
Chemical Bonding and Dynamic Structural Fluxionality of a Boron-Based Na5B7 Sandwich Cluster
by
 , and
, and
Peng-Fei Han
1,
Ying-Jin Wang
2,
Lin-Yan Feng
1,2,
Shu-Juan Gao
1,
Qiang Sun
3 and
Hua-Jin Zhai
1,* 1
Nanocluster Laboratory, Institute of Molecular Science, Shanxi University, Taiyuan 030006, China
2
Department of Chemistry, Xinzhou Teachers University, Xinzhou 034000, China
3
Center for Applied Physics and Technology, School of Materials Science and Engineering, Peking University, Beijing 100871, China
*
Author to whom correspondence should be addressed.
Molecules 2023, 28(7), 3276; https://doi.org/10.3390/molecules28073276
Submission received: 15 March 2023
/
Revised: 3 April 2023
/
Accepted: 5 April 2023
/
Published: 6 April 2023
(This article belongs to the Special Issue Aromatic Inorganic and Metallic Compounds)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Doping alkali metals into boron clusters can effectively compensate for the intrinsic electron deficiency of boron and lead to interesting boron-based binary clusters, owing to the small electronegativity of the former elements. We report on the computational design of a three-layered sandwich cluster, Na5B7, on the basis of global-minimum (GM) searches and electronic structure calculations. It is shown that the Na5B7 cluster can be described as a charge-transfer complex: [Na4]2+[B7]3−[Na]+. In this sandwich cluster, the [B7]3− core assumes a molecular wheel in shape and features in-plane hexagonal coordination. The magic 6π/6σ double aromaticity underlies the stability of the [B7]3− molecular wheel, following the (4n + 2) Hückel rule. The tetrahedral Na4 ligand in the sandwich has a [Na4]2+ charge-state, which is the simplest example of three-dimensional aromaticity, spherical aromaticity, or superatom. Its 2σ electron counting renders σ aromaticity for the ligand. Overall, the sandwich cluster has three-fold 6π/6σ/2σ aromaticity. Molecular dynamics simulation shows that the sandwich cluster is dynamically fluxional even at room temperature, with a negligible energy barrier for intramolecular twisting between the B7 wheel and the Na4 ligand. The Na5B7 cluster offers a new example for dynamic structural fluxionality in molecular systems.
1. Introduction
Due to the electron deficiency of boron with three valence electrons (2s22p1), boron and its relevant compounds and clusters have rich and unique structures, as well as unconventional chemical bonding [1,2,3,4,5,6,7,8]. Recent experimental and theoretical studies have explored the structures and bonding of a wide range of boron-based clusters. It is now known that elemental boron clusters assume two-dimensional (2D) planar or quasi-planar geometries with up to 40 atoms for anions, extended 2D sheet structures (borophenes), and three-dimensional (3D) structures such as borospherenes [8,9,10,11,12,13]. Chemical bonding in these clusters is governed by π/σ aromaticity, antiaromaticity, and conflicting aromaticity, in which electron delocalization is essential in order to compensate for the intrinsic electron deficiency of boron.
Boron-based cluster nanomachines represent a new research direction in physical chemistry, in which small clusters demonstrate dynamic structural fluxionality [14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31]. Such unique dynamic behaviors are also dictated by boron’s electron deficiency. Researchers continuously designed and reported a series of pure boron clusters with dynamic fluxionality, such as B19−, B13+, B182−, B20−, B11−/0, and B15+ [14,15,16,17,18,19,20,21,22]. In subsequent studies, researchers discovered that doping or mixing low electronegativity metals to form boron-based clusters is an effective way toward the rational design of metal-doped boron-based nanosystems with dynamic fluxionality [23,24,25,26,27,28,29,30,31]. For example, a binary Mg2B8 cluster was computationally designed as a “nanocompass”, in which an Mg2 needle was shown to rotate freely on the B8 baseplate [23]. By doping alkali metals to boron clusters, a series of boron-based sandwich clusters (B7Li4+, Na6B7−, and Na8B7+) were lately studied theoretically, which also show dynamic fluxionality [29,30]. In 2017, Zhai and coworkers discovered two virtually isoenergetic triple-layered and helix-type structures for a Be6B11− cluster [31]. The former sandwich structure demonstrated dual dynamic rotation/twisting modes of structural fluxionality, akin to an earth–moon system at the nanoscale.
Alkali metal elements are clearly an ideal choice for doping due to their small electronegativities, which would allow precise tuning of the number of valence electrons in a boron-based alloy cluster, one at a time. The purpose of this work is to explore how the structure of a binary Na-B cluster depends on its ratio of Na versus B components, what underlies the stability of such a cluster structure, and whether new examples can be offered for dynamic structural fluxionality. To this end, we have reached a binary Na5B7 cluster. Computational global-minimum (GM) structure searches reveal a sandwich-type geometry for this cluster, which features a B7 molecular wheel sandwiched in between a Na4 tetrahedron and an isolated Na atom. Chemical bonding analysis suggests double 6π/6σ aromaticity for the B7 molecular wheel, as well as 2σ aromaticity for the tetrahedral Na4 ligand. In other words, the sandwich Na5B7 cluster has collectively three-fold 6π/6σ/2σ aromaticity. The sandwich shape allows the precise number of electrons to be transferred from the Na4 and Na ligands to the B7 core, so that the whole sandwich system can be stabilized via electrostatics. The cluster is faithfully formulated as [Na4]2+[B7]3−[Na]+; that is, a charge-transfer complex. The unique sandwich shape of the cluster facilitates dynamic structural fluxionality, as confirmed in molecular dynamics simulations. This work also highlights the robustness of [B7]3− as a potential inorganic ligand.
2. Results
2.1. Global-Minimum and Transition-State Structures
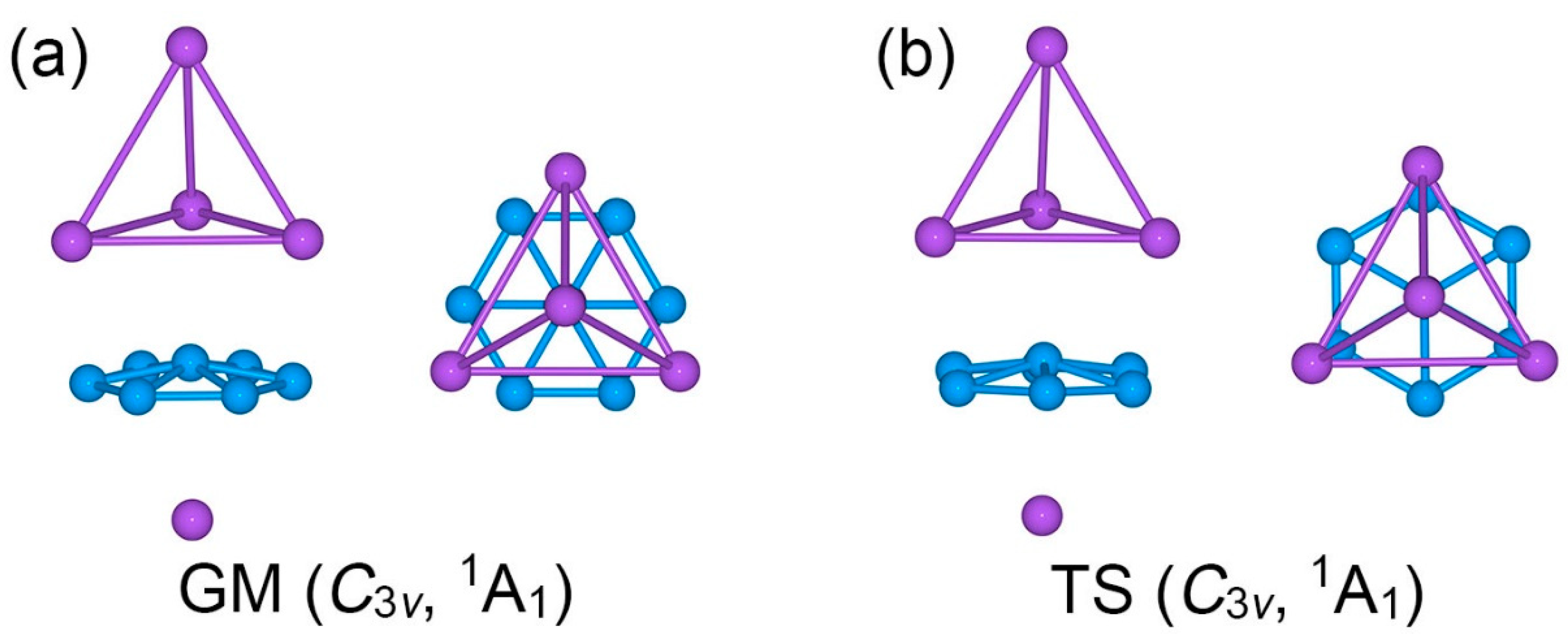
The optimized low-lying structures of Na5B7 cluster at the PBE0/6-311 + G* level are shown in the Supplementary Materials (Figure S1). The GM Na5B7 cluster is identified herein using the global searches and electronic structure calculations at three levels of theory. The Cartesian coordinates for GM C3v (1A1) Na5B7 cluster at the PBE0/6-311 + G* level are presented in Table S1. The PBE0/6-311 + G* method has been widely used for boron-based clusters [8,31,32]. The comparative B3LYP/6-311 + G* data for top candidate structures serve to check for the consistency of different functionals in terms of geometries and energetics. The single-point CCSD(T) calculations on the basis of optimized PBE0/6-311 + G* geometries allow benchmarking of the energetics. All three levels of theory confirm C3v (1A1) structure (Figure 1a) as the GM of Na5B7 cluster.
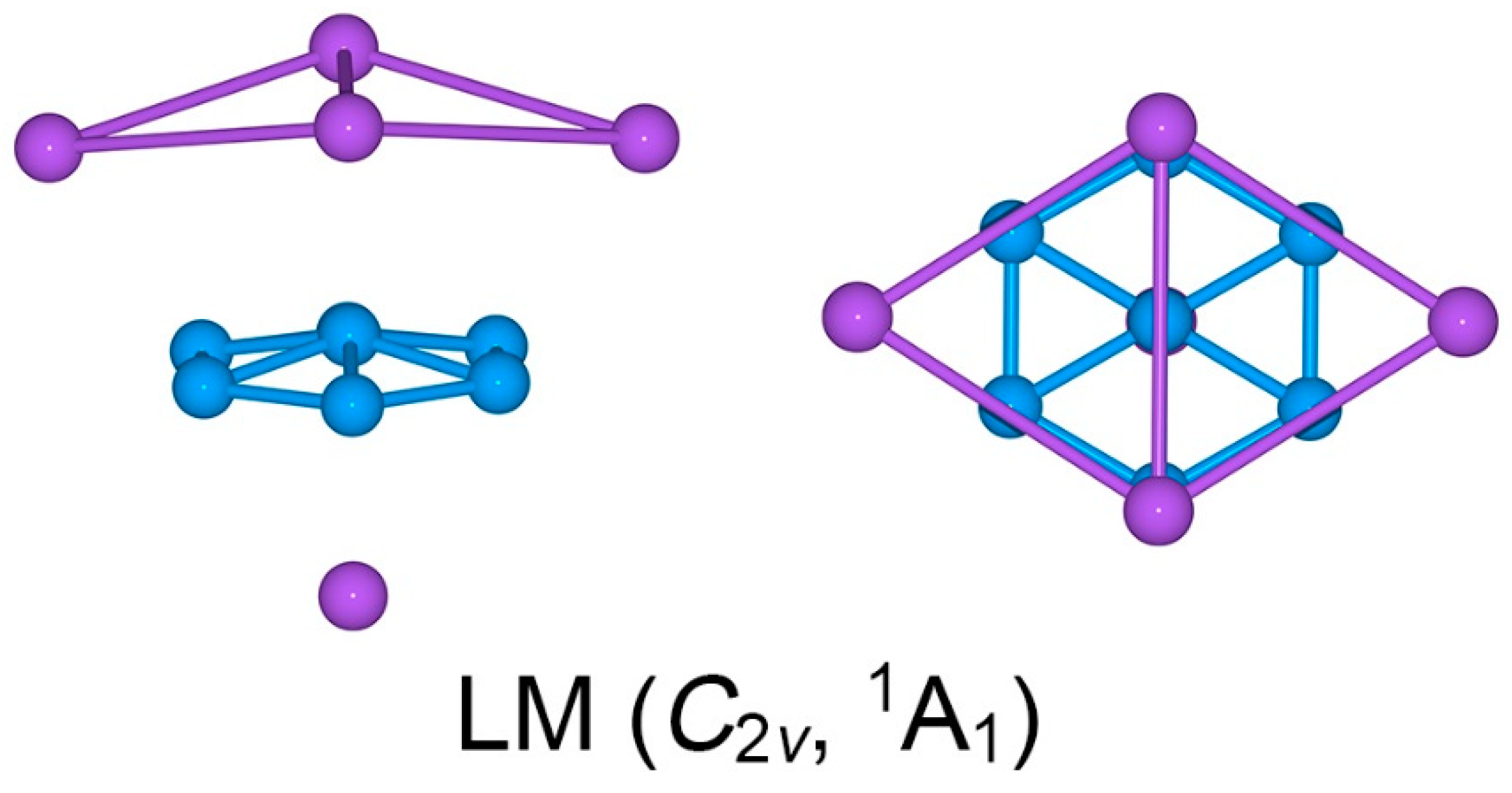
Among the low-lying isomers is a local-minimum (LM) C2v (1A1) structure (Figure S1), which is about 0.2 eV above the GM cluster. The remaining isomers are substantially higher in energy. Considering that adequate intramolecular charge transfers can take place from Na to B, the triplet-state structures are relatively unimportant for the present cluster system with an even number of 26 valence electrons. Indeed, our calculations show that the corresponding triplet-state structures for the top 12 singlet-state isomers in Figure S1 are 0.82–2.20 eV higher than the GM cluster at the PBE0/6-311 + G* level. Specifically, as illustrated in Figure S2, the lowest-lying triplet-state geometry of Na5B7 cluster, C1 (3A), lies 0.82 eV higher above the GM structure. Other triplet-state geometries are even higher in energy, by up to 2.20 eV (relative to the singlet GM cluster), for the 12th isomeric triplet structure. Thus, the C3v (1A1) cluster (Figure 1a) is reasonably well defined on its potential energy surface as the real GM structure. Alternative structures, either singlets or triplets, are not energetically competitive, except for an LM C2v (1A1) structure as mentioned above.
The top- and side-views of GM C3v (1A1) Na5B7 cluster are presented in Figure 1a. Basically, it is a three-layered sandwich cluster. The boron component forms a quasi-planar B7 molecular wheel as the core layer of the sandwich. It features six-fold in-plane coordination for the central B site. On the other hand, the Na5 component is divided into a Na4 tetrahedron and an isolated Na atom. The two Na-based layers serve as ligands for the B7 molecular wheel. Specifically, three Na atoms in the Na4 tetrahedron are each situated on a B–B edge. The GM sandwich cluster may be referred to as the staggered conformation.
Twisting the Na4 tetrahedron against the B7 molecular wheel by 30°, one readily reaches another C3v (1A1) structure, which turns out to be a transition state (TS), as depicted in Figure 1b. The TS structure is referred to herein as the eclipsed conformation. The closest low-lying structure, LM C2v (1A1), is also a sandwich (Figure 2). It differs from the GM structure in the top Na4 ligand. The Na4 ligand in the LM cluster is distorted into a roof-like (or rhombic) structure, so that only two Na atoms are coordinated to the B–B edges. The distortion alters the nature of bonding in the Na4 unit (vide infra).
2.2. Bond Distances, Wiberg Bond Indices, and Natural Atomic Charges
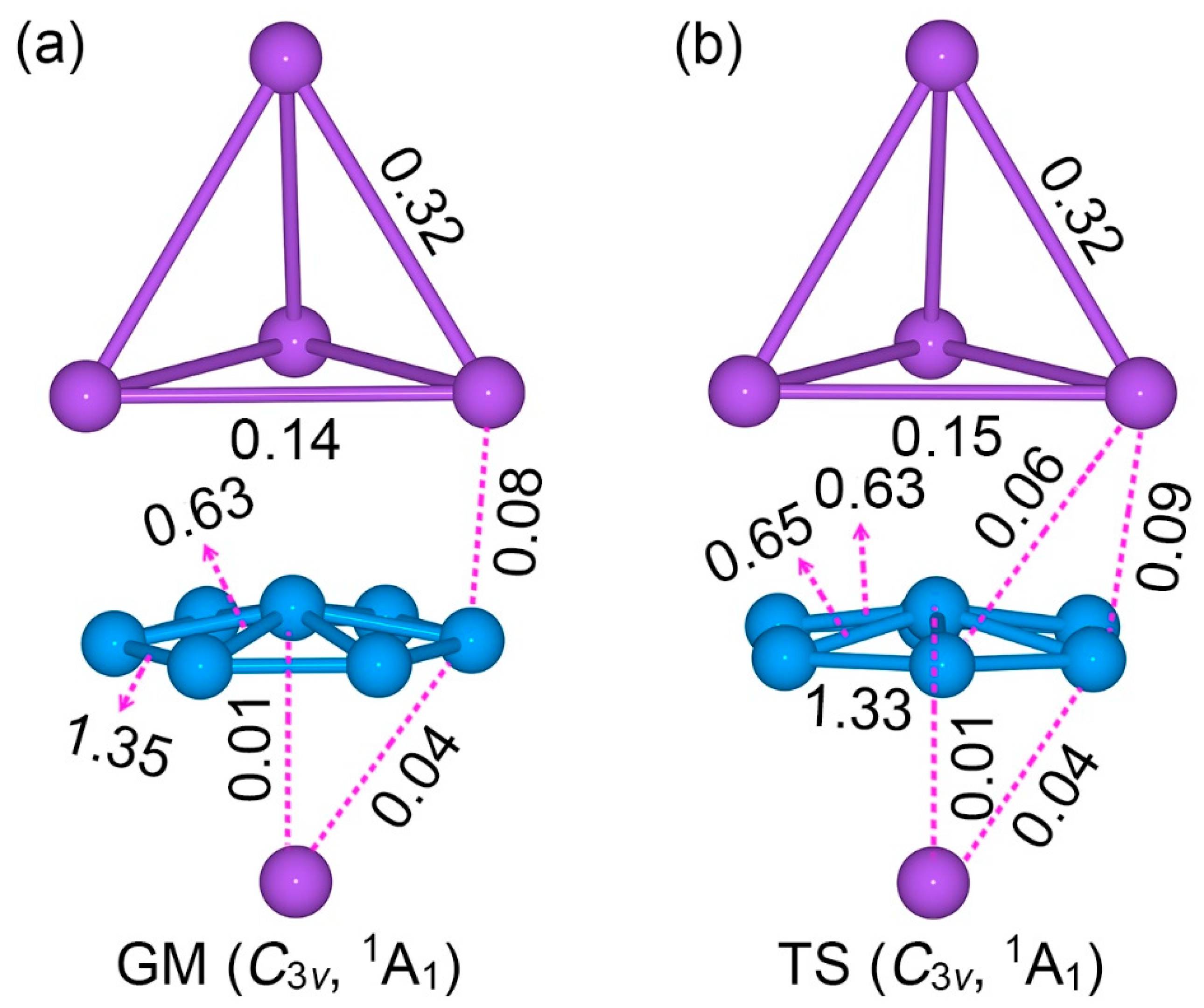
The calculated bond distances, Wiberg bond indices (WBIs), and natural atomic charges for GM and TS structures of the Na5B7 cluster are shown in Figure S3, Figure 3, and Figure S4, respectively. The three sets of data are generally coherent with each other. In particular, the GM and TS structures are quite similar, either qualitatively or quantitatively. Therefore, we shall primarily describe the GM cluster only.
In the GM cluster, the B7 wheel has uniform B–B distances for the peripheral links (1.61 Å) and radial ones (1.64 Å). These distances are shorter than the single bond (upper limit: 1.70 Å) [33]. It is invaluable to compare these with bare B7 cluster, whose peripheral and radial distances are 1.56–1.62 and 1.69–1.76 Å, respectively [34]. Thus, the peripheral B6 ring in GM Na5B7 cluster seems to be expanded, despite the fact that the radial B–B distances are clearly shortened. The above structural data are in line with a negatively charged B7 wheel in GM Na5B7 cluster.
The Na–Na distances for Na4 tetrahedron in the GM structure are 3.60/3.64 Å, whose shape also differs distinctly from that of a bare Na4 cluster. The latter assumes a rhombic D2h structure [35]. The above Na–Na distances are far longer than a Na–Na single bond (3.10 Å) [33]. The tetrahedral Na4 ligand is not in a neutral state (vide infra). The shortest B–Na distance is 2.51 Å, which is longer than the recommended value for a B–Na σ single bond. The calculated WBIs fully support the above assignments (Figure 3a). The peripheral and radial B–B links have WBI values of 1.35 and 0.63, respectively. The Na–Na edges in the Na4 ligand have WBIs of 0.14–0.32, which are collectively consistent with a delocalized four-center two-electron (4c-2e) σ bond.
According to the above analysis, sandwich Na5B7 cluster has quite substantial intramolecular charge-transfers in between the B7 core and the Na4/Na ligands. The calculated natural atomic charges offer a quantitative picture (Figure S4a). Basically, each B atom in the B7 wheel has a negative charge from −0.27 to −0.35 |e|. Overall, the B7 wheel carries a total charge of −2.37 |e|. The tetrahedral Na4 ligand has a collective charge of +1.49 |e|, whereas the Na ligand carries a charge of +0.87 |e|. Clearly, the sandwich cluster can be formulated as a charge-transfer [Na4]2+[B7]3−[Na]+ complex. A similar analysis of the structure, WBIs, and natural atomic charges of the LM structure of Na5B7 cluster may be performed (see Figure S5).
We have also run structural optimization for a bare B73− trianion cluster at the PBE0/6-311 + G* level. The trianionic nature observed suggests that this should at most be considered as a model cluster. The interatomic distances may tend to expand to some extent, due to intramolecular Coulomb repulsion associated with three extra charges. The calculated bond distances and WBIs of the model B73− disk cluster are shown in Figure S6. The bond distances of the peripheral B–B links and radial ones are only 0.02 and 0.04 Å longer than those in the GM Na5B7 cluster, respectively. The WBIs of the peripheral B–B links and the radial ones show very minor variations relative to those in GM Na5B7 cluster, by 0.03 and 0.02, respectively. The central and peripheral B atoms in the model B73− cluster have negative charges of −0.30 and −0.45 |e|, respectively, which are close to those in the GM Na5B7 cluster. Overall, the central B atom in model B73− cluster is slightly above the B6 ring plane by 0.40 Å, as compared to 0.26 Å in GM Na5B7 cluster. The sandwich GM Na5B7 cluster moderately flattens the B7 wheel, which is intuitively expected and not surprising. The above comparison between the bare B73− model cluster and the B wheel in Na5B7 cluster further confirms the [B7]3− nature of the B wheel in GM Na5B7 cluster. It is stressed that the [B7]3− nature in GM Na5B7 cluster represents a solid conclusion on the basis of the structural data, natural bond orbital (NBO) analysis, and in particular the canonical molecular orbital (CMO) analysis and adaptive natural density partitioning (AdNDP) results. This conclusion stands firmly even without any calculations on a bare B73− model cluster. The latter calculations are merely an extra computational effort, which is a secondary part in the present paper.
3. Methods
The GM structure and low-lying isomers of Na5B7 cluster were explored by computer global searches using the Coalescence Kick (CK) algorithm [36,37], which were also aided by manual structure constructions. About 3000 stationary points were probed on the potential energy surface. The Gaussian 09 program was used subsequently to reoptimize the structures at the PBE0/6-311 + G* level [38,39,40]. To verify the reliability in terms of energetics, the top candidate structures were further assessed at the B3LYP/6-311 + G* and single-point CCSD(T)/6-311 + G*//PBE0/6-311 + G* levels [41]. Vibrational frequencies were calculated at the same density-functional theory (DFT) levels, that is, PBE0 and B3LYP, to ensure that the reported low-lying structures are true minima on the potential energy surface of the system, unless specifically stated otherwise.
Chemical bonding was elucidated using the CMO analysis, as well as the AdNDP analysis [42]. The AdNDP results were visualized using the Molekel 5.4.0.8 program [43]. The WBIs and charge distribution were calculated by NBO analysis [44] at the PBE0/6-311 + G* level. Born–Oppenheimer molecular dynamics (BOMD) simulations [45] were performed at PBE0/6-31G to demonstrate the structural fluxionality of the system.
4. Discussion
4.1. Chemical Bonding
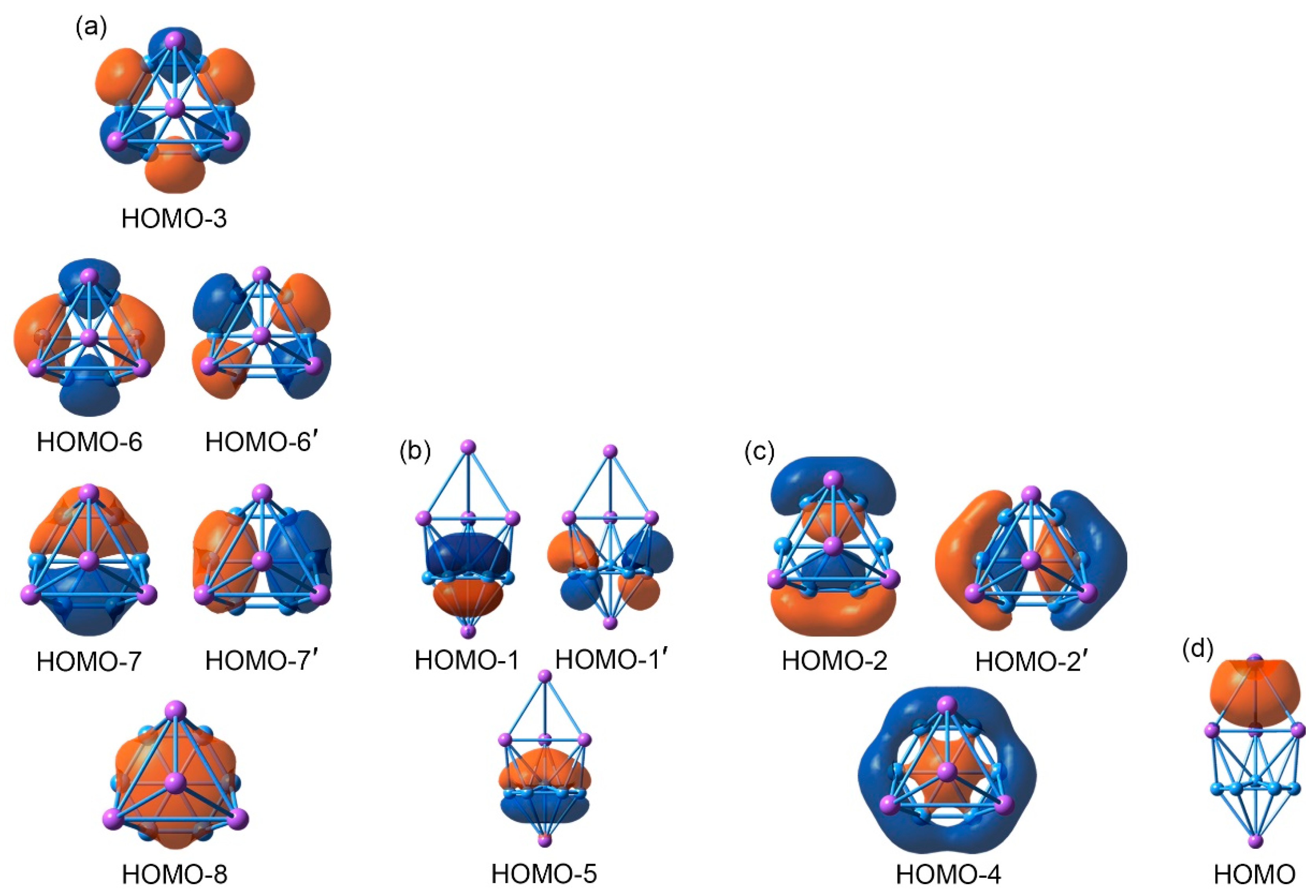
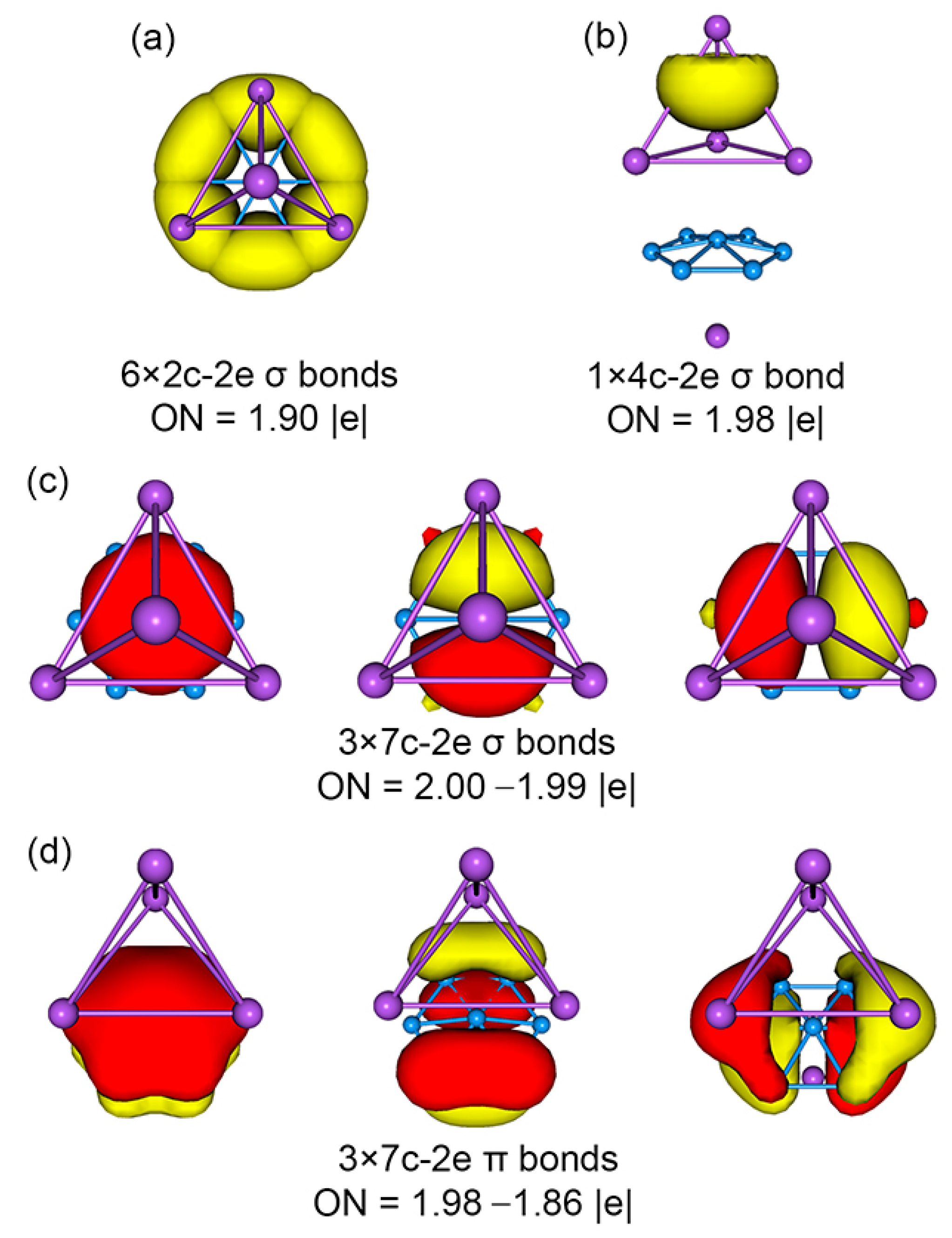
An in-depth chemical bonding analysis is essential toward understanding the stability, unique structure, and dynamic fluxionality of the title Na5B7 cluster. The CMO analysis is fundamental for this purpose. The Na5B7 cluster has 26 valence electrons. Their occupied CMOs are depicted in Figure 4. Of these 13 CMOs, six σ CMOs in subset (a) are primarily composed of B 2s atomic orbitals (AOs) of the peripheral B6 ring. These CMOs show from 0, 1, 2 to 3 nodal planes, including two degenerate pairs in the middle. Following the orbital construction principles, they can be recombined and localized as six Lewis-type two-center, two-electron (2c-2e) σ single bonds, one for each B–B edge.
Subset (b) in Figure 4 shows three π CMOs on the B7 wheel. Owing to the six-fold symmetry of the wheel, this π sextet cannot be localized as Lewis-type π bonds, akin to that in benzene, thus rendering π aromaticity for the sandwich cluster. The magic 6π electron counting conforms to the (4n + 2) Hückel rule. Likewise, the three σ CMOs in subset (c) have similar spatial distribution compared to those in the π sextet, except that the former CMOs are σ in nature. Again, the σ sextet is intrinsically delocalized and cannot be reduced to Lewis-type σ single bonds. It is, therefore, imperative to claim σ aromaticity for the sandwich as well, following the (4n + 2) Hückel rule.
Lastly, subset (d) shows only one σ CMO, which is clouded on the Na4 ligand. It is 4c-2e in nature and cannot be localized as one Lewis-type Na–Na σ single bond. The delocalized nature of these 2σ electrons renders three-dimensional σ aromaticity for the tetrahedral Na4 ligand and, hence, the sandwich cluster. Overall, the sandwich Na5B7 cluster collectively features three-fold π/σ aromaticity with the 6π/6σ/2σ electron counting.
The bonding picture is perfectly borne out from the AdNDP analysis. The AdNDP scheme of sandwich GM Na5B7 cluster is presented in Figure 5. The above analysis suggests that there is relatively minor covalent bonding between the three layers of the sandwich, and the cluster is indeed a charge-transfer complex. Similar CMO and AdNDP analyses can be conducted for the TS Na5B7 cluster, which are relatively straightforward and easy to understand. The relevant data are presented in Figures S7 and S8.
4.2. Dynamic Structural Fluxionality
The sandwich GM Na5B7 cluster, like numerous boron-based clusters reported in the recent literature [15,21,22,23,24,26,30,31,46,47], exhibits intriguing molecular dynamics properties. This is apparent on the basis of the close similarity between GM and TS structures in terms of the structures (Figure 3) and bonding (Figure 4 and Figure 5; Figures S7 and S8). In particular, the unique three-fold 6π/6σ/2σ aromaticity (see Section 4.1) underlies the dynamic fluxionality of GM Na5B7 cluster.
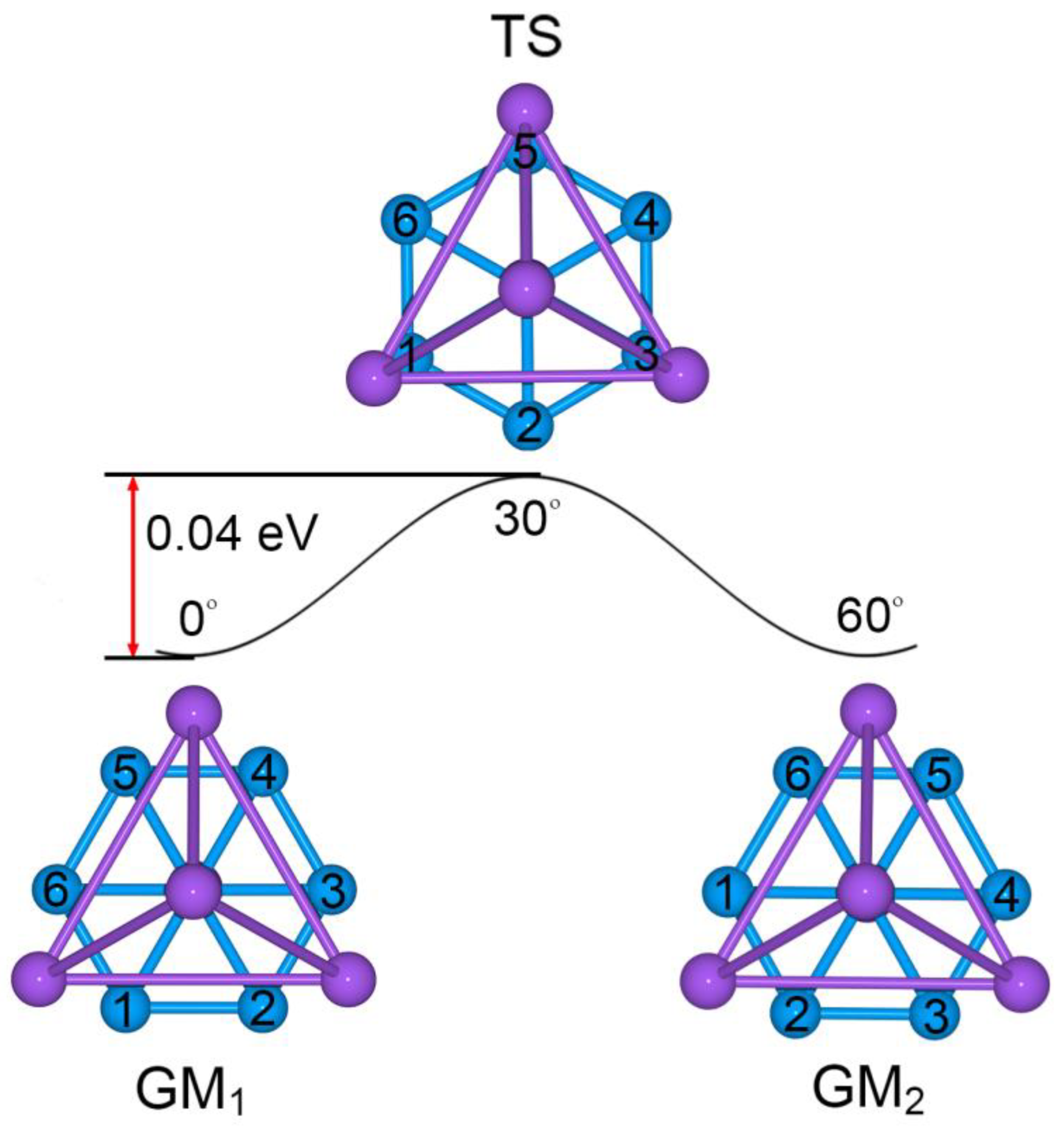
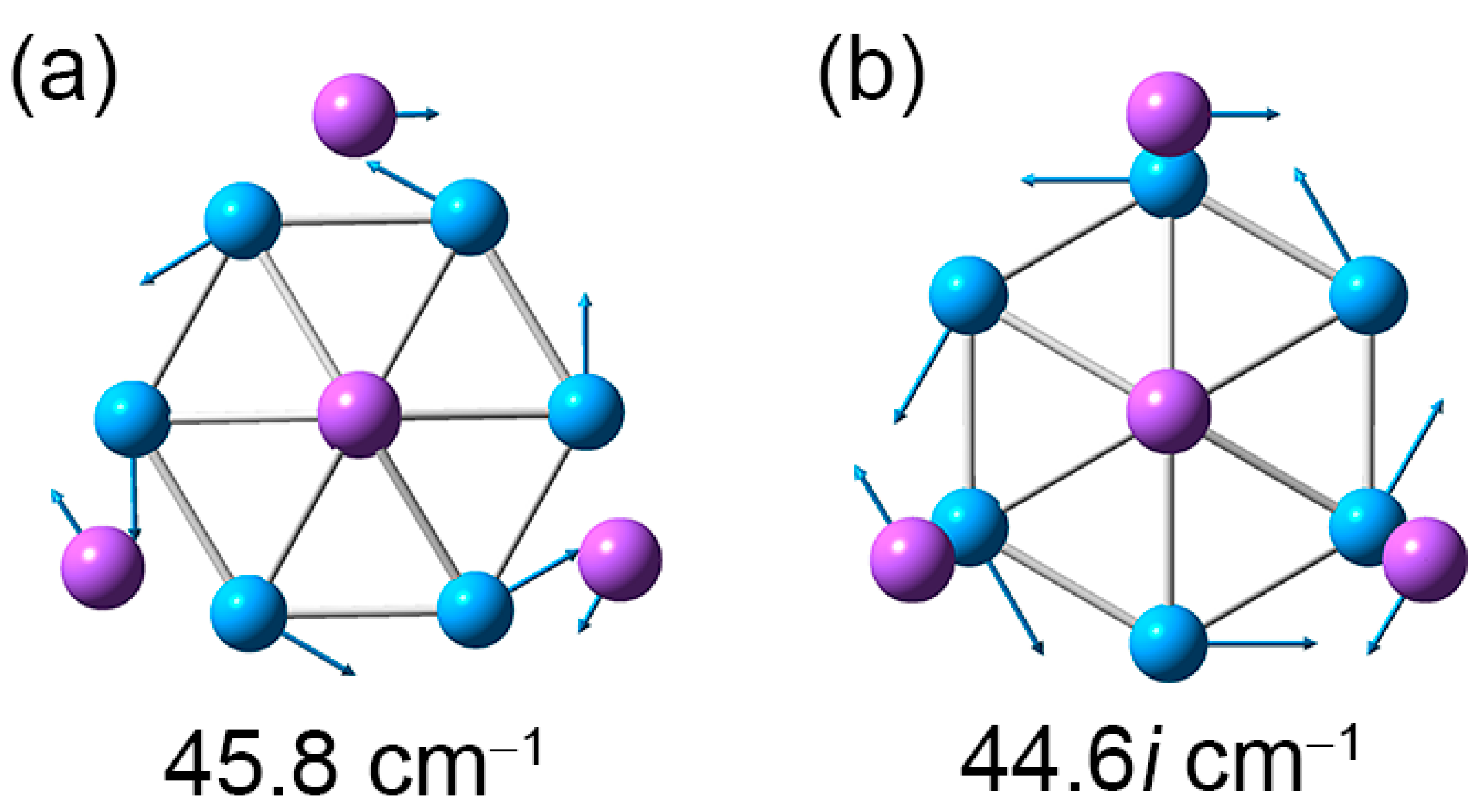
As shown in Figure 6, the GM and TS geometries of sandwich Na5B7 cluster are connected straightforwardly. Starting from GM (labeled as “GM1”) and twisting the B7 wheel clockwise relative to the Na4 tetrahedron by 30°, one readily reaches the TS structure. Further rotation in the same direction by 30° results in recovery of the GM structure (labeled as “GM2”). The energy barrier between the GM and TS structures is 0.04 eV at the PBE0/6-311 + G* level, which is relatively minor considering the anticipated strong electrostatic interaction between the B7 wheel and the Na4/Na ligands in the sandwich. The calculated soft vibrational modes of 45.8 and 44.6i cm−1, respectively, for the GM and TS structures are in line with the intramolecular rotation of the sandwich cluster (Figure 7).
To further validate the dynamic fluxionality of Na5B7 cluster, a BOMD simulation was performed at a selected temperature of 300 K. Specifically, the BOMD simulation was performed using the Hessian-based predictor-corrector method [48] at an initial setup temperature of 300 K, which was run for a time span of 50 ps (10,000 steps) at the PBE0/6-31G level. We took the GM geometry as the initial structure and simulated the dynamic evolution process. The vibrational sampling temperature was shown to be 298 K, which is close to the setting temperature. Before being recalculated analytically, the Bofill update method was used to update the Hessian evaluation for five steps. A trajectory step size of 1.0 amu1/2 bohr was used in the whole simulation process, and the total number of trajectories was 1. The maximum point for each trajectory was 200,000. Despite the variation in potential and kinetic energies during the BOMD process, the total energy remained constant, which was conserved to 10−6 hartree during the simulation process. A short movie extracted from the BOMD data is presented in the Supplementary Materials, which vividly demonstrates the intriguing structural dynamics of the cluster (see Video S1). In short, the title cluster is dynamically fluxional even at near room temperature. We comment here that the BOMD simulation represents a relatively minor part in this study. It is not intended to provide any information with accuracy. It offers molecular dynamics information of the system qualitatively, rather than quantitatively.
4.3. On the Low-Lying LM Structure: The Importance of Three-Fold π/σ Aromaticity for GM Na5B7 Cluster
Our exploration of the potential energy surface of Na5B7 cluster shows that only two structures, GM (C3v, 1A1) and LM (C2v, 1A1), are close in energy, within about 0.2 eV at all three levels of theory (Figure S1). Other isomeric structures are substantially higher in energy, by at least 0.9 eV. Therefore, it is invaluable to understand the LM cluster, which is depicted in Figure 2. Basically, the LM and GM structures differ in the upper Na4 ligand. While the Na4 ligand in GM cluster is a slightly distorted tetrahedron (3.60 versus 3.64 Å; Figure S3a), it becomes a roof-like, quasi-planar ligand (or a rhombic ligand) in the LM cluster (Figure S5a). In the LM cluster, the roof-like Na4 ligand appears to interact with the B7 wheel primarily via two Na atoms along the longer diagonal, which allows optimal intramolecular charge transfer (Figure S5c). As a consequence, chemical bonding in the roof-like Na4 ligand is dominated by σ bonding along the shorter diagonal. The shorter Na2 diagonal in the highest occupied molecular orbital (HOMO) contributes to 66% of the whole Na4 ligand (Figures S9d and S10b). In other words, chemical bonding in the roof-like Na4 ligand is largely a Lewis-type 2c-2e σ bond in nature, at least in the zeroth-order picture.
In contrast, the 2σ framework in GM cluster is truly delocalized in a three-dimensional manner, which offers extra stabilization to the sandwich GM cluster. As a model system, one can evaluate a free-standing Na42+ dication cluster in its tetrahedral Td and rhombic D2h geometries (Figure S11). At the PBE0 level, the Td cluster is 0.21 eV more stable than its rhombic D2h isomer. This energetic difference is comparable to those between the GM and LM structures of Na5B7 cluster (Figure S1). Thus, 2σ aromaticity is clearly a decisive factor that helps distinguish between the GM Na5B7 cluster from its LM isomer.
The tetrahedral Na4 ligand, in its specific [Na4]2+ charge state, is the simplest polyhedral structure. Hence it can be considered the simplest example of “spherical” aromaticity. The 2σ electron counting also conforms to the 2(n + 1)2 rule for spherical aromaticity. Alternatively, the tetrahedral Na4 ligand may be viewed as the simplest superatom [49,50,51].
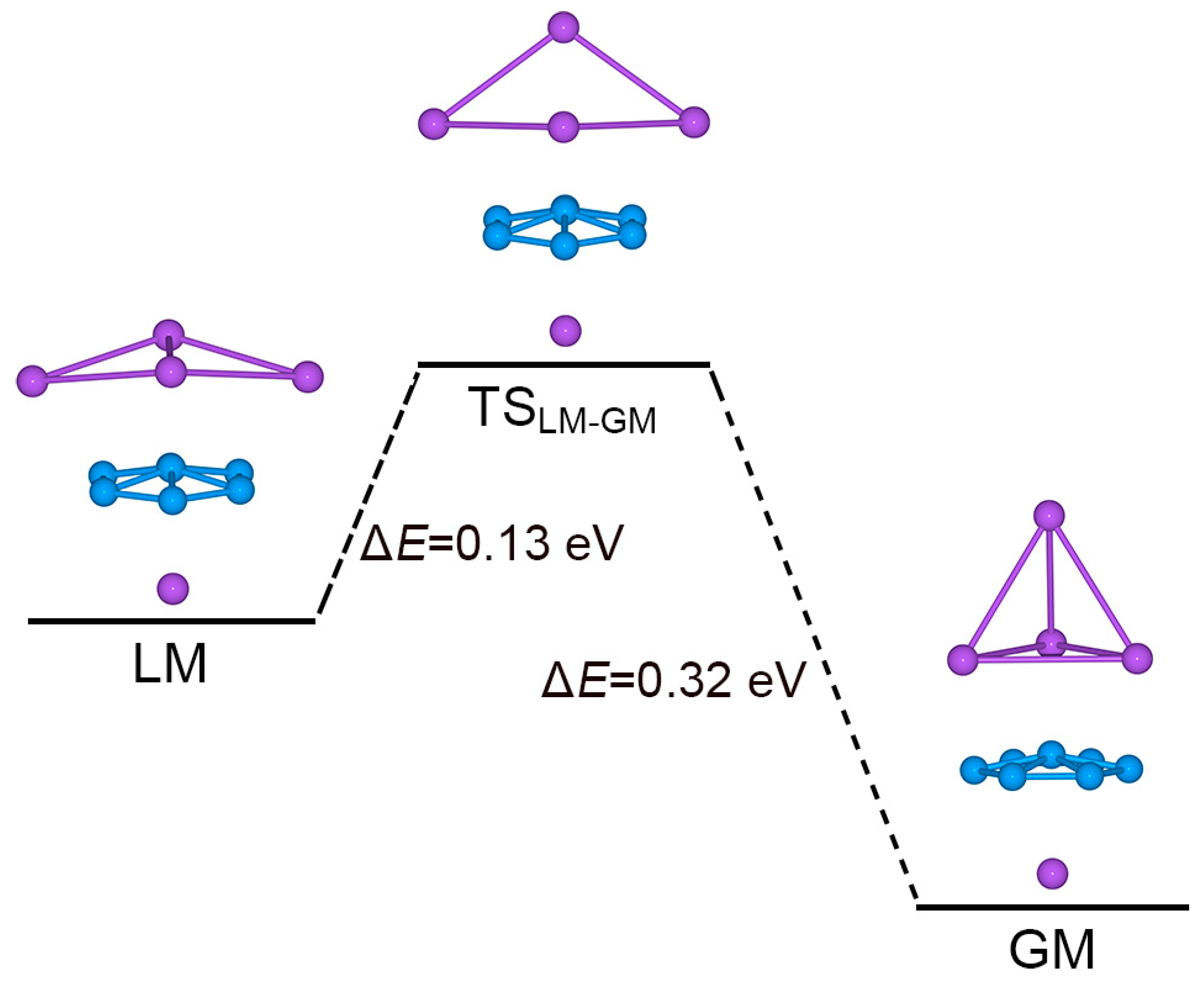
Interestingly, one can directly connect the GM and LM structures via a TS geometry, TSLM–GM, as shown in Figure 8. The TSLM–GM is located at only 0.13 eV above LM at PBE0. The TSLM–GM structure has a soft imaginary mode of 53.0i cm−1. The GM cluster lies 0.19 eV below LM, as well as 0.32 eV below TSLM–GM. According to this energy diagram, the GM cluster is relatively robust against isomerization. Our electronic structure calculations also indicate that the energy gap of GM Na5B7 cluster (2.89 eV; see Figure 9), between its HOMO and lowest unoccupied molecular orbital (LUMO), is significantly larger than that in its LM isomer (0.25 eV), thus demonstrating the electronic robustness of sandwich GM Na5B7 cluster. Indeed, the sizable energy gap in Figure 9 is a strong indication that a [B7]3− motif in binary Na5B7 cluster is appropriate, which favors a closed-shell configuration in the triply charged anionic state. The same reason underlies the nature of a charge-transfer [Na4]2+[B7]3−[Na]+ complex for GM Na5B7 cluster, as well as its unique sandwich geometry.
5. Conclusions
We have computationally designed a boron-based alloy Na5B7 cluster that assumes unique three-layered sandwich structure. The cluster may be described as a charge-transfer [Na4]2+[B7]3−[Na]+ complex, which is composed of a quasi-planar B7 wheel as core, as well as a Na4 tetrahedron and an isolated Na atom as its two ligand layers at the top and at the bottom. Chemical bonding analysis indicates magic 6π/6σ double aromaticity for the [B7]3− wheel and three-dimensional 2σ aromaticity for the tetrahedral [Na4]2+ ligand. The latter is also the simplest example of three-dimensional or spherical aromaticity, as well as the simplest superatom. Collectively, the Na5B7 cluster has three-fold 6π/6σ/2σ aromaticity, which underlies its thermodynamic stability. The same mechanism facilitates the intriguing dynamic structural fluxionality for the sandwich cluster, even at near room temperature. This work also highlights the structural and electronic robustness of the [B7]3− molecular wheel as a potential inorganic ligand.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28073276/s1. Table S1: Cartesian coordinates for global-minimum (GM) and transition-state (TS) structures of Na5B7 cluster at PBE0/6-311 + G*; Figure S1: Alternative optimized structures of Na5B7 cluster at PBE0; Figure S2: Selected triplet-state structures of Na5B7 cluster; Figures S3 and S4: Calculated bond distances and natural atomic charges for GM and TS structures of Na5B7 cluster; Figure S5: Bond distances, Wiberg bond indices (WBIs), and natural atomic charges for a local minimum (LM) structure; Figure S6: Calculated bond distances and WBIs of model B73− cluster at PBE0/6-311 + G*; Figures S7–S10: CMOs and AdNDP schemes of TS and LM structures of Na5B7 cluster; Figure S11: Relative energies for tetrahedral versus rhombic structures of a free-standing Na42+ model cluster at PBE0; Video S1: A short movie extracted from the BOMD simulation for Na5B7 cluster at 300 K.
Author Contributions
Conceptualization, H.-J.Z.; methodology, P.-F.H., Y.-J.W. and H.-J.Z.; software, P.-F.H.; validation, P.-F.H., Y.-J.W., L.-Y.F., S.-J.G., Q.S. and H.-J.Z.; investigation, P.-F.H. and Y.-J.W.; writing—original draft preparation, P.-F.H.; writing—review and editing, H.-J.Z.; supervision, H.-J.Z. and Q.S.; funding acquisition, H.-J.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (21873058, 21573138, and 22173053) and the Natural Science Foundation of Shanxi Province (201801D121103 and 20210302123439).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lipscomb, W.N. The Boranes and Their Relatives. Science 1977, 196, 1047–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oger, E.; Crawford, N.R.M.; Kelting, R.; Weis, P.; Kappes, M.M.; Ahlrichs, R. Boron cluster cations: Transition from planar to cylindrical structures. Angew. Chem. Int. Ed. 2007, 46, 8503–8506. [Google Scholar] [CrossRef] [PubMed]
- Zhai, H.-J.; Kiran, B.; Li, J.; Wang, L.-S. Hydrocarbon analogues of boron clusters—Planarity, aromaticity and antiaromaticity. Nat. Mater. 2003, 2, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Zhai, H.-J.; Alexandrova, A.N.; Birch, K.A.; Boldyrev, A.I.; Wang, L.-S. Hepta- and octacoordinate boron in molecular wheels of eight- and nine-atom boron clusters: Observation and confirmation. Angew. Chem. Int. Ed. 2003, 42, 6004–6008. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Zhao, Y.-F.; Li, W.-L.; Jian, T.; Chen, Q.; You, X.-R.; Ou, T.; Zhao, X.-Y.; Zhai, H.-J.; Li, S.-D.; et al. Observation and characterization of the smallest borospherene: B28− and B28. J. Chem. Phys. 2016, 144, 064307. [Google Scholar] [CrossRef] [Green Version]
- Kiran, B.; Bulusu, S.; Zhai, H.-J.; Yoo, S.; Zeng, X.-C.; Wang, L.-S. Photoelectron spectroscopy of aromatic compound clusters of the B12 all-boron benzene: B12Au− and B12(BO)−. Proc. Natl. Acad. Sci. USA 2005, 102, 961–964. [Google Scholar] [CrossRef] [Green Version]
- Li, W.-L.; Chen, Q.; Tian, W.-J.; Bai, H.; Zhao, Y.-F.; Hu, H.-S.; Li, J.; Zhai, H.-J.; Li, S.-D.; Wang, L.-S. The B35 cluster with a double-hexagonal vacancy: A new and more flexible structural motif for borophene. J. Am. Chem. Soc. 2014, 136, 12257–12260. [Google Scholar] [CrossRef]
- Zhai, H.-J.; Zhao, Y.-F.; Li, W.-L.; Chen, Q.; Bai, H.; Hu, H.-S.; Piazza, Z.A.; Tian, W.-J.; Lu, H.-G.; Wu, Y.-B.; et al. Observation of an all-boron fullerene. Nat. Chem. 2014, 6, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.-J.; Zhang, J.; Zhong, Q.; Li, W.-B.; Li, S.; Li, H.; Cheng, P.; Meng, S.; Chen, L.; Wu, K.-H. Experimental realization of two-dimensional boron sheets. Nat. Chem. 2016, 8, 563–568. [Google Scholar] [CrossRef] [Green Version]
- Mannix, A.J.; Zhou, X.-F.; Kiraly, B.; Wood, J.D.; Alducin, D.; Myers, B.D.; Liu, X.-L.; Fisher, B.L.; Santiago, U.; Guest, J.R.; et al. Synthesis of borophenes: Anisotropic, two-dimensional boron polymorphs. Science 2015, 350, 1513–1516. [Google Scholar] [CrossRef] [Green Version]
- Sergeeva, A.P.; Zubarev, D.Y.; Zhai, H.-J.; Boldyrev, A.I.; Wang, L.-S. A photoelectron spectroscopic and theoretical study of B16− and B162−: An all-boron naphthalene. J. Am. Chem. Soc. 2008, 130, 7244–7246. [Google Scholar] [CrossRef]
- Alexandrova, A.N.; Boldyrev, A.I.; Zhai, H.-J.; Wang, L.-S. All-boron aromatic clusters as potential new inorganic ligands and building blocks in chemistry. Coord. Chem. Rev. 2006, 250, 2811–2866. [Google Scholar] [CrossRef]
- Gu, F.-L.; Yang, X.-M.; Tang, A.-C.; Jiao, H.-J.; Schleyer, P.V.R. Structure and stability of B13+ clusters. J. Comput. Chem. 1998, 19, 203–214. [Google Scholar] [CrossRef]
- Huang, W.; Sergeeva, A.P.; Zhai, H.-J.; Averkiev, B.B.; Wang, L.-S.; Boldyrev, A.I. A concentric planar doubly π-aromatic B19− cluster. Nat. Chem. 2010, 2, 202–206. [Google Scholar] [CrossRef]
- Jimeénez-Halla, J.O.C.; Islas, R.; Heine, T.; Merino, G. B19−: An aromatic Wankel motor. Angew. Chem. Int. Ed. 2010, 49, 5668–5671. [Google Scholar] [CrossRef]
- Fowler, J.E.; Ugalde, J.M. The curiously stable B13+ cluster and its neutral and anionic counterparts: The advantages of planarity. J. Phys. Chem. A 2000, 104, 397–403. [Google Scholar] [CrossRef]
- Aihara, J.I. B13+ is highly aromatic. J. Phys. Chem. A 2001, 105, 5486–5489. [Google Scholar] [CrossRef]
- Martínez-Guajardo, G.; Sergeeva, A.P.; Boldyrev, A.I.; Heine, T.; Ugalde, J.M.; Merino, G. Unravelling phenomenon of internal rotation in B13+ through chemical bonding analysis. Chem. Commun. 2011, 47, 6242–6244. [Google Scholar] [CrossRef]
- Moreno, D.; Pan, S.; Martínez-Guajardo, G.; Zeonjuk, L.L.; Islas, R.; Osorio, E.; Chattaraj, P.K.; Heine, T.; Merino, G. B182−: A quasi-planar bowl member of the Wankel motor family. Chem. Commun. 2014, 50, 8140–8143. [Google Scholar] [CrossRef]
- Tai, T.-B.; Ceulemans, A.; Nguyen, M.T. Disk aromaticity of the planar and fluxional anionic boron clusters B20−/2−. Chem. Eur. J. 2012, 18, 4510–4512. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Zhao, X.-Y.; Chen, Q.; Zhai, H.-J.; Li, S.-D. B11−: A moving subnanoscale tank tread. Nanoscale 2015, 7, 16054–16060. [Google Scholar] [CrossRef]
- Wang, Y.-J.; You, X.-R.; Chen, Q.; Feng, L.-Y.; Wang, K.; Ou, T.; Zhao, X.-Y.; Zhai, H.-J.; Li, S.-D. Chemical bonding and dynamic fluxionality of a B15+ cluster: A nanoscale double-axle tank tread. Phys. Chem. Chem. Phys. 2016, 18, 15774–15782. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Feng, L.-Y.; Guo, J.-C.; Zhai, H.-J. Dynamic Mg2B8 cluster: A nanoscale compass. Chem. Asian J. 2017, 12, 2899–2903. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.-Y.; Luo, X.-M.; Tian, X.-X.; Lu, H.-G.; Li, S.-D. NiB10, NiB11−, NiB12, and NiB13+: Half-sandwich complexes with the universal coordination bonding pattern of σ plus π double delocalization. J. Clust. Sci. 2019, 30, 115–121. [Google Scholar] [CrossRef]
- Li, W.-L.; Jian, T.; Chen, X.; Li, H.-R.; Chen, T.-T.; Luo, X.-M.; Li, S.-D.; Li, J.; Wang, L.-S. Observation of a metal-centered B2-Ta@B18− tubular molecular rotor and a perfect Ta@B20− boron drum with the record coordination number of twenty. Chem. Commun. 2017, 53, 1587–1590. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Li, H.-R.; Zhao, X.-Y.; Lu, X.-Q.; Mu, Y.-W.; Lu, H.-G.; Li, S.-D. Fluxional bonds in planar B19−, tubular Ta©B20−, and cage-like B39−. J. Comput. Chem. 2019, 40, 966–970. [Google Scholar] [CrossRef] [Green Version]
- Popov, I.A.; Li, W.-L.; Piazza, Z.A.; Boldyrev, A.I.; Wang, L.-S. Complexes between planar boron clusters and transition metals: A photoelectron spectroscopy and ab initio study of CoB12− and RhB12−. J. Phys. Chem. A 2014, 118, 8098–8105. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Moreno, D.; Osorio, E.; Castro, A.C.; Pan, S.; Chattaraj, P.K.; Heine, T.; Merino, G. Structure and bonding of IrB12−: Converting a rigid boron B12 platelet to a Wankel motor. RSC Adv. 2016, 6, 27177–27182. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Feng, L.-Y.; Zhai, H.-J. Divide and stack up: Boron-based sandwich cluster as a subnanoscale propeller. Chem. Asian J. 2019, 14, 2945–2949. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Feng, L.-Y.; Zhai, H.-J. Sandwich-type Na6B7− and Na8B7+ clusters: Charge-transfer complexes, four-fold π/σ aromaticity, and dynamic fluxionality. Phys. Chem. Chem. Phys. 2019, 21, 18338–18345. [Google Scholar] [CrossRef]
- Guo, J.-C.; Feng, L.-Y.; Wang, Y.-J.; Jalife, S.; Vásquez-Espinal, A.; Cabellos, J.L.; Pan, S.; Merino, G.; Zhai, H.-J. Coaxial triple-layered versus helical Be6B11− clusters: Dual structural fluxionality and multifold aromaticity. Angew. Chem. Int. Ed. 2017, 56, 10174–10177. [Google Scholar] [CrossRef]
- Fagiani, M.R.; Song, X.W.; Petkov, P.; Debnath, S.; Gewinner, S.; Schöllkopf, W.; Heine, T.; Fielicke, A.; Asmis, K.R. Structure and fluxionality of B13+ probed by infrared photodissociation spectroscopy. Angew. Chem. Int. Ed. 2017, 56, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Pyykkö, P. Additive covalent radii for single-, double-, and triple-bonded molecules and tetrahedrally bonded crystals: A summary. J. Phys. Chem. A 2015, 119, 2326–2337. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, A.N.; Boldyrev, A.I.; Zhai, H.-J.; Wang, L.-S. Electronic structure, isomerism, and chemical bonding in B7− and B7. J. Phys. Chem. A 2004, 108, 3509–3517. [Google Scholar] [CrossRef]
- Bonačić-Koutecký, V.; Fantucci, P.; Koutecký, J. Quantum chemistry of small clusters of elements of groups Ia, Ib, and IIa: Fundamental concepts, predictions, and interpretation of experiments. Chem. Rev. 1991, 91, 1035–1108. [Google Scholar] [CrossRef]
- Saunders, M. Stochastic search for isomers on a quantum mechanical surface. J. Comput. Chem. 2004, 25, 621–626. [Google Scholar] [CrossRef]
- Bera, P.P.; Sattelmeyer, K.W.; Saunders, M.; Schaefer III, H.F.; Schleyer, P.v.R. Mindless chemistry. J. Phys. Chem. A 2006, 110, 4287–4290. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. GAUSSIAN 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Scuseria, G.E.; Janssen, C.L.; Schaefer III, H.F. An efficient reformulation of the closed-shell coupled cluster single and double excitation (CCSD) equations. J. Chem. Phys. 1988, 89, 7382–7387. [Google Scholar] [CrossRef]
- Zubarev, D.Y.; Boldyrev, A.I. Developing paradigms of chemical bonding: Adaptive natural density partitioning. Phys. Chem. Chem. Phys. 2008, 10, 5207–5217. [Google Scholar] [CrossRef] [PubMed]
- Varetto, U. Molekel 5.4.0.8; Swiss National Supercomputing Center: Manno, Switzerland, 2009. [Google Scholar]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. QUICKSTEP: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef] [Green Version]
- Erhardt, S.; Frenking, G.; Chen, Z.-F.; Schleyer, P.v.R. Aromatic boron wheels with more than one carbon atom in the center: C2B8, C3B93+, and C5B11+. Angew. Chem. Int. Ed. 2005, 44, 1078–1082. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Guo, J.-C.; Zhai, H.-J. Why nanoscale tank treads move? Structures, chemical bonding, and molecular dynamics of a doped boron cluster B10C. Nanoscale 2017, 9, 9310–9316. [Google Scholar] [CrossRef] [PubMed]
- Millam, J.M.; Bakken, V.; Chen, W.; Hase, W.L.; Schlegel, H.B. Ab initio classical trajectories on the Born-Oppenheimer surface: Hessian-based integrators using fifth-order polynomial and rational function fits. J. Chem. Phys. 1999, 111, 3800–3805. [Google Scholar] [CrossRef]
- Luo, Z.-X.; Castleman, A.W. Special and general superatoms. Acc. Chem. Res. 2014, 47, 2931–2940. [Google Scholar] [CrossRef] [PubMed]
- Reber, A.C.; Khanna, S.N. Superatoms: Electronic and geometric effects on reactivity. Acc. Chem. Res. 2017, 50, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Jena, P.; Sun, Q. Super atomic clusters: Design rules and potential for building blocks of materials. Chem. Rev. 2018, 118, 5755–5870. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Optimized geometries of (a) C3v (1A1) global minimum (GM) and (b) C3v (1A1) transition state (TS) of Na5B7 cluster at the PBE0/6-311 + G* level. Both top- and side-views are presented.
Figure 1.
Optimized geometries of (a) C3v (1A1) global minimum (GM) and (b) C3v (1A1) transition state (TS) of Na5B7 cluster at the PBE0/6-311 + G* level. Both top- and side-views are presented.

Figure 2.
Optimized geometry of C2v (1A1) local minimum (LM) of the Na5B7 cluster at the PBE0/6-311 + G* level. Both top- and side-views are presented.
Figure 2.
Optimized geometry of C2v (1A1) local minimum (LM) of the Na5B7 cluster at the PBE0/6-311 + G* level. Both top- and side-views are presented.

Figure 3.
Wiberg bond indices (WBIs) for (a) GM and (b) TS structures of Na5B7 cluster at the PBE0/6-311 + G* level, as obtained from the natural bond orbital (NBO) analyses.
Figure 3.
Wiberg bond indices (WBIs) for (a) GM and (b) TS structures of Na5B7 cluster at the PBE0/6-311 + G* level, as obtained from the natural bond orbital (NBO) analyses.

Figure 4.
Pictures of the canonical molecular orbitals (CMOs) of GM C3v (1A1) structure of the Na5B7 cluster. (a) Six σ CMOs for peripheral two-center two-electron (2c-2e) B–B Lewis σ bonds in the B7 wheel. (b) Three delocalized π CMOs. (c) Three delocalized σ CMOs. (d) One delocalized σ CMO over the Na4 tetrahedron. Subsets (b) through (d) collectively render three-fold 6π/6σ/2σ aromaticity for the cluster.
Figure 4.
Pictures of the canonical molecular orbitals (CMOs) of GM C3v (1A1) structure of the Na5B7 cluster. (a) Six σ CMOs for peripheral two-center two-electron (2c-2e) B–B Lewis σ bonds in the B7 wheel. (b) Three delocalized π CMOs. (c) Three delocalized σ CMOs. (d) One delocalized σ CMO over the Na4 tetrahedron. Subsets (b) through (d) collectively render three-fold 6π/6σ/2σ aromaticity for the cluster.

Figure 5.
Chemical bonding scheme of C3v (1A1) GM structure of the Na5B7 cluster on the basis of adaptive natural density partitioning (AdNDP) analysis. Occupation numbers (ONs) are shown.
Figure 5.
Chemical bonding scheme of C3v (1A1) GM structure of the Na5B7 cluster on the basis of adaptive natural density partitioning (AdNDP) analysis. Occupation numbers (ONs) are shown.

Figure 6.
Structural evolution of Na5B7 cluster during the dynamic rotation. The B7 wheel is assumed to rotate clockwise with respect to the Na4 tetrahedron. The energy barrier is evaluated to be only 0.04 eV at the PBE0/6-311 + G* level.
Figure 6.
Structural evolution of Na5B7 cluster during the dynamic rotation. The B7 wheel is assumed to rotate clockwise with respect to the Na4 tetrahedron. The energy barrier is evaluated to be only 0.04 eV at the PBE0/6-311 + G* level.

Figure 7.
Displacement vectors for soft vibrational modes of Na5B7 cluster. (a) Soft mode for the GM structure. (b) Imaginary soft mode for the TS structure.
Figure 7.
Displacement vectors for soft vibrational modes of Na5B7 cluster. (a) Soft mode for the GM structure. (b) Imaginary soft mode for the TS structure.

Figure 8.
Evolution from the LM structure to the GM structure of Na5B7 cluster. The TSLM-GM structure is a transition state that connects LM and GM. Relative energies are shown in eV at the PBE0/6-311 + G* level.
Figure 8.
Evolution from the LM structure to the GM structure of Na5B7 cluster. The TSLM-GM structure is a transition state that connects LM and GM. Relative energies are shown in eV at the PBE0/6-311 + G* level.

Figure 9.
Orbital energy diagram of GM Na5B7 cluster at the PBE0/6-311 + G* level.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Han, P.-F.; Wang, Y.-J.; Feng, L.-Y.; Gao, S.-J.; Sun, Q.; Zhai, H.-J. Chemical Bonding and Dynamic Structural Fluxionality of a Boron-Based Na5B7 Sandwich Cluster. Molecules 2023, 28, 3276. https://doi.org/10.3390/molecules28073276
AMA Style
Han P-F, Wang Y-J, Feng L-Y, Gao S-J, Sun Q, Zhai H-J. Chemical Bonding and Dynamic Structural Fluxionality of a Boron-Based Na5B7 Sandwich Cluster. Molecules. 2023; 28(7):3276. https://doi.org/10.3390/molecules28073276
Chicago/Turabian StyleHan, Peng-Fei, Ying-Jin Wang, Lin-Yan Feng, Shu-Juan Gao, Qiang Sun, and Hua-Jin Zhai. 2023. "Chemical Bonding and Dynamic Structural Fluxionality of a Boron-Based Na5B7 Sandwich Cluster" Molecules 28, no. 7: 3276. https://doi.org/10.3390/molecules28073276