
Selenonium Salt as a Catalyst for Nucleophilic Substitution Reactions in Water: Synthesis of Thiocyanites and Selenocyanates



Abstract
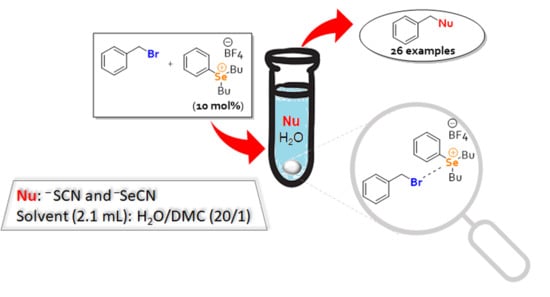
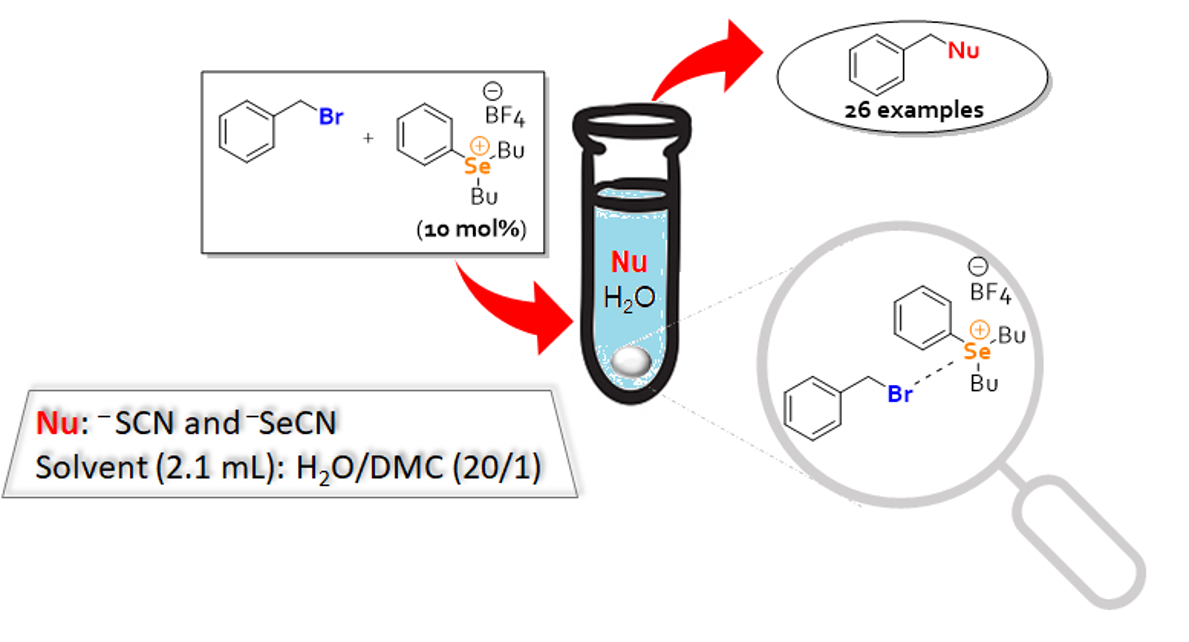
:
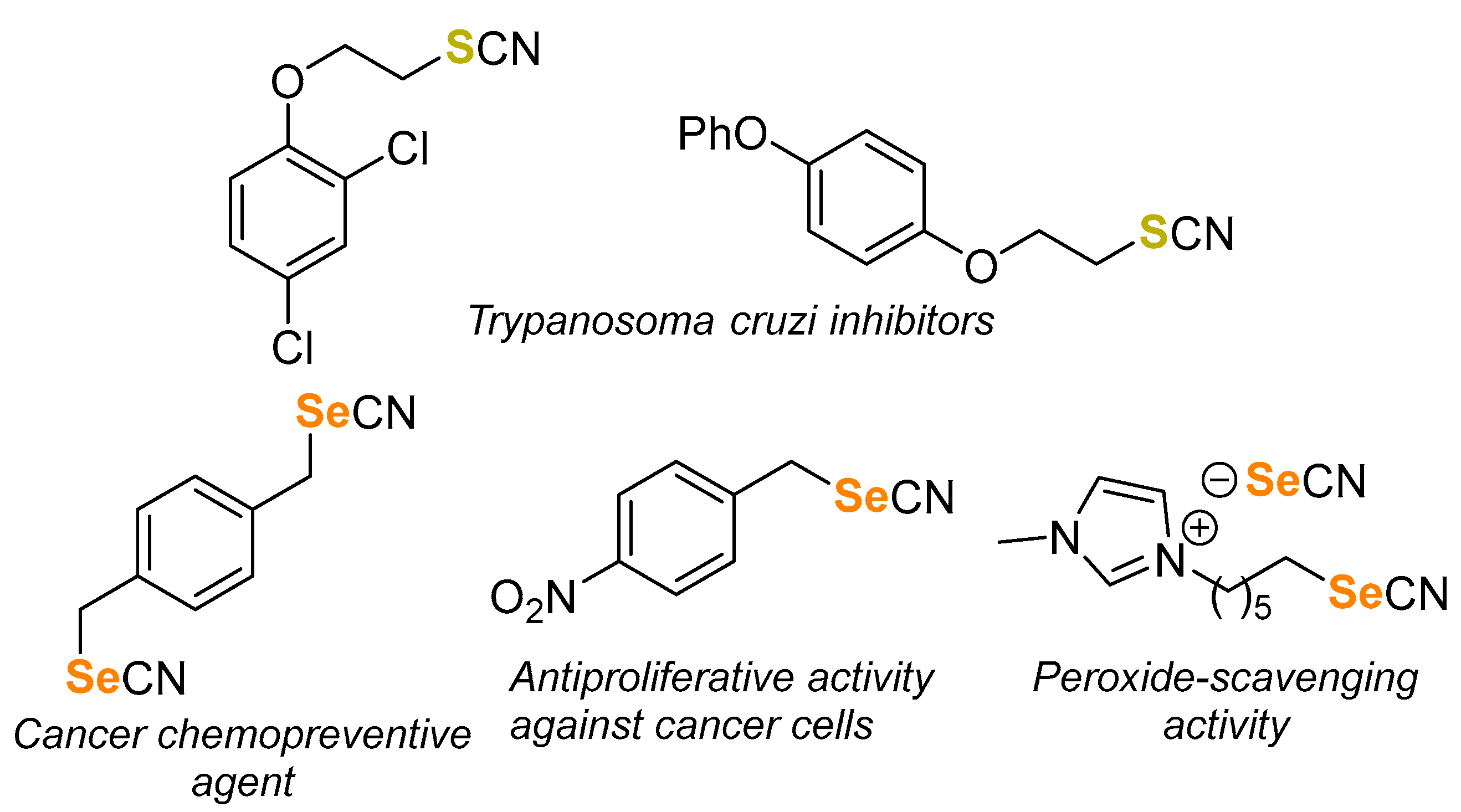
1. Introduction
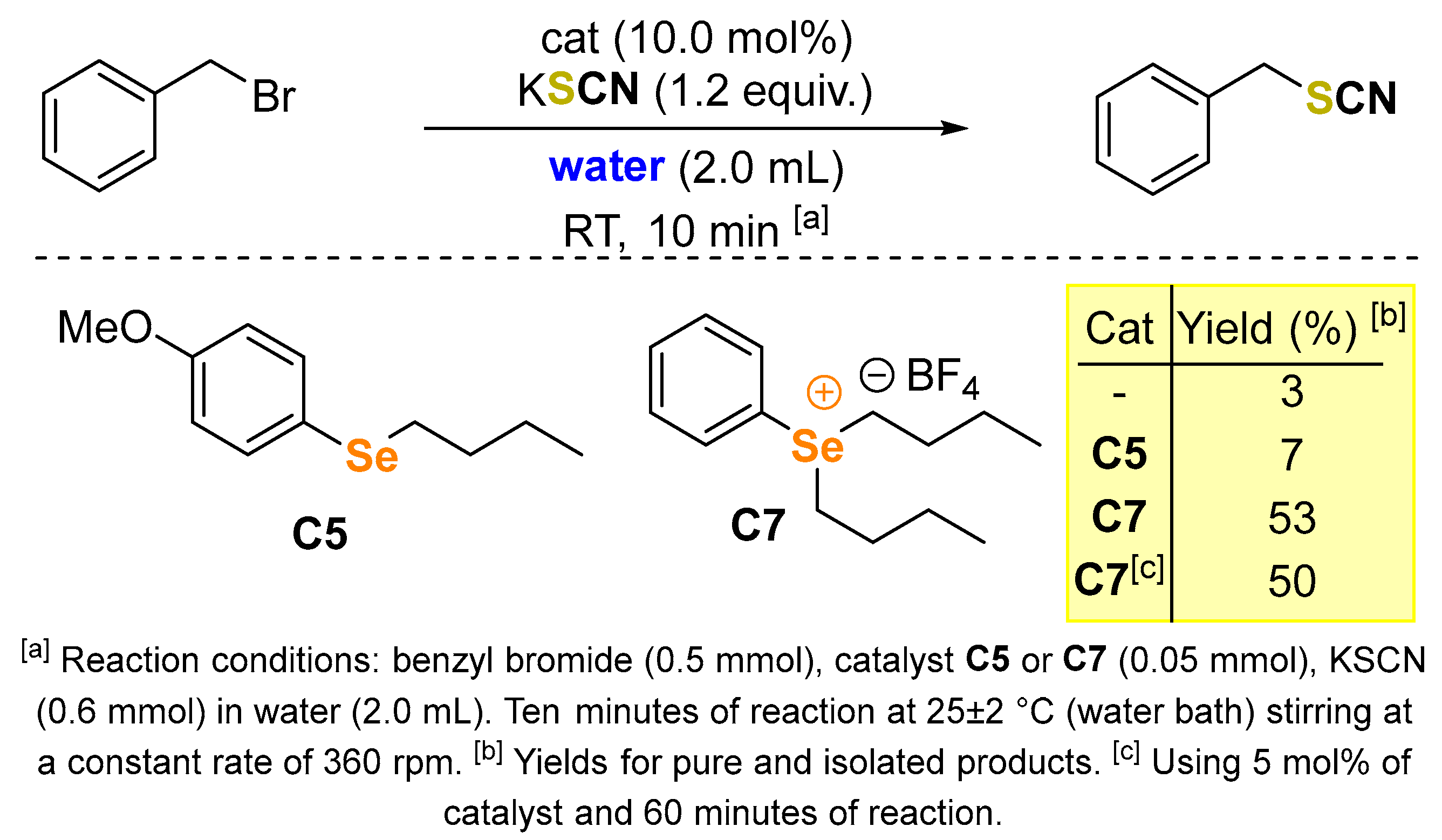
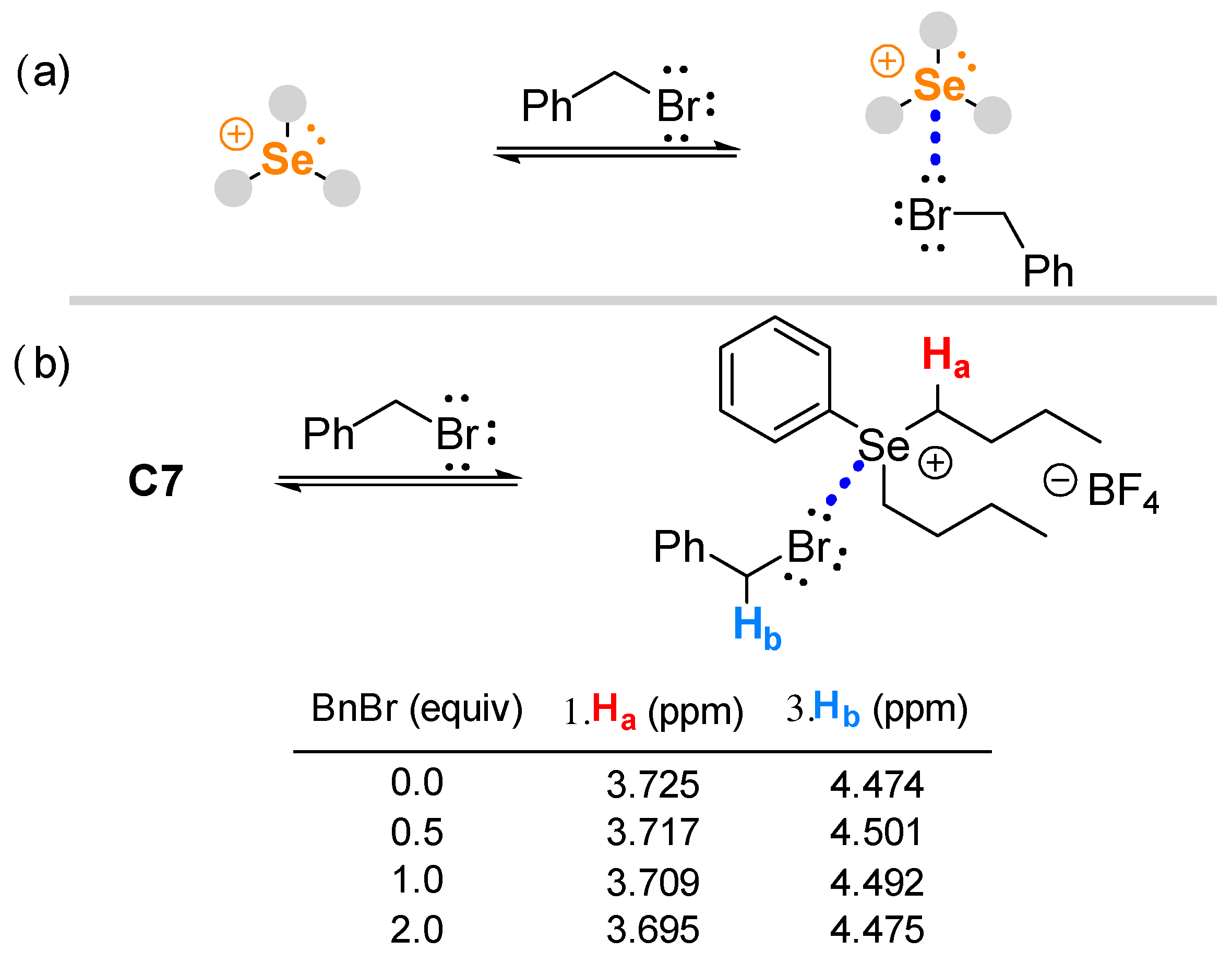
2. Results and Discussion
3. Materials and Methods
3.1. General Remarks
3.2. Synthesis of Catalysts C1–5
3.3. Synthesis of Catalyst 4-(butylselanyl)benzoic Acid (C6)
3.4. Synthesis of Catalyst Dibutyl(phenyl)selenonium Tetrafluoroborate (C7)
3.5. Synthesis of Thio- and Selenocyanates
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Clark, T.; Hennemann, M.; Murray, S.J.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291. [Google Scholar] [CrossRef] [PubMed]
- Wonner, P.; Vogel, L.; Duser, M.; Gomes, L.; Kniep, F.; Mallick, B.; Werz, D.B.; Huber, S.M. Carbon–Halogen Bond Activation by Selenium-Based Chalcogen Bonding. Angew. Chem. Int. Ed. 2017, 56, 12009. [Google Scholar] [CrossRef] [Green Version]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The σ-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166. [Google Scholar] [CrossRef] [PubMed]
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the chalcogen bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889. [Google Scholar] [CrossRef]
- Zhou, B.; Gabbaï, F.P. Redox-controlled chalcogen-bonding at tellurium: Impact on Lewis acidity and chloride anion transport properties. Chem. Sci. 2020, 11, 7495. [Google Scholar] [CrossRef]
- Zhou, B.; Gabbaï, F.P. Lewis Acidic Telluronium Cations: Enhanced Chalcogen-Bond Donor Properties and Application to Transfer Hydrogenation Catalysis. Organometallics 2021, 40, 2371. [Google Scholar] [CrossRef]
- Novikov, A.S.; Bolotin, D.S. Halonium, chalconium, and pnictonium salts as noncovalent organocatalysts: A computational study on relative catalytic activity. Org. Biomol. Chem. 2022, 20, 7632. [Google Scholar] [CrossRef]
- Lenardão, E.J.; Mendes, S.R.; Ferreira, P.C.; Perin, G.; Silveira, C.C.; Jacob, R.G. Selenium- and tellurium-based ionic liquids and their use in the synthesis of octahydroacridines. Tetrahedron Lett. 2006, 47, 7439. [Google Scholar] [CrossRef]
- Lenardão, E.J.; Borges, E.L.; Mendes, S.R.; Perin, G.; Jacob, R.G. Selenonium ionic liquid as an efficient catalyst for the synthesis of thioacetals under solvent-free conditions. Tetrahedron Lett. 2008, 49, 1919. [Google Scholar] [CrossRef]
- Lenardão, E.J.; Feijó, J.O.; Thurow, S.; Perin, G.; Jacob, R.G.; Silveira, C.C. Selenonium ionic liquid as efficient catalyst for the Baylis–Hillman reaction. Tetrahedron Lett. 2009, 50, 5215. [Google Scholar] [CrossRef]
- He, X.; Wang, X.; Tse, Y.-L.; Ke, Z.; Yeung, Y.-Y. Applications of Selenonium Cations as Lewis Acids in Organocatalytic Reactions. Angew. Chem. Int. Ed. 2018, 57, 12869. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Wang, X.; Tse, Y.-L.; Ke, Z.; Yeung, Y.-Y. Bis-selenonium Cations as Bidentate Chalcogen Bond Donors in Catalysis. ACS Catal. 2021, 11, 12632. [Google Scholar] [CrossRef]
- Okuno, K.; Nakamura, T.; Shirakawa, S. Asymmetric Catalysis of Chiral Bifunctional Selenides and Selenonium Salts Bearing a Urea Group. Asian J. Org. Chem. 2021, 10, 655. [Google Scholar] [CrossRef]
- Il’in, M.V.; Novikov, A.S.; Bolotin, D.S. Sulfonium and Selenonium Salts as Noncovalent Organocatalysts for the Multicomponent Groebke–Blackburn–Bienaymé Reaction. J. Org. Chem. 2022, 87, 10199. [Google Scholar] [CrossRef]
- Liao, L.; Zhao, X. Modern Organoselenium Catalysis: Opportunities and Challenges. Synlett 2021, 32, 1262. [Google Scholar] [CrossRef]
- Ángel, A.Y.B.; Campos, P.R.O.; Alberto, E.E. Synthetic application of chalcogenonium salts: Beyond sulfonium. Org. Biomol. Chem. 2023, 21, 223. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.V.; Wang, C.-Q.; Simi, B.; Rodriguez, J.G.; Cooma, I.; El-Bayoumy, K.; Reddy, B.S. Chemoprevention of Colon Cancer by a Glutathione Conjugate of 1,4-Phenylenebis(methylene)selenocyanate, a Novel Organoselenium Compound with Low Toxicity. Cancer Res. 2001, 61, 3647. [Google Scholar]
- Banerjee, K.; Padmavathi, G.; Bhattarcherjee, D.; Saha, S.; Kunnumakkara, A.B.; Bhabak, K.P. Potent anti-proliferative activities of organochalcogenocyanates towards breast cancer. Org. Biomol. Chem. 2018, 16, 8769. [Google Scholar] [CrossRef]
- Banerjee, K.; Bhattarcherjee, D.; Mahato, S.K.; Sufian, A.; Bhabak, K.P. Selenocyanates: Ionic Organoselenium Compounds with Efficient Peroxide Scavenging Activities. Inorg. Chem. 2021, 60, 12984. [Google Scholar] [CrossRef]
- Szajnman, S.H.; Yan, W.; Bailey, B.N.; Docampo, R.; Elhalem, E.; Rodriguez, J.B. Design and Synthesis of Aryloxyethyl Thiocyanate Derivatives as Potent Inhibitors of Trypanosoma cruzi Proliferation. J. Med. Chem. 2000, 43, 1826. [Google Scholar] [CrossRef]
- Alcolea, V.; Pérez-Silanes, S. Selenium as an interesting option for the treatment of Chagas disease: A review. Eur. J. Med. Chem. 2020, 206, 112673. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Hernández, M.; Alcolea, V.; Pérez-Silanes, S. Potential of sulfur-selenium isosteric replacement as a strategy for the development of new anti-chagasic drugs. Acta Tropica 2022, 233, 106547. [Google Scholar] [CrossRef] [PubMed]
- Castanheiro, T.; Suffert, J.; Donnard, M.; Gulea, M. Recent advances in the chemistry of organic thiocyanates. Chem. Soc. Rev. 2016, 45, 494. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Lin, J.-H.; Xiao, J.-C. HCF2Se/HCF2S Installation by Tandem Substitutions from Alkyl Bromides. J. Org. Chem. 2021, 86, 13153. [Google Scholar] [CrossRef] [PubMed]
- Karmaker, P.G.; Huo, F. Organic Selenocyanates: Rapid Advancements and Applicationsin the Field of Organic Chemistry. Asian J. Org. Chem. 2022, 11, e202200226. [Google Scholar] [CrossRef]
- Alfuth, J.; Jeannin, O.; Fourmigué, M. Topochemical, Single-Crystal-to-Single-Crystal [2+2] Photocycloadditions Driven by Chalcogen-Bonding Interactions. Angew. Chem. Int. Ed. 2022, 61, e202206249. [Google Scholar] [CrossRef]
- Azaroon, M.; Kiasat, A.R. Crown ether functionalized magnetic hydroxyapatite as eco-friendly microvessel inorganic-organic hybrid nanocatalyst in nucleophilic substitution reactions: An approach to benzyl thiocyanate, cyanide, azide and acetate derivatives. Appl. Organomet. Chem. 2018, 32, e4046. [Google Scholar] [CrossRef]
- Goodajdar, B.M.; Akbari, F.; Davarpanah, J. PEG-DIL-based MnCl42−: A novel phase transfer catalyst for nucleophilic substitution reactions of benzyl halides. Appl. Organomet. Chem. 2018, 33, 4647. [Google Scholar] [CrossRef]
- Shi, X.-L.; Chen, Y.; Hu, Q.; Meng, H.; Duan, P. Fiber-Supported Poly(quaternaryammonium Bromide)s as Supported-Phase Transfer Catalysts in the Spinning Basket Reactor. Ind. Eng. Chem. Res. 2018, 57, 7450. [Google Scholar] [CrossRef]
- Shen, J.-C.; Jiang, W.-L.; Guo, W.-D.; Qi, Q.-Y.; Ma, D.-L.; Lou, X.; Shen, M.; Hu, B.; Yang, H.-B.; Zhao, X. A rings-in-pores net: Crown ether-based covalent organic frameworks for phase-transfer catalysis. Chem. Commun. 2020, 56, 595. [Google Scholar] [CrossRef]
- Melillo, A.; Kiani, A.; Schettini, R.; Acocella, M.R. Carbon black intercalation compound as catalyst for unprecedent phase-transfer-catalyzed nucleophilic substitution (SN2) in water. Mol. Catal. 2023, 537, 112951. [Google Scholar] [CrossRef]
- Guo, W.; Tan, W.; Zhao, M.; Zheng, L.; Tao, K.; Chen, D.; Fan, X. Direct Photocatalytic S–H Bond Cyanation with Green “CN” Source. J. Org. Chem. 2018, 83, 6580. [Google Scholar] [CrossRef]
- Wu, D.; Duan, Y.; Liang, K.; Yin, H.; Chen, F.-X. AIBN-initiated direct thiocyanation of benzylic sp3 C–H with N-thiocyanatosaccharin. Chem. Commun. 2021, 57, 9938. [Google Scholar] [CrossRef]
- Zhao, X.; Ji, L.; Gao, Y.; Sun, T.; Qiao, J.; Li, A.; Lu, K. Visible-Light-Promoted Selenocyanation of Cyclobutanone Oxime Esters Using Potassium Selenocyanate. J. Org. Chem. 2021, 86, 11399. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Li, C.; Wang, C.; Zhang, H.; Cao, Z.-Y. (1-Selenocyanatoethyl)benzene: A Selenocyanation Reagent for Site-Selective Selenocyanation of Inert Alkyl C(sp3)–H Bonds. Org. Lett. 2021, 23, 7156. [Google Scholar] [CrossRef]
- Tao, S.; Jiang, L.; Du, Y. Electrophilic Oxythio/selenocyanation of o-Alkenyl Benzoic Acids: Divergent Synthesis of Thio/selenocyanated Isobenzofuranones and Isocoumarins. Asian J. Org. Chem. 2022, 11, e202200595. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.-Z.; Liu, Y.-J.; Yan, X.-X.; Yan, Y.-H.; Chao, S.-J.; Shang, X.; Ni, T.; Zhou, P.-X. Synthesis of Isoquinolylselenocyanates and Quinolylselenocyanates via Electrophilic Selenocyanogen Cyclization Induced by Pseudohalogen (SeCN)2 Generated in situ. Adv. Synth. Catal. 2022, 364, 187. [Google Scholar] [CrossRef]
- Banliat, A.D.; Grollier, K.; Damond, A.; Billard, T.; Dagousset, G.; Magnier, E.; Pégot, B. Solvent free nucleophilic selenocyanation with [bmim][SeCN]. Direct access to perfluoroalkylselenide compounds. Tetrahedron 2021, 101, 132507. [Google Scholar] [CrossRef]
- Martins, N.S.; Ángel, A.Y.B.; Anghinoni, J.M.; Lenardão, E.J.; Barcellos, T.; Alberto, E.E. From Stoichiometric Reagents to Catalytic Partners: Selenonium Salts as Alkylating Agents for Nucleophilic Displacement Reactions in Water. Adv. Synth. Catal. 2021, 364, 87. [Google Scholar] [CrossRef]
- Alder, C.M.; Hayler, J.D.; Henderson, R.K.; Redman, A.M.; Shukla, L.; Shuster, L.E.; Sneddon, H.F. Updating and further expanding GSK’s solvent sustainability guide. Green Chem. 2016, 18, 3879. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, P.; Yan, T.; Cai, M. A highly efficient heterogeneous ruthenium(III)-catalyzed reaction of diaryl diselenides with alkyl halides leading to unsymmetrical diorganyl selenides. J. Organomet. Chem. 2017, 840, 38. [Google Scholar] [CrossRef]
- Saba, S.; Botteselle, G.V.; Godoi, M.; Frizon, T.E.A.; Galetto, F.Z.; Rafique, J.; Braga, A.L. Copper-catalyzed synthesis of unsymmetrical diorganyl chalcogenides (Te/Se/S) from boronic acids under solvent-free conditions. Molecules 2017, 22, 1367. [Google Scholar] [CrossRef] [Green Version]
- Redon, S.; Vanelle, P. Nucleophilic Selenocyanation from Selenium Dioxide. Synthesis 2023, 55, 510. [Google Scholar] [CrossRef]
- Jiang, C.; Chen, P.; Liu, G. Copper-catalyzed benzylic C–H bond thiocyanation: Enabling late-stage diversifications. CCS Chem. 2021, 3, 1884. [Google Scholar] [CrossRef]
- Nasim, M.J.; Witek, K.; Kincses, A.; Abdin, A.Y.; Zeslawska, E.; Marc, M.A.; Gajdacs, M.; Spengler, G.; Nitek, W.; Latacz, G.; et al. Pronounced activity of aromatic selenocyanates against multidrug resistant ESKAPE bacteria. New J. Chem. 2019, 43, 6021. [Google Scholar] [CrossRef] [Green Version]
- Kiasat, A.R.; Badri, R.; Sayyahi, S. A facile and convenient method for synthesis of alkyl thiocyanates under homogeneous phase transfer catalyst conditions. Chin. Chem. Lett. 2008, 19, 1301. [Google Scholar] [CrossRef]
- Iwaoka, M.; Katsuda, T.; Komatsu, H.; Tomoda, S. Experimental and theoretical studies on the nature of weak nonbonded interactions between divalent selenium and halogen atoms. J. Org. Chem. 2005, 70, 321. [Google Scholar] [CrossRef] [PubMed]
- Bound, D.J.; Bettadaiah, B.K.; Srinivas, P. Microwave-assisted synthesis of alkyl thiocyanates. Synth. Commun. 2013, 43, 1138. [Google Scholar] [CrossRef]
- Mokhtari, B.; Azadi, R.; Rahmani-Nezhad, S. In situ-generated N-thiocyanatosuccinimide (NTS) as a highly efficient reagent for the one-pot thiocyanation or isothiocyanation of alcohols. Tetrahedron Lett. 2009, 50, 6588. [Google Scholar] [CrossRef]
- Priestap, H.A. Effect of some benzyl thiocyanate analogs on tetracycline production. J. Antibiot. 1987, 40, 1341. [Google Scholar] [CrossRef] [Green Version]
- Drabek, J. The synthesis of pyrethrum’s synergists with methylene-and ethylenedioxybenzene. Chem. Pap. 1956, 10, 357–367. [Google Scholar]
- Begines, P.; Sevilla-Horrillo, L.; Puerta, A.; Puckett, R.; Bayort, S.; Lagunes, I.; Maya, I.; Padrón, J.M.; López, O.; Fernández-Bolaños, J.G. Masked phenolic-selenium conjugates: Potent and selective antiproliferative agents overcoming P-gp resistance. Pharmaceuticals 2020, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Rad, M.N.S. A simple, one-pot and phosphine-free procedure for thiocyanation of alcohols Using N-(p-toluenesulfonyl) imidazole (TsIm). J. Chem. Res. 2016, 40, 583. [Google Scholar] [CrossRef]
- Lam, L.K.T.; Ahmed, N. Organoselenium Compounds for Cancer Chemoprevention. U.S. Patent 2002/01652.15A1, 7 November 2002. [Google Scholar]
- Muthyala, M.K.; Choudhary, S.; Kumar, A. Synthesis of ionic liquid-supported hypervalent iodine reagent and its application as a ‘catch and release’ reagent for α-substituted acetophenones. RSC Adv. 2014, 4, 14297. [Google Scholar] [CrossRef]
- Xiao, J.-A.; Li, Y.-C.; Cheng, X.-L.; Chen, W.-Q.; Cui, J.-G.; Huang, Y.-M.; Huang, J.; Xiao, Q.; Su, W.; Yang, H. Selenocyanobenziodoxolone: A practical electrophilic selenocyanation reagent and its application for solid-state synthesis of α-carbonyl selenocyanates. Org. Chem. Front. 2019, 6, 1967. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||
| Entry | Catalyst | Yield of Benzyl Thiocyanate (%) [b] |
| 1 | - | 17 |
| 2 | C1 | 25 |
| 3 | C2 | 25 |
| 4 | C3 | 20 |
| 5 | C4 | 32 |
| 6 | C5 | 37 |
| 7 | C6 | 28 |
 | |||
| Entry | C7 (mol%) | KSCN (equiv.) | Yield (%) [b] |
| 1 | 0.0 | 1.2 | 5 ± 2 |
| 2 | 10.0 | 1.2 | 41 ± 1 |
| 3 | 10.0 | 2.0 | 86 ± 2 |
| 4 [c] | 10.0 | 2.0 | 85 ± 2 |
| 5 | 5.0 | 2.0 | 37 ± 2 |
| 6 | 2.5 | 2.0 | 13 ± 2 |
 | |||||
| Entry | Time (min) | Product | Yield (%) [b] | Product | Yield (%) [b] |
| 1 | 10 |  | 86 |  | 94 |
| 2 | 10 |  | 86 |  | 97 |
| 3 | 10 |  | 86 |  | 87 |
| 4 | 10 |  | 84 |  | 74 |
| 5 | 10 |  | 61 |  | 82 |
| 6 | 10 |  | 76 |  | 52 |
| 7 | 10 |  | 68 |  | 82 |
| 8 | 60 |  | 87 |  | 69 |
| 9 | 60 |  | 92 |  | 80 |
| 10 | 60 |  | 60 |  | 81 |
| 11 | 10 |  | 37 |  | 41 |
| 12 | 10 |  | 64 |  | 72 |
| 13 | 10 |  | 60 |  | 70 |
| 14 | 24 h |  | 0 |  | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ángel, A.Y.B.; Campos, P.R.O.; Alberto, E.E. Selenonium Salt as a Catalyst for Nucleophilic Substitution Reactions in Water: Synthesis of Thiocyanites and Selenocyanates. Molecules 2023, 28, 3056. https://doi.org/10.3390/molecules28073056
Ángel AYB, Campos PRO, Alberto EE. Selenonium Salt as a Catalyst for Nucleophilic Substitution Reactions in Water: Synthesis of Thiocyanites and Selenocyanates. Molecules. 2023; 28(7):3056. https://doi.org/10.3390/molecules28073056
Chicago/Turabian StyleÁngel, Alix Y. Bastidas, Philipe Raphael O. Campos, and Eduardo E. Alberto. 2023. "Selenonium Salt as a Catalyst for Nucleophilic Substitution Reactions in Water: Synthesis of Thiocyanites and Selenocyanates" Molecules 28, no. 7: 3056. https://doi.org/10.3390/molecules28073056