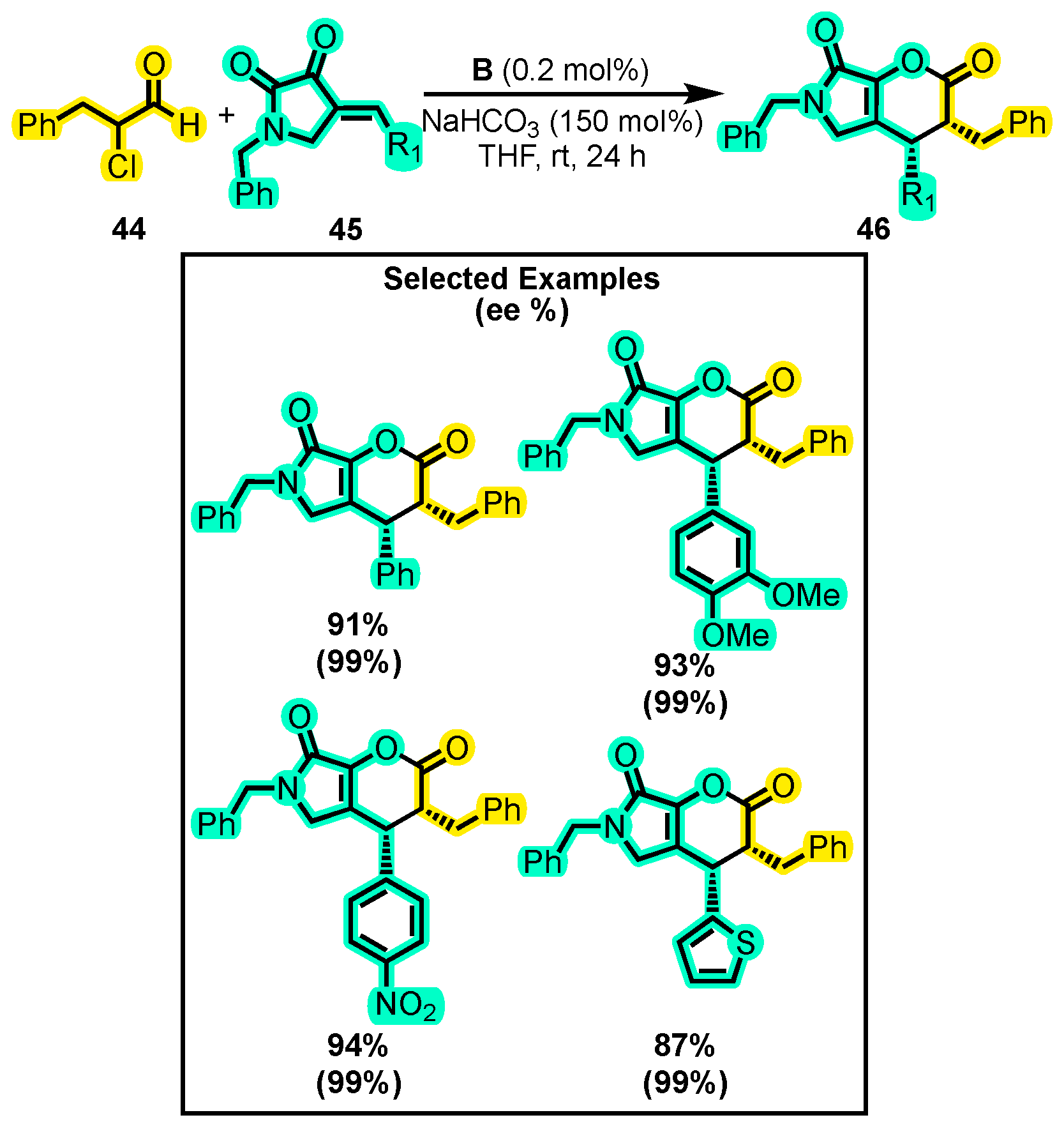
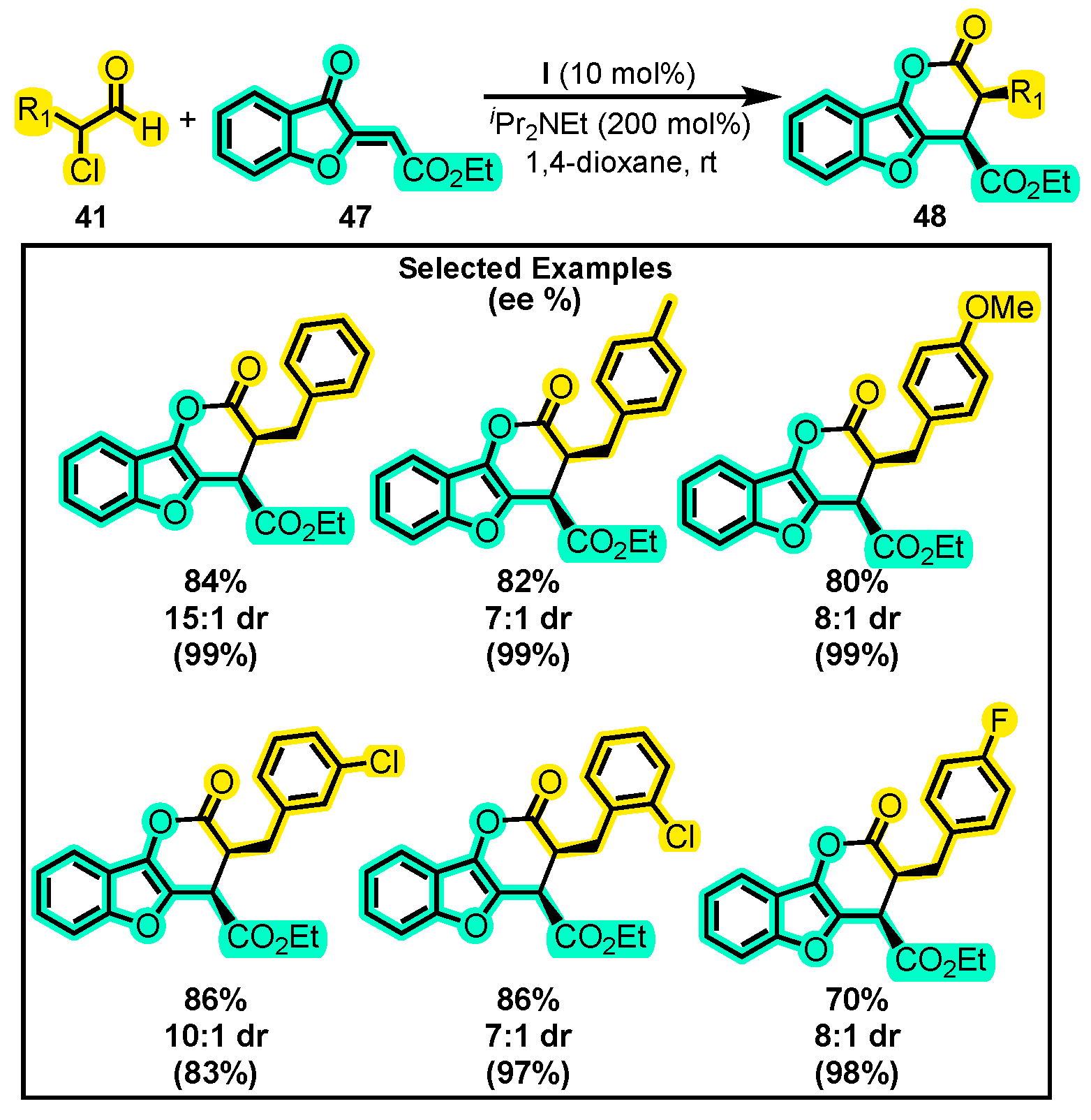
- Li, Y.; Chen, K.; Zhang, Y.; Sun, D.; Ye, S. N-Heterocyclic Carbene-Catalyzed [4+2] Cyclization of α-Chloroaldehydes and Aurones: Highly Enantioselective Synthesis of Benzofuran-Fused Dihydropyran-2-Ones. Chin. Chem. Lett. 2018, 29, 1209–1211. [Google Scholar] [CrossRef]
- Sheppard, T.D. Strategies for the Synthesis of 2,3-Dihydrobenzofurans. J. Chem. Res. 2011, 35, 377–385. [Google Scholar] [CrossRef]
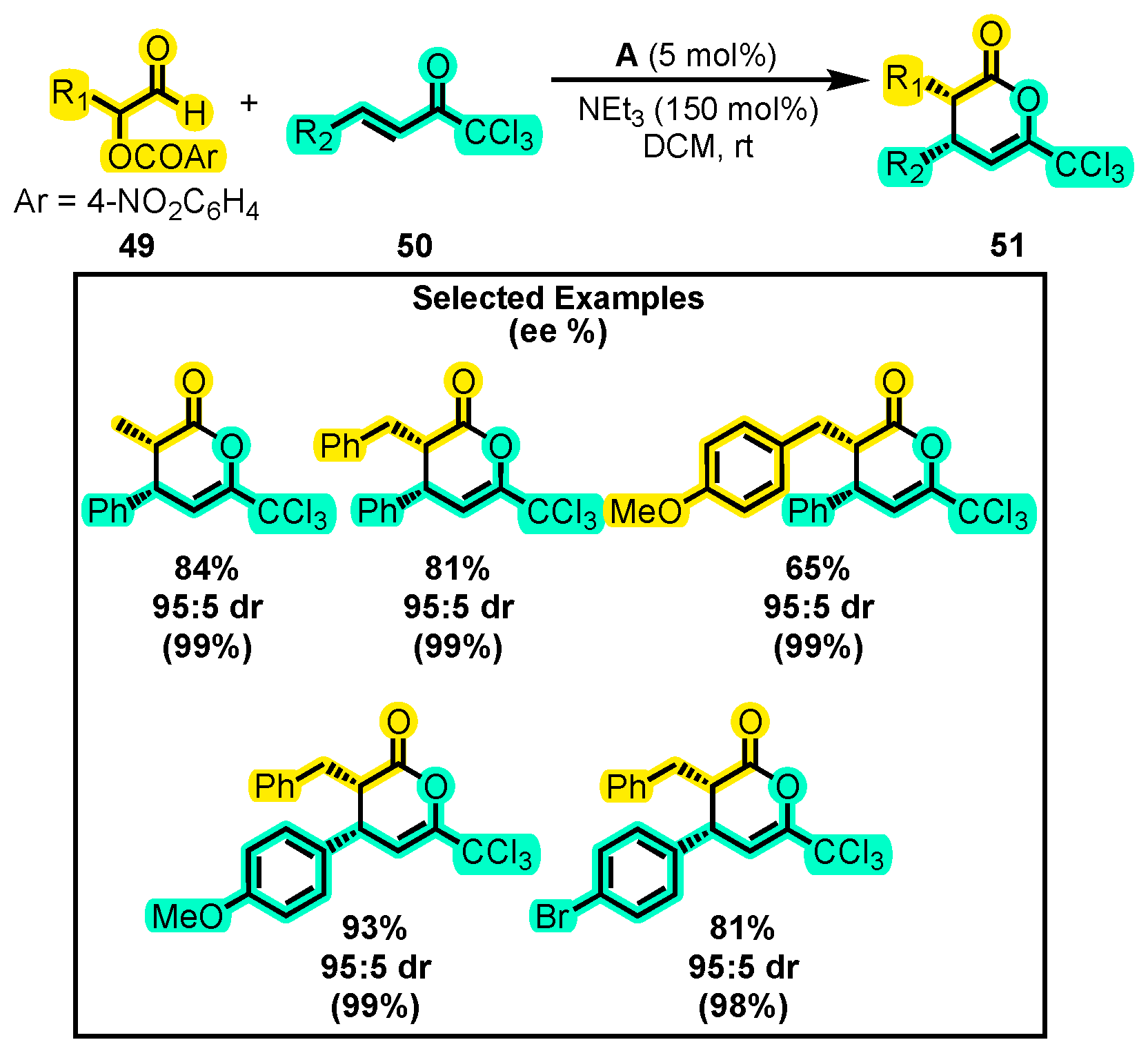
- Attaba, N.; Taylor, J.E.; Slawin, A.M.Z.; Smith, A.D. Enantioselective NHC-Catalyzed Redox [4+2]-Hetero-Diels-Alder Reactions Using α,β-Unsaturated Trichloromethyl Ketones as Amide Equivalents. J. Org. Chem. 2015, 80, 9728–9739. [Google Scholar] [CrossRef] [PubMed]
- Morrill, L.C.; Stark, D.G.; Taylor, J.E.; Smith, S.R.; Squires, J.A.; D’Hollander, A.C.A.; Simal, C.; Shapland, P.; O’Riordan, T.J.C.; Smith, A.D. Organocatalytic Michael Addition-Lactonisation of Carboxylic Acids Using α,β-Unsaturated Trichloromethyl Ketones as α,β-Unsaturated Ester Equivalents. Org. Biomol. Chem. 2014, 12, 9016–9027. [Google Scholar] [CrossRef]
- Taylor, J.E.; Davies, A.T.; Douglas, J.J.; Churchill, G.; Smith, A.D. Enantioselective NHC-Catalysed Redox [4+2]-Hetero-Diels-Alder Reactions Using α-Aroyloxyaldehydes and Unsaturated Ketoesters. Tet. Asymm. 2017, 28, 355–366. [Google Scholar] [CrossRef]
- Juhl, K.; Jørgensen, K.A. The First Organocatalytic Enantioselective Inverse-Electron-Demand Hetero-Diels-Alder Reaction. Angew. Chem. Int. Ed. 2003, 42, 1498–1501. [Google Scholar] [CrossRef] [PubMed]
- Katakam, N.K.; Kim, Y.; Headley, A.D. A Highly Enantioselective [4+2] Cycloaddition Involving Aldehydes and β,γ-Unsaturated-α-Keto Esters. Tet. Asymm. 2017, 28, 1591–1595. [Google Scholar] [CrossRef]
- Sinha, D.; Perera, S.; Zhao, J.C.G. Highly Enantioselective Inverse-Electron-Demand Hetero-Diels-Alder Reactions Catalyzed by Modularly Designed Organocatalysts. Chem. Eur. J. 2013, 19, 6976–6979. [Google Scholar] [CrossRef]
- Wang, J.; Yu, F.; Zhang, X.; Ma, D. Enantioselective Assembly of Substituted Dihydropyrones via Organocatalytic Reaction in Water Media. Org. Lett. 2008, 10, 2561–2564. [Google Scholar] [CrossRef] [PubMed]
- Prieto, L.; Sánchez-Díez, E.; Uria, U.; Reyes, E.; Carrillo, L.; Vicario, J.L. Catalytic Generation of Donor-Acceptor Cyclopropanes under N-Heterocyclic Carbene Activation and Their Stereoselective Reaction with Alkylideneoxindoles. Adv. Synth. Catal. 2017, 359, 1678–1683. [Google Scholar] [CrossRef]
- Hao, X.; Lin, L.; Tan, F.; Yin, C.; Liu, X.; Feng, X. Ligand Control of Diastereodivergency in Asymmetric Inverse Electron Demand Diels-Alder Reaction. ACS Catal. 2015, 5, 6052–6056. [Google Scholar] [CrossRef]
- Kappe, T.; Oliver Kappe, C.; Wentrup, C.; Färber, G. Direct Observation and Trapping of a Heterocyclic A-Oxo Ketene: 3-Carbonyl-1,3-dihydro-1-methyl-2H-indol-2-one. Chem. Ber. 1993, 126, 2357–2360. [Google Scholar] [CrossRef]
- Ren, Q.; Li, M.; Yuan, L. Expeditious Assembly of Fused Dihydropyranones via N-Heterocyclic Carbene-Catalyzed Tandem Michael Addition/Lactonization. Org. Biomol. Chem. 2017, 15, 1329–1333. [Google Scholar] [CrossRef]
- Choura, E.; Ncir, M.; Maalej, E.; Marco-Contelles, J.; Ismaili, L.; Chabchoub, F. New Method for the Synthesis of 4-Aryl-3,4-Dihydro-2H-Benzo[g]Chromene-2,5,10-Triones. Synth. Commun. 2019, 49, 2834–2839. [Google Scholar] [CrossRef]
- Hamama, W.S.; Hassanien, A.E.; Zoorob, H.H. Concise Synthesis and Pharmacological Applications of New α-Lapachone Analogues. J. Heterocycl. Chem. 2018, 55, 282–290. [Google Scholar] [CrossRef]
- Garkavtsev, I.; Chauhan, V.P.; Wong, H.K.; Mukhopadhyay, A.; Glicksman, M.A.; Peterson, R.T.; Jain, R.K. Dehydro-α-Lapachone, a Plant Product with Antivascular Activity. Proc. Natl. Acad. Sci. USA 2011, 108, 11596–11601. [Google Scholar] [CrossRef]
- Sun, W.; Frost, B.; Liu, J. Oleuropein, Unexpected Benefits! Oncotarget 2017, 8, 17409. [Google Scholar] [CrossRef]
- Rigakou, A.; Diamantakos, P.; Melliou, E.; Magiatis, P. S-(E)-Elenolide: A New Constituent of Extra Virgin Olive Oil. J. Sci. Food Agric. 2019, 99, 5319–5326. [Google Scholar] [CrossRef]
- Vedachalam, S.; Murugesh, N.; Chakraborty, P.; Karvembu, R.; Liu, X.W. NHC Catalyzed Enantioselective Coates-Claisen Rearrangement: A Rapid Access to the Dihydropyran Core for Oleuropein Based Secoiridoids. New J. Chem. 2018, 42, 1832–1839. [Google Scholar] [CrossRef]
- Kirmse, W. 100 Years of the Wolff Rearrangement. Eur. J. Org. Chem. 2002, 2002, 2193–2256. [Google Scholar] [CrossRef]
- Wang, C.; Wang, Z.; Yang, J.; Shi, S.H.; Hui, X.P. Sequential Visible-Light and N-Heterocyclic Carbene Catalysis: Stereoselective Synthesis of Tetrahydropyrano [2,3-b]Indoles. Org. Lett. 2020, 22, 4440–4443. [Google Scholar] [CrossRef] [PubMed]
- Zaragozá, R.J.; Aurell, M.J.; González-Cardenete, M.A. A Theoretical Study on NHC-Catalysed Enantioselective Cycloaddition of Ketenes and 3-Aroylcoumarins: Mechanism and Enantioselectivity. Org. Biomol. Chem. 2018, 16, 5474–5482. [Google Scholar] [CrossRef] [PubMed]
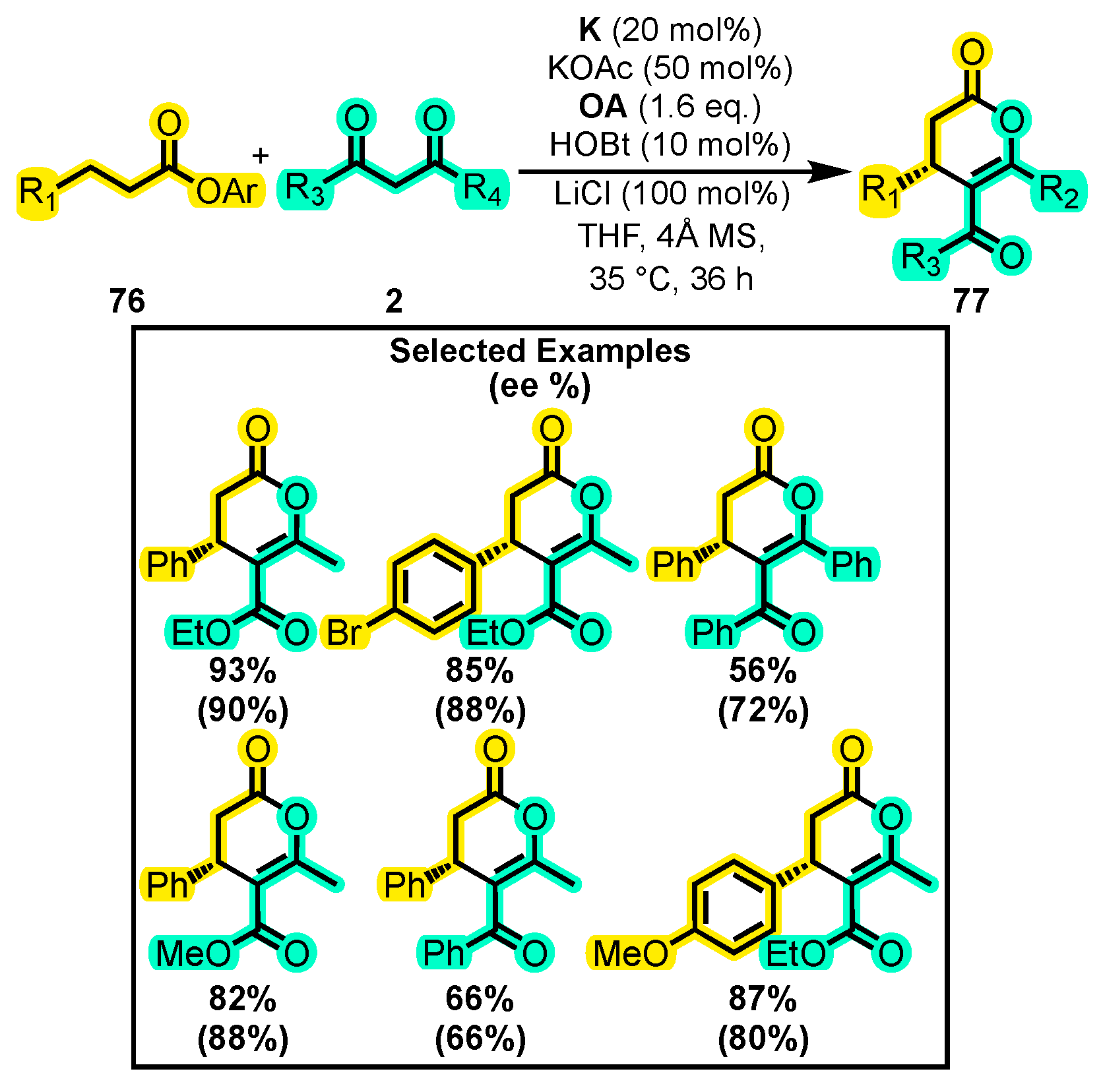
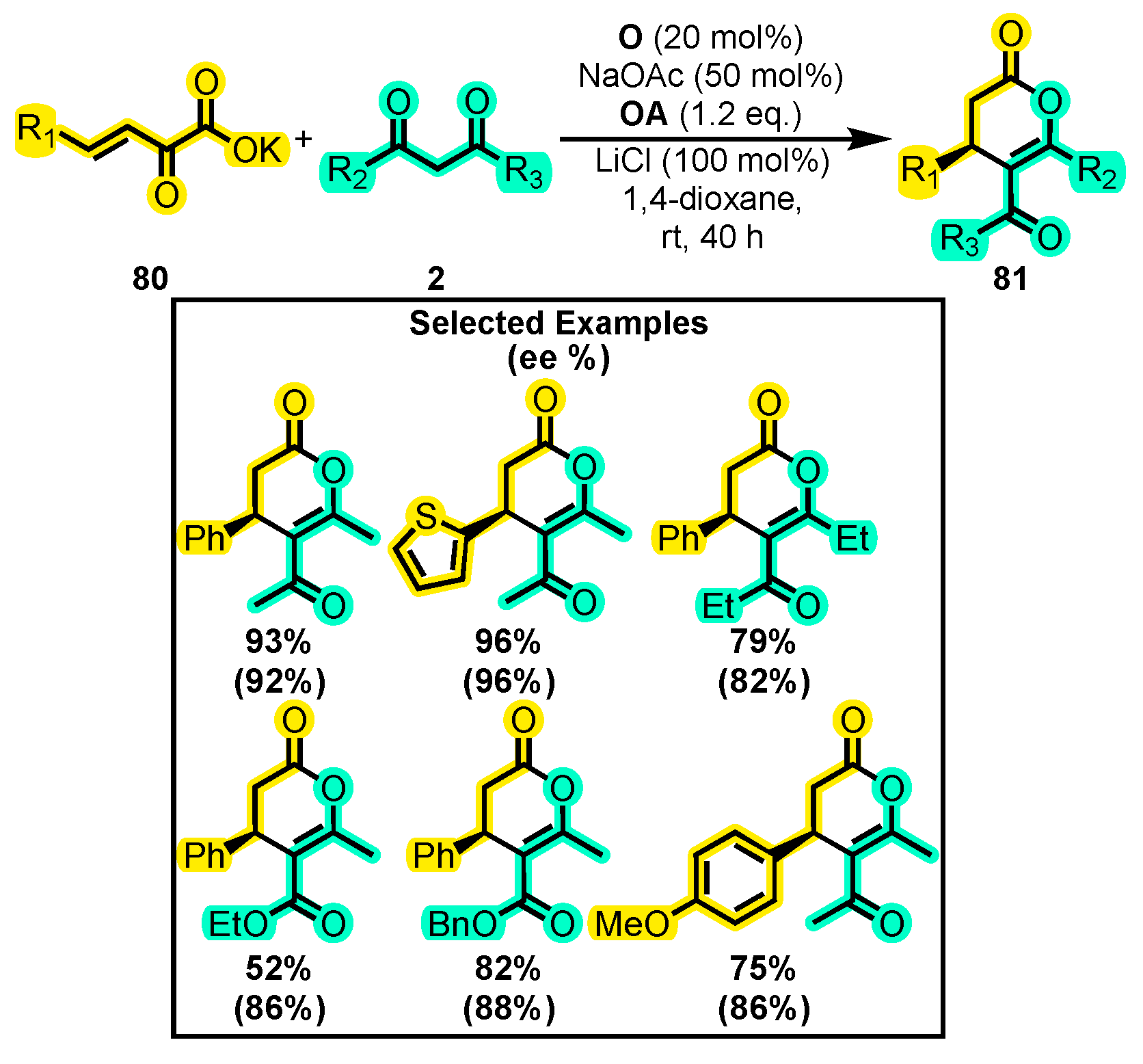
- Que, Y.; Lu, Y.; Wang, W.; Wang, Y.; Wang, H.; Yu, C.; Li, T.; Wang, X.S.; Shen, S.; Yao, C. An Enantioselective Assembly of Dihydropyranones through an NHC/LiCl-Mediated in Situ Activation of α,β-Unsaturated Carboxylic Acids. Chem. Asian J. 2016, 11, 678–681. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, J.; Cao, J.; Fang, C.; Zhu, J.; Lu, T.; Du, D. N-Heterocyclic Carbene-Mediated Formal [3+3] Annulation of Isatin-Derived α, β-Unsaturated Acids: Access to Functionalized 3,4′-Spirooxindole δ-Lactones. Tetrahedron 2017, 73, 3249–3254. [Google Scholar] [CrossRef]
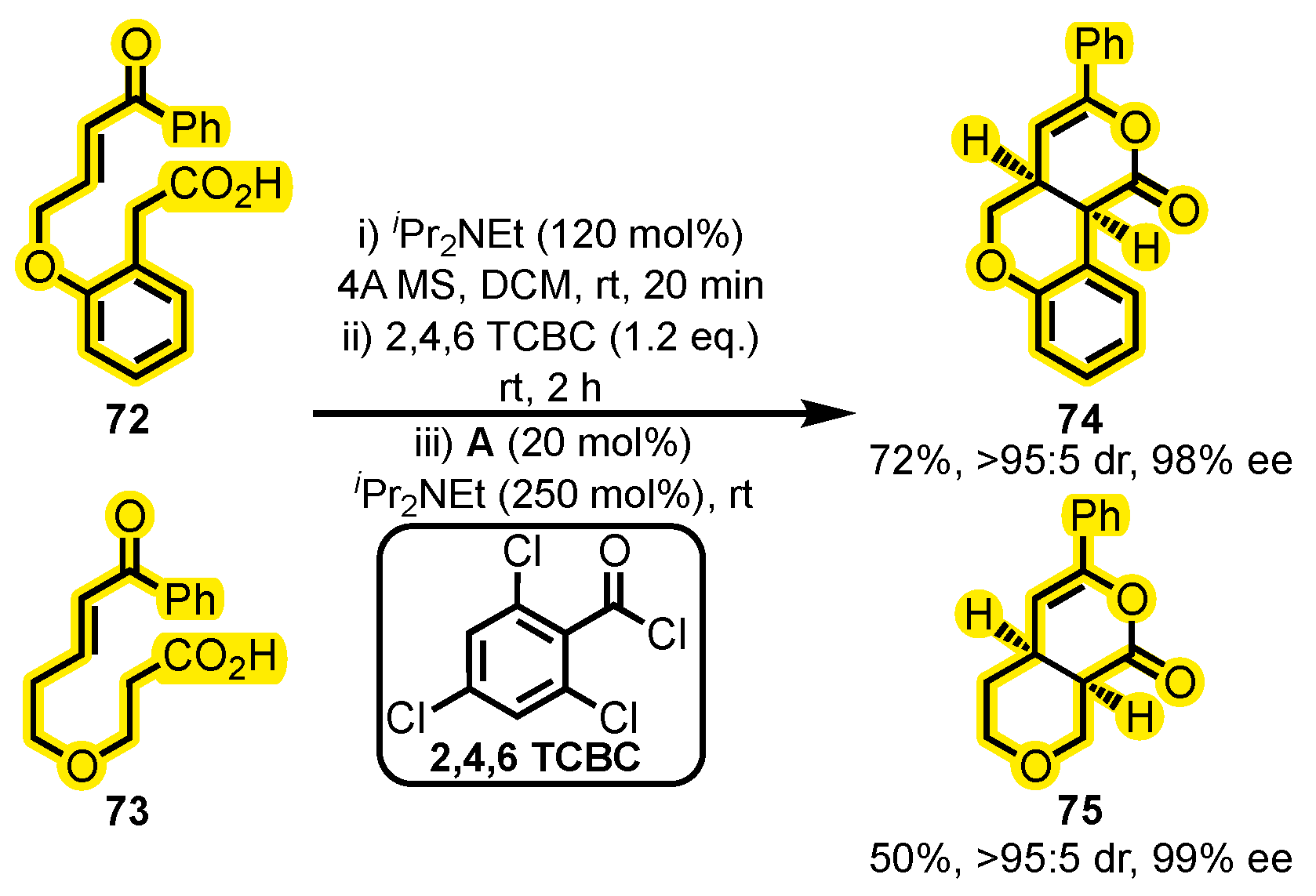
- Attaba, N.; Smith, A.D. NHC-Catalysed Enantioselective Intramolecular Formal [4+2] Cycloadditions Using Carboxylic Acids as Azolium Enolate Precursors. Tetrahedron 2020, 76, 130835. [Google Scholar] [CrossRef]
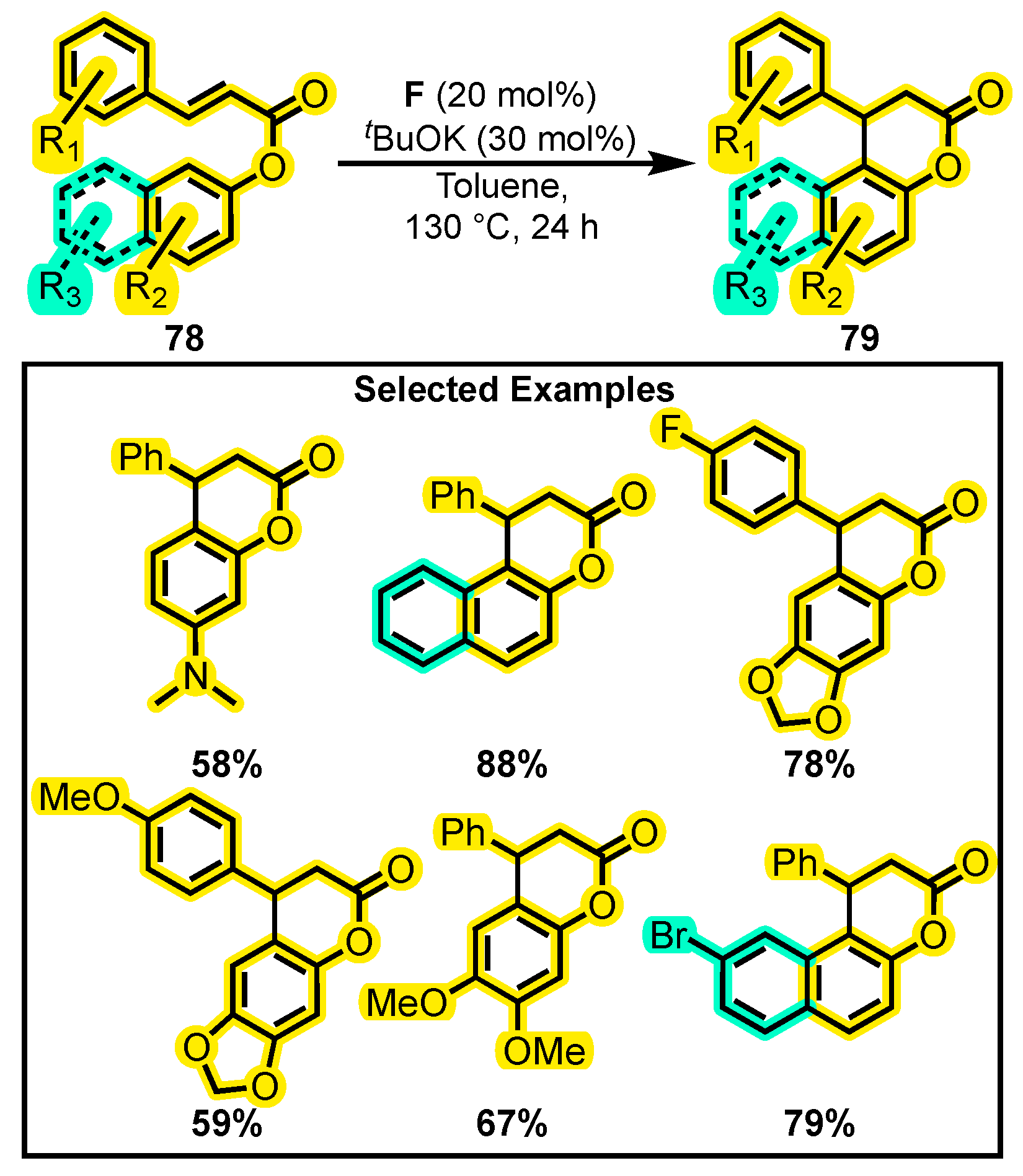
- Neyyappadath, R.M.; Cordes, D.B.; Slawin, A.M.Z.; Smith, A.D. 6-Exo-Trig Michael Addition-Lactonizations for Catalytic Enantioselective Chromenone Synthesis. Chem. Commun. 2017, 53, 2555–2558. [Google Scholar] [CrossRef]
- Zhou, W.; Ni, C.; Chen, J.; Wang, D.; Tong, X. Enantioselective Synthesis of 4H-Pyran via Amine-Catalyzed Formal (3 + 3) Annulation of δ-Acetoxy Allenoate. Org. Lett. 2017, 19, 1890–1893. [Google Scholar] [CrossRef]
- Wang, X.B.; Yang, C.S.; Luo, J.G.; Zhang, C.; Luo, J.; Yang, M.H.; Kong, L.Y. Experimental and Theoretical Calculation Studies on the Structure Elucidation and Absolute Configuration of Calyxins from Alpinia Katsumadai. Fitoterapia 2017, 119, 121–129. [Google Scholar] [CrossRef]
- Liu, B.; Wang, W.; Huang, R.; Yan, J.; Wu, J.; Xue, W.; Yang, S.; Jin, Z.; Chi, Y.R. Direct Activation of β-sp3-Carbons of Saturated Carboxylic Esters as Electrophilic Carbons via Oxidative Carbene Catalysis. Org. Lett. 2018, 20, 260–263. [Google Scholar] [CrossRef]
- Zheng, J.; Zhu, K.; Yuan, Y.; Cheng, J. N-Heterocyclic Carbene/Potassium Ion Cooperatively Catalyzed Phenylation Reactions of α,β-Unsaturated Acylazoliums. Eur. J. Org. Chem. 2016, 2016, 4569–4576. [Google Scholar] [CrossRef]
- Gao, Y.; Ma, Y.; Xu, C.; Li, L.; Yang, T.; Sima, G.; Fu, Z.; Huang, W. Potassium 2-Oxo-3-Enoates as Effective and Versatile Surrogates for α, β-Unsaturated Aldehydes in NHC-Catalyzed Asymmetric Reactions. Adv. Synth. Catal. 2018, 360, 479–484. [Google Scholar] [CrossRef]
- Zhu, S.Y.; Zhang, H.; Ma, Q.W.; Liu, D.; Hui, X.P. Oxidative NHC Catalysis: Direct Activation of β-sp3 Carbons of Saturated Acid Chlorides. Chem. Commun. 2019, 55, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Gillard, R.; Jahanbakhsh, A.; Breugst, M.; Lupton, D.W. Enantioselective N-Heterocyclic Carbene Catalysis via Acyl Azolium without Exogenous Oxidants. ACS Catal. 2020, 10, 11791–11796. [Google Scholar] [CrossRef]
- Ni, Q.; Xiong, J.; Song, X.; Raabe, G.; Enders, D. NHC-Catalyzed Activation of α,β-Unsaturated N-Acyltriazoles: An Easy Access to Dihydropyranones. Chem. Commun. 2015, 51, 14628–14631. [Google Scholar] [CrossRef] [PubMed]
- Guisado-Barrios, G.; Soleilhavoup, M.; Bertrand, G. 1 H-1,2,3-Triazol-5-Ylidenes: Readily Available Mesoionic Carbenes. Acc. Chem. Res. 2018, 51, 3236–3244. [Google Scholar] [CrossRef]
- Kano, T.; Tokuda, O.; Maruoka, K. Synthesis of a Biphenyl-Based Axially Chiral Amino Acid as a Highly Efficient Catalyst for the Direct Asymmetric Aldol Reaction. Tet. Lett. 2006, 47, 7423–7426. [Google Scholar] [CrossRef]
- Lombardo, M.; Easwar, S.; Pasi, F.; Trombinia, C. The Ion Tag Strategy as a Route to Highly Efficient Organocatalysts for the Direct Asymmetric Aldol Reaction. Adv. Synth. Catal. 2009, 351, 276–282. [Google Scholar] [CrossRef]
- Rodríguez, B.; Bolm, C. Thermal Effects in the Organocatalytic Asymmetric Mannich Reaction. J. Org. Chem. 2006, 71, 2888–2891. [Google Scholar] [CrossRef]
- Rueping, M.; Antonchick, A.P.; Theissmann, T. Remarkably Low Catalyst Loading in Brønsted Acid Catalyzed Transfer Hydrogenations: Enantioselective Reduction of Benzoxazines, Benzothiazines, and Benzoxazinones. Angew. Chem. Int. Ed. 2006, 45, 6751–6755. [Google Scholar] [CrossRef]
- Wiesner, M.; Upert, G.; Angelici, G.; Wennemers, H. Enamine Catalysis with Low Catalyst Loadings—High Efficiency via Kinetic Studies. J. Am. Chem. Soc. 2010, 132, 6–7. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, P.H.; Li, Y.; Lauridsen, V.H.; Jørgensen, D.K.B.; Palazzo, T.A.; Meazza, M.; Jørgensen, K.A. Organocatalytic Formation of Chiral Trisubstituted Allenes and Chiral Furan Derivatives. Angew. Chem. Int. Ed. 2018, 57, 10661–10665. [Google Scholar] [CrossRef]
- He, M.; Struble, J.R.; Bode, J.W. Highly Enantioselective Azadiene Diels—Alder Reactions Catalyzed by Chiral. J. Am. Chem. Soc. 2006, 128, 8418–8420. [Google Scholar] [CrossRef] [PubMed]
- Das, T.K.; Biju, A.T. A Method for the Analysis of Free Carbenes Present after NHC-Organocatalyzed Transformations. Eur. J. Org. Chem. 2017, 2017, 4500–4506. [Google Scholar] [CrossRef]







































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales-Manrique, C.; Baquero, E.A.; Guevara-Pulido, J. Recent Advances in the Synthesis of 3,4-Dihydropyran-2-Ones Organocatalyzed by N-Heterocyclic Carbenes. Molecules 2023, 28, 3743. https://doi.org/10.3390/molecules28093743
Morales-Manrique C, Baquero EA, Guevara-Pulido J. Recent Advances in the Synthesis of 3,4-Dihydropyran-2-Ones Organocatalyzed by N-Heterocyclic Carbenes. Molecules. 2023; 28(9):3743. https://doi.org/10.3390/molecules28093743
Chicago/Turabian StyleMorales-Manrique, Camilo, Edwin A. Baquero, and James Guevara-Pulido. 2023. "Recent Advances in the Synthesis of 3,4-Dihydropyran-2-Ones Organocatalyzed by N-Heterocyclic Carbenes" Molecules 28, no. 9: 3743. https://doi.org/10.3390/molecules28093743