
Structural (XRD) Characterization and an Analysis of H-Bonding Motifs in Some Tetrahydroxidohexaoxidopentaborate(1-) Salts of N-Substituted Guanidinium Cations †

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Thermal and Spectroscopic Properties
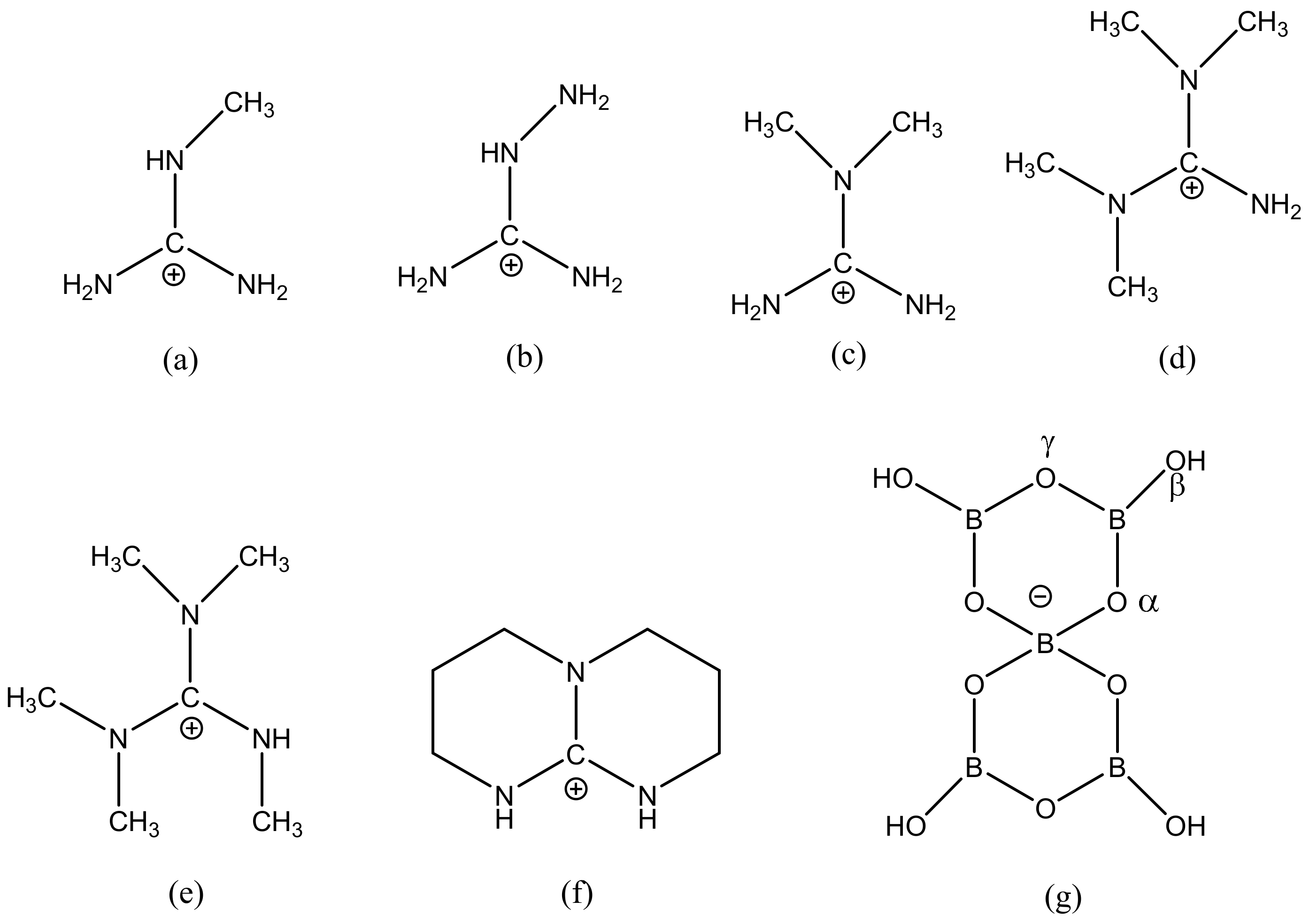
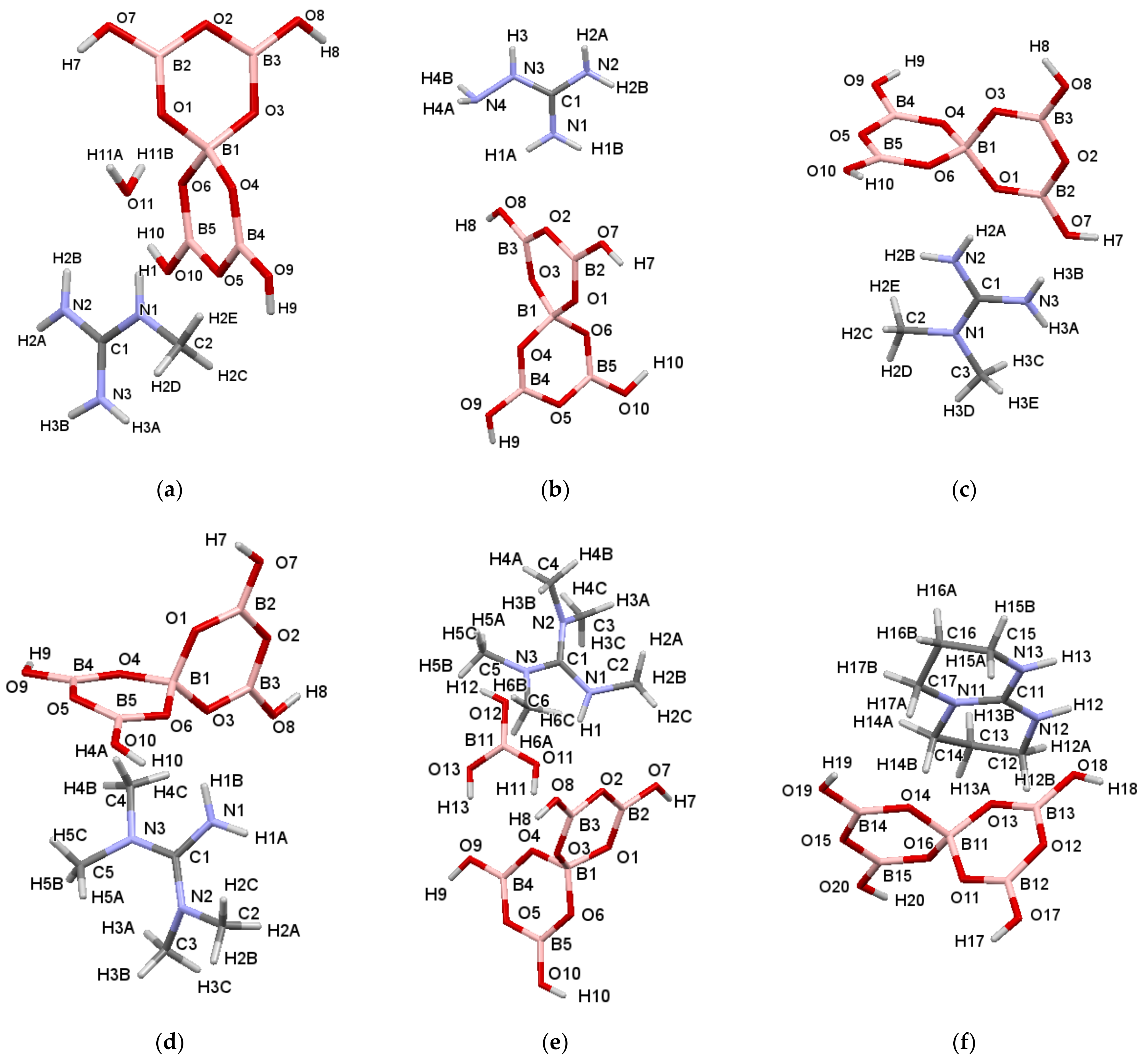
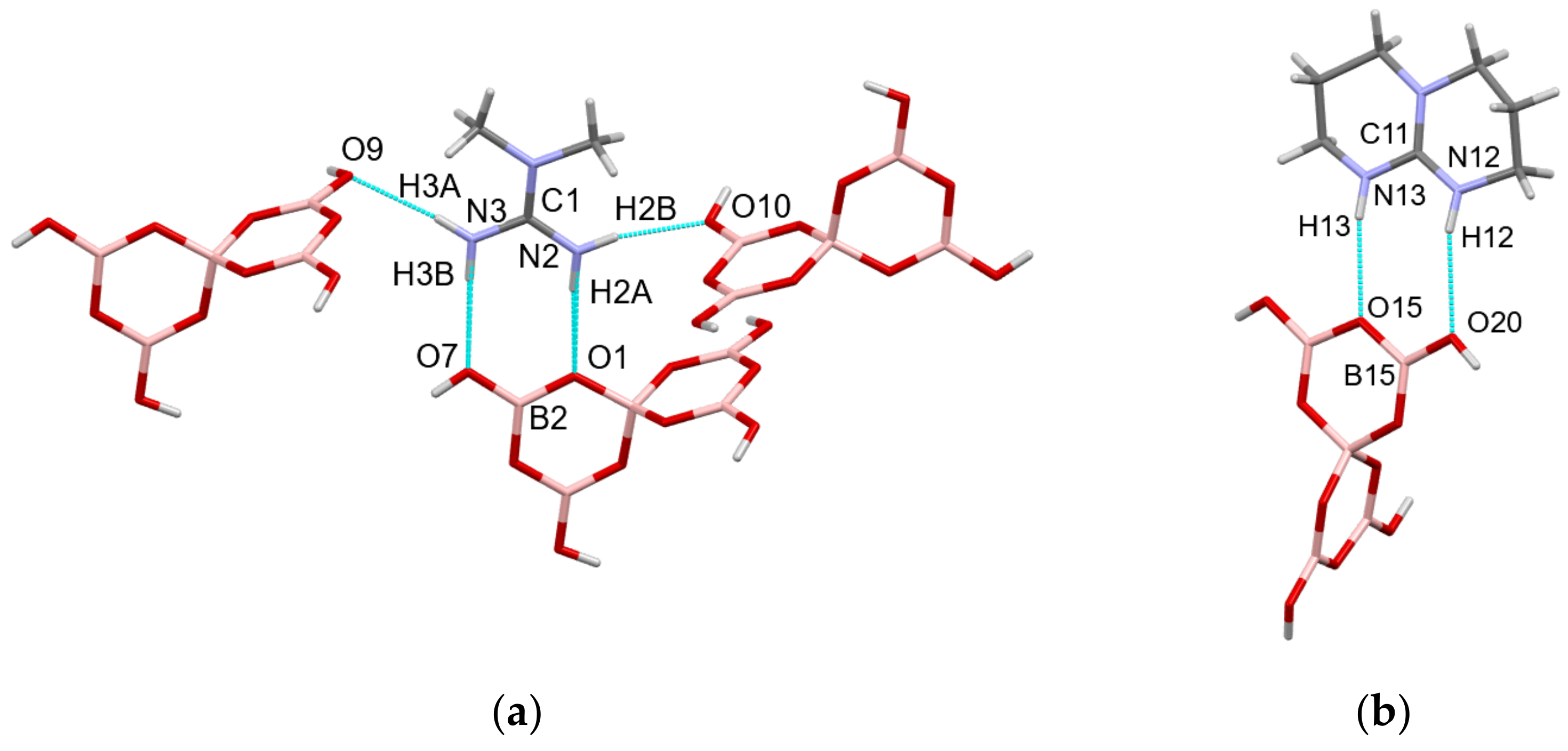
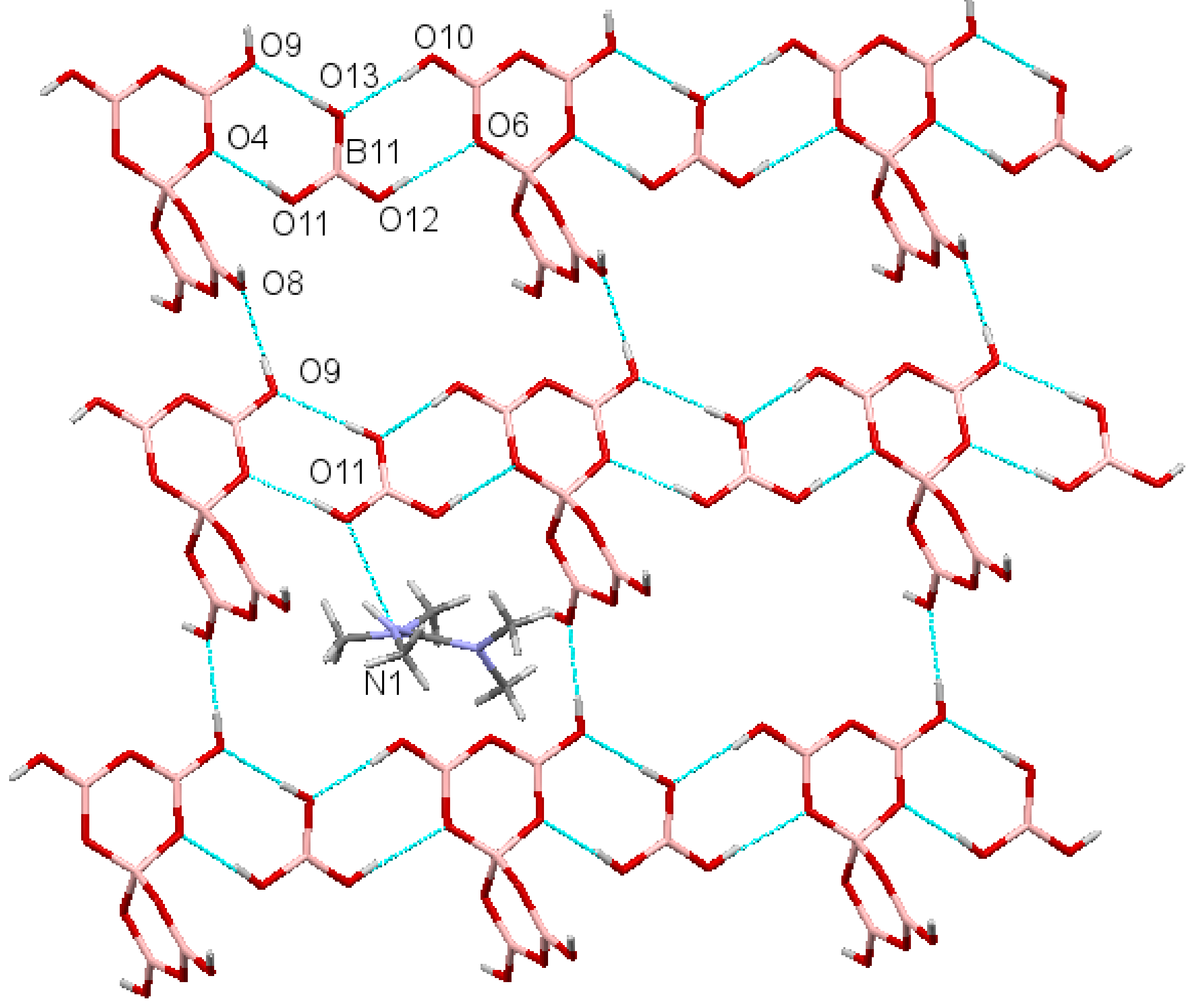
2.3. Single-Crystal XRD Studies
2.4. Discussion on H-Bonding in Guanidinium Polyborates
3. Materials and Experimental Methods
3.1. General
3.2. X-ray Crystallography
3.3. Preparation of [C(NH2)2(NHMe)][B5O6(OH)4]·H2O (1)
3.4. Preparation of [C(NH2)2(NH{NH2})][B5O6(OH)4] (2)
3.5. Preparation of [C(NH2)2(NMe2)][B5O6(OH)4] (3)
3.6. XRD Data for [C(NH2)(NMe2)2][B5O6(OH)4] (4)
3.7. Preparation of [C(NHMe)(NMe2)2]I
3.8. Preparation of [C(NHMe)(NMe2)2][B5O6(OH)4]·B(OH)3 (5)
3.9. Preparation of [TBDH][B5O6(OH)4] (6)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Farmer, J.B. Metal borates. Adv. Inorg. Chem Radiochem. 1982, 25, 187–237. [Google Scholar]
- Christ, C.L.; Clark, J.R. A crystal-chemical classification of borate structures with emphasis on hydrated borates. Phys. Chem. Miner. 1977, 2, 59–87. [Google Scholar] [CrossRef]
- Heller, G. A survey of structural types of borates and polyborates. Top. Curr. Chem. 1986, 131, 39–98. [Google Scholar]
- Becker, P. Borate materials in nonlinear optics. Adv. Mater. 1998, 10, 979–992. [Google Scholar] [CrossRef]
- Burns, P.C.; Grice, J.D.; Hawthorne, F.C. Borate minerals I. Polyhedral clusters and fundamental building blocks. Can. Mineral. 1995, 33, 1131–1151. [Google Scholar]
- Grice, J.D.; Burns, P.C.; Hawthorne, F.C. Borate minerals II. A hierarchy of structures based upon the borate fundamental building block. Can. Mineral. 1999, 37, 731–762. [Google Scholar]
- Touboul, M.; Penin, N.; Nowogrocki, G. Borates: A survey of main trends concerning crystal chemistry, polymorphism and dehydration process of alkaline and pseudo-alkaline borates. Solid State Sci. 2003, 5, 1327–1342. [Google Scholar] [CrossRef]
- Schubert, D.M.; Smith, R.A.; Visi, M.Z. Studies of crystalline non-metal borates. Glass Technol. 2003, 44, 63–70. [Google Scholar]
- Schubert, D.M.; Knobler, C.B. Recent studies of polyborate anions. Phys. Chem. Glasses Eur. J. Glass Sci. Technol. B 2009, 50, 71–78. [Google Scholar]
- Beckett, M.A. Recent Advances in crystalline hydrated borates with non-metal or transition-metal complex cations. Coord. Chem. Rev. 2016, 323, 2–14. [Google Scholar] [CrossRef] [Green Version]
- Xin, S.-S.; Zhou, M.-H.; Beckett, M.A.; Pan, C.-Y. Recent advances in crystalline oxidopolyborate complexes of d-block or p-block metals: Structural aspects, synthesis, and physical properties. Molecules 2021, 26, 3815. [Google Scholar] [CrossRef]
- Schubert, D.M. Borates in industrial use. Struct. Bond. 2003, 105, 1–40. [Google Scholar]
- Schubert, D.M. Boron oxide, boric acid, and borates. In Kirk-Othmer Encyclopedia of Chemical Technology, 5th ed.; John and Wiley and Sons: Hoboken, NJ, USA, 2011; pp. 1–68. [Google Scholar]
- Schubert, D.M. Hydrated zinc borates and their industrial use. Molecules 2019, 24, 2419. [Google Scholar] [CrossRef] [Green Version]
- Beckett, M.A.; Brellocks, B.; Chizhevsky, I.T.; Damhus, T.; Hellwich, K.-H.; Kennedy, J.D.; Laitinen, R.; Powell, W.H.; Rabinovich, D.; Vinas, C.; et al. Nomenclature for boranes and related species (IUPAC Recommendations 2019). Pure Appl. Chem. 2020, 92, 355–381. [Google Scholar] [CrossRef] [Green Version]
- Shaw, J.W.; Grayson, D.H.; Rozas, I. Topics in Heterocyclic Chemistry; Springer: Berlin/Heidelberg, Germany, 2015; pp. 1–51. [Google Scholar]
- Weakley, T.J.R. Guanidinium tetraborate(2-) dihydrate, (CH6N3)2[B4O5(OH)4]·2H2O. Acta Cryst. 1985, C41, 377–399. [Google Scholar] [CrossRef] [Green Version]
- Dhatchaiyini, M.K.; Rajasekar, G.; Mohideen, M.N.; Bhaskaran, A. Investigation on structural, spectral, optical, thermal studies and third order NLO properties of guanidinium pentaborate monohydrate single crystal. J. Mol. Sci. 2020, 1210, 128065. [Google Scholar] [CrossRef]
- Schubert, D.M.; Visi, M.Z.; Knobler, C.B. Guanidinium and imidazolium borates containing the first examples of an isolated nonaborate oxoanion: [B9O12(OH)6](3−). Inorg. Chem. 2000, 39, 2250–2251. [Google Scholar] [CrossRef]
- Rosenheim, A.; Lyser, F. Über Polyborate in wäßriger lösung. (Zur kenntnis der iso-und heteropolysäuren. XVII. Mitteilung). Z. Anorg. Allg. Chem. 1921, 119, 1–38. [Google Scholar] [CrossRef] [Green Version]
- Bowden, G.H. Supplement to Mellor’s Comprehensive Treatise on Inorganic and Theoretical Chemistry; Boron, Part A: Boron-Oxygen Compounds; Longman Group Ltd.: London, UK, 1980; Volume 5. [Google Scholar]
- Wood, G.L.; Janik, J.F.; Visi, M.Z.; Schubert, D.M.; Paine, R.T. New borate precursors for boron nitride powder synthesis. Chem Mater. 2005, 17, 1855–1859. [Google Scholar] [CrossRef]
- Blondeau, P.; Segura, M.; Perez-Fernandez, R.; de Mendoza, J. Molecular recognition of oxoanions based on guanidinium receptors. Chem. Soc. Rev. 2007, 26, 198–210. [Google Scholar] [CrossRef]
- Abrahams, B.F.; Haywood, M.G.; Robson, R. Guanidinium ion as a symmetrical template in the formation of cubic hydrogen-bonded borate networks with the boracite topology. J. Am. Chem. Soc. 2005, 127, 816–817. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R. Supramolecular synthons in crystal engineering—A new organic synthesis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Gavezzotti, A. Supramolecular synthons: Validation and ranking of intermolecular interaction energies. Cryst. Growth Des. 2012, 12, 5873–5877. [Google Scholar] [CrossRef]
- Salentine, G. High-field 11B NMR of alkali borate. Aqueous polyborate equilibria. Inorg. Chem. 1983, 22, 3920–3924. [Google Scholar] [CrossRef]
- Anderson, J.L.; Eyring, E.M.; Whittaker, M.P. Temperature jump rate studies of polyborate formation in aqueous boric acid. J. Phys. Chem. 1964, 68, 1128–1132. [Google Scholar] [CrossRef]
- Corbett, P.T.; Leclaire, J.; Vial, L.; West, K.R.; Wietor, J.-L.; Sanders, J.K.M.; Otto, S. Dynamic combinatorial chemistry. Chem. Rev. 2006, 106, 3652–3711. [Google Scholar] [CrossRef]
- Sola, J.; Lafuente, M.; Atcher, J.; Alfonso, I. Constitutional self-selection from dynamic combinatorial libraries in aqueous solution through supramolecular interactions. Chem. Commun. 2014, 50, 4564–4566. [Google Scholar] [CrossRef] [Green Version]
- Beckett, M.A.; Horton, P.N.; Hursthouse, M.B.; Knox, D.A.; Timmis, J.L. Structural (XRD) and thermal (DSC, TGA) and BET analysis of materials derived from non-metal cation pentaborate salts. Dalton Trans. 2010, 39, 3944–3951. [Google Scholar] [CrossRef] [PubMed]
- Visi, M.Z.; Knobler, C.B.; Owen, J.J.; Khan, M.I.; Schubert, D.M. Structures of self-assembled nonmetal borates derived from α,ω-diaminoalkanes. Cryst. Growth Des. 2006, 6, 538–545. [Google Scholar] [CrossRef]
- Beckett, M.A.; Coles, S.J.; Davies, R.A.; Horton, P.N.; Jones, C.L. Pentaborate(1−) salts templated by substituted pyrrolidinium cations: Synthesis, structural characterization, and modelling of solid-state H-bond interactions by DFT calculations. Dalton Trans. 2015, 44, 7032–7040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonner, O.D.; Jordan, C.F. The infrared and Raman spectra of guanidinium salts. SpectroChim. Acta Part A Mol. Spectrosc. 1976, 32, 1243–1246. [Google Scholar] [CrossRef]
- Li, J.; Xia, S.; Gao, S. FT-IR and Raman spectroscopic study of hydrated borates. Spectrochim. Acta 1995, 51, 519–532. [Google Scholar]
- Beckett, M.A.; Horton, P.N.; Coles, S.J.; Kose, D.A.; Kreuziger, A.-M. Structural and thermal studies of non-metal cation pentaborate salts with cations derived from 1,5-diazobicyclo[4.3.0]non-5-ene, 1,8-diazobicyclo[5.4.0]undec-7-ene and 1,8-bis(dimethylamino)naphthalene. Polyhedron 2012, 38, 157–161. [Google Scholar] [CrossRef]
- Drozd, M. Molecular structure and infrared spectra of guanidinium cation a combined theoretical and spectroscopic study. Mat. Sci. Eng. B 2007, 6, 20–28. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic chemistry. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Durka, K.; Jarzembska, K.N.; Kaminski, R.; Lulinski, S.; Serwatowski, J.; Wozniak, K. Structure and energetic landscapes of fluorinated 1,4-phenylboronic acids. Cryst. Growth Design 2012, 12, 3720–3734. [Google Scholar] [CrossRef]
- Heller, G. Die hydrolyse von borsäuretrimethylester in gegenwart organischer basen. J. Inorg. Nucl. Chem. 1968, 30, 2743–2754. [Google Scholar] [CrossRef]
- Beckett, M.A.; Coles, S.J.; Horton, P.N.; Jones, C.L. Polyborate anions partnered with large non-metal cations: Triborate(1-), pentaborate(1-) and heptaborate(2-) salts. Eur. J. Inorg. Chem. 2017, 2017, 4510–4518. [Google Scholar] [CrossRef] [Green Version]
- Freyhardt, C.C.; Wiebcke, M.; Felsche, J.; Englehardt, G. N(nPr4)[B5O6(OH)4][B(OH)3]2 and N(nBu4)[B5O6(OH)4][B(OH)3]2: Clathrates with a diamondoid arrangement of hydrogen bonded pentaborate anions. J. Inclusion Phenom. Mol. Recogn. Chem. 1994, 18, 161–175. [Google Scholar] [CrossRef]
- Beckett, M.A.; Horton, P.N.; Hursthouse, M.B.; Timmis, J.L.; Varma, K.S. Templated heptaborate and pentaborate salts of cyclo-alkylammonium cations: Structural and thermal properties. Dalton Trans. 2012, 41, 4396–4403. [Google Scholar] [CrossRef]
- Hass, D.J.; Harris, R.R.; Mills, H.H. The crystal structure of guanidinium chloride. Acta Cryst. 1965, 19, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.; Wermuth, U.D. Three-dimensional hydrogen-bonded structures in guanidinium salts of the monocyclic dicarboxylic acids rac-trans-cyclohexane-1,2-dicarboxylic acid (2:1, anhydrous) isophthalic (1:1, monohydrate) and terephthalic acid (2:1, trihydrate). Acta Cryst. Sect. C 2010, C66, o575–o580. [Google Scholar] [CrossRef] [Green Version]
- Vadivel, S.; Sultan, B.A.; Samad, S.A.; Shunmuganarayanan, A.; Muthu, R. Synthesis, structural elucidation, thermal, mechanical, linear and nonlinear optical properties of hydrogen bonded organic single crystal guanidinium propionate for optoelectronic device application. Chem Phys. Lett. 2018, 707, 165–171. [Google Scholar] [CrossRef]
- Russell, V.A.; Etter, M.C.; Ward, M.D. Layered materials by design: Structural enforcement by hydrogen bonding in guanidinium alkane- and arene-sulfonates. J. Am. Chem Soc. 1994, 116, 1941–1952. [Google Scholar] [CrossRef]
- Schug, K.A.; Lindner, W. Noncovalent Binding between Guanidinium and Anionic Groups: Focus on Biological- and Synthetic-Based Arginine/Guanidinium Interactions with Phosph[on]ate and Sulf[on]ate Residues. Chem. Rev. 2005, 105, 67–113. [Google Scholar] [CrossRef]
- Thomas, M.; Lagones, T.A.; Judd, M.; Morshedi, M.; White, N.G. Hydrogen bond-driven self-assembly between amidiniums and carboxylates: A combined molecular dynamics, NMR ppectroscopy, and single crystal X-ray diffraction study. Chem. Asian J. 2017, 12, 1587. [Google Scholar] [CrossRef] [PubMed]
- Avdeeva, V.V.; Malinina, E.A.; Vologzhanina, A.V.; Sivaev, I.B.; Kuznetsov, N.T. Formation of oxidoborates in destruction of the [B11H14]− anion promoted by transition metals. Inorg. Chim. Acta 2020, 509, 119693. [Google Scholar] [CrossRef]
- CrysAlisPro Software System; Rigaku Oxford Diffraction; Agilent Technologies UK Ltd: Yarnton, UK, 2019.
- Sheldrick, G.M. ShelXT—intergrated space-group and crystal structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Hooft, R.; Nonius, B.V. COLLECT, Data Collection Software; Nonius BV: Delft, The Netherlands, 1998. [Google Scholar]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Meth. Enzymol. 1997, 276, 307–326. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, A64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with ShelXL. Acta Cryst. 2015, C27, 3–8. [Google Scholar]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beckett, M.A.; Coles, S.J.; Horton, P.N.; Rixon, T.A. Structural (XRD) Characterization and an Analysis of H-Bonding Motifs in Some Tetrahydroxidohexaoxidopentaborate(1-) Salts of N-Substituted Guanidinium Cations. Molecules 2023, 28, 3273. https://doi.org/10.3390/molecules28073273
Beckett MA, Coles SJ, Horton PN, Rixon TA. Structural (XRD) Characterization and an Analysis of H-Bonding Motifs in Some Tetrahydroxidohexaoxidopentaborate(1-) Salts of N-Substituted Guanidinium Cations. Molecules. 2023; 28(7):3273. https://doi.org/10.3390/molecules28073273
Chicago/Turabian StyleBeckett, Michael A., Simon J. Coles, Peter N. Horton, and Thomas A. Rixon. 2023. "Structural (XRD) Characterization and an Analysis of H-Bonding Motifs in Some Tetrahydroxidohexaoxidopentaborate(1-) Salts of N-Substituted Guanidinium Cations" Molecules 28, no. 7: 3273. https://doi.org/10.3390/molecules28073273