3.1. Chemistry
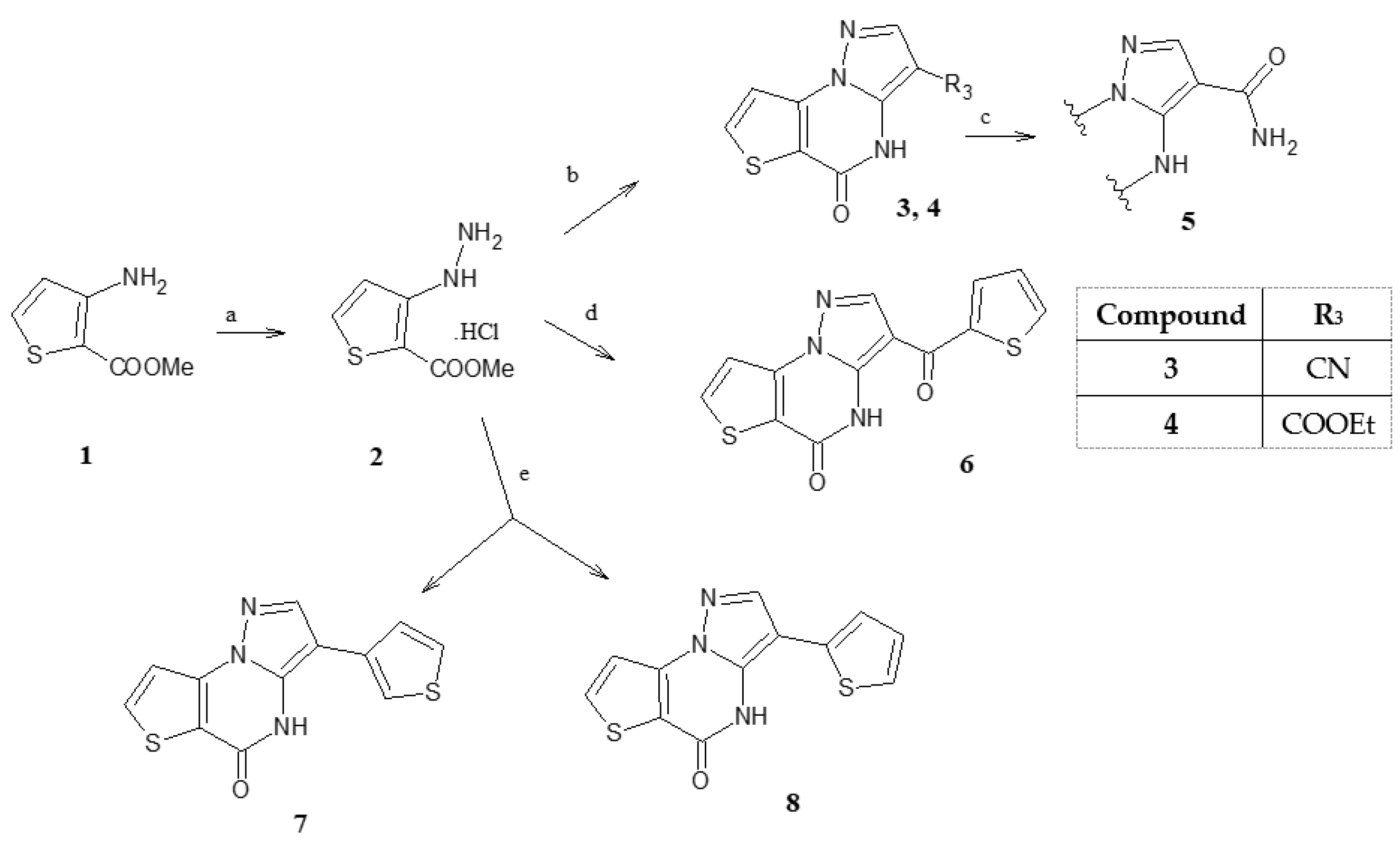
General procedure for the synthesis of compounds 3,4,6-
8. To a solution of 2 (1 mmol, 0.170 g) in DMF abs. and sodium acetate (1.3 mmol) was added 2-ethoxymethylenmalononitrile or ethyl-2-cyano-3-ethoxyacrylate, 3-(dimethylamino)- 2-(thien-2-carbonyl)acrylonitrile [
15] (1.12 mmol) to obtain
3,
4 and
6. The solvent was AcOH when 3-oxo-2-(thien-3-yl)propionitrile and 3-oxo-2-(thien-2-yl)propionitrile [
16] were used to obtain
7 and
8. The reaction was refluxed for three hours, and after cooling, the addition of water and ice gave a precipitate that was filtered and purified by a suitable solvent
5-Oxo-4,5-dihydropyrazolo[1,5-a]thieno[2,3-e]pyrimidine-3-carbonitrile (3). From 2 and 2-ethoxymethylenmalononitrile. Recrystallized by i-propanol, yield 60%, cream crystals, mp > 300 °C. TLC: toluene/ethyl acetate/methanol 8:2:1.5 v/v/v (Rf: 0.4); 1H-NMR (400 MHz, DMSO-d6) δ 13.47 (bs, 1H, NH, exch.); 8.28 (m, 2H, H-2 and H-7); 7.67 (d, 1H, H-8, J = 5.2 Hz). 13C-NMR (100 MHz, DMSO-d6) δ 163.70, 156.56, 148.45, 144.35, 138.87, 135.86, 129.09, 117.24, 115.45. Anal. C9H4N4OS (C, H, N).
Ethyl 5-oxo-4,5-dihydropyrazolo[1,5-a]thieno[2,3-e]pyrimidine-3-carboxylate (4). From 2 and ethyl 2-cyano-3-ethoxyacrylate. Recrystallized by ethanol, yield 58%, cream crystals, mp 215–216 °C. TLC: toluene/ethyl acetate/methanol 8:2:1.5 v/v/v (Rf: 0.5); 1H-NMR (400 MHz, DMSO-d6) δ 11.74 (bs, 1H, NH, exch.); 8.35 (d, 1H, H-7, J = 5.2 Hz); 8.17 (s, 1H, H-2); 7.68 (d, 1H, H-8, J = 5.2 Hz); 4.29 (q, 2H, CH2, J = 7.2 Hz); 1.29 (t, 3H, CH3, J = 7.2 Hz). 1H-NMR (400 MHz, CDCl3) δ 9.63 (bs, 1H, NH, exch.); 8.08 (s, 1H, H-2); 7.93 (d, 1H, H-7, J = 5.2 Hz); 7.67 (d, 1H, H-8, J = 5.2 Hz); 4.39 (q, 2H, CH2, J = 6.8 Hz); 1.41 (t, 3H, CH3, J = 6.8 Hz). 13C-NMR (100 MHz, DMSO-d6) δ 165.29, 161.50, 156.56, 144.35, 143.95, 135.87, 133.26, 129.05, 117.25, 60.94, 14.15. Anal. C11H9N3O3S (C, H, N).
3-(Thiophene-2-carbonyl)pyrazolo[1,5-a]thieno[2,3-e]pyrimidin-5(4H)-one (6). From
2 and 3-(dimethylamino)-2-(thien-2-carbonyl)acrylonitrile [
15]. Recrystallized by ethanol, yield 95%, cream crystals, mp > 300 °C. TLC: toluene/ethyl acetate/methanol 8:2:1.5
v/
v/
v (Rf: 0.7);
1H-NMR (400 MHz, DMSO-d
6) δ 11.59 (bs, 1H, NH, exch.); 8.63 (s, 1H, H-2); 8.37 (d, 1H, H-7,
J = 5.2 Hz); 8.17 (s, 1H, H-5′); 8.05 (d, 1H, H-3′,
J = 4.4 Hz); 7.74 (d, 1H, H-8,
J = 5.2 Hz); 7.29 (m, 1H, H-4′).
13C-NMR (100 MHz, DMSO-d
6) δ 157.90, 142.76, 142.25, 138.37, 134.86, 133.09, 129.39, 117.34. Anal. C
13H
7N
3O
2S
2 (C, H, N).
3-(Thiophene-3-yl)pyrazolo[1,5-a]thieno[2,3-e]pyrimidin-5(4H)-one (7). From
2 and 3-oxo-2-(thien-3-yl)propionitrile [
16]. Recrystallized by ethanol, yield 94%, cream crystals, mp 274–276 °C. TLC: toluene/ethyl acetate/methanol 8:2:1.5
v/
v/
v (Rf: 0.7);
1H-NMR (400 MHz, DMSO-d
6) δ 12.06 (bs, 1H, NH, exch.); 8.29 (d, 1H, H-7,
J = 5.2 Hz); 8.18 (s, 1H, H-2); 7.86 (s, 1H, H-2′); 7.67 (d, 1H, H-8,
J = 5.2 Hz); 7.06 (m, 1H, H-4′); 7.52 (m, 1H, H-5′).
13C-NMR (100 MHz, DMSO-d
6) δ 165.10, 144.35, 141.71, 139.60, 129.95, 128.30, 127.90, 124.75, 117.20. Anal. C
12H
7N
3OS
2 (C, H, N).
3-(Thiophene-2-yl)pyrazolo[1,5-a]thieno[2,3-e]pyrimidin-5(4H)-one (8). From
2 and 3-oxo-2-(thien-2-yl)propionitrile [
16]. Recrystallized by ethanol, yield 89%, green light crystals, mp 248–250 °C. TLC: toluene/ethyl acetate/methanol 8:2:1.5
v/
v/
v (Rf: 0.7);
1H-NMR (400 MHz, DMSO-d
6) δ 12.18 (bs, 1H, NH, exch.); 8.30 (d, 1H, H-7,
J = 5.2 Hz); 8.00 (s, 1H, H-2); 7.67 (d, 1H, H-8,
J = 5.2 Hz); 7.46 (d, 1H, H-5′,
J = 4.8 Hz); 7.40 (d, 1H, H-3′,
J = 2.8 Hz); 7.11 (dd, 1H, H-4′,
J1 = 4.8 Hz,
J2 = 4.0 Hz).
13C-NMR (100 MHz, DMSO-d
6) δ 157.90, 141.71, 137.66, 128.40, 124.61, 117.19Anal. C
12H
7N
3OS
2 (C, H, N).
5-Oxo-4,5-dihydropyrazolo[1,5-a]thieno[2,3-e]pyrimidine-3-carboxamide (5). Compound 3 (0.23 mmol, 0.05 g) was suspended in H2SO4 conc., 1 mL and heated at 80 °C under stirring. After the starting material disappeared in TLC (toluene/ethyl acetate/methanol 8:2:1.5 v/v/v, as eluent, Rf: 0.2), the reaction was stopped; the addition of ice/water gave a precipitate which was filtered and purified by recrystallization with ethanol. Yield 92%, white crystals, mp > 300 °C. TLC: 1H-NMR (400 MHz, DMSO-d6) δ 10.75 (bs, 1H, NH, exch.); 8.33 (d, 1H, H-7, J = 4.8 Hz); 8.28 (s, 1H, H-2); 7.83 (bs, 1H, CONH, exch.); 7.68 (d, 1H, H-8, J = 4.8 Hz); 7.32 (bs, 1H, CONH, exch.).13C-NMR (100 MHz, DMSO-d6) δ 165.22, 163.70, 156.56, 150.55, 144.35, 143.91, 135.85, 132.06, 129.00. Anal. C9H6N4O2S (C, H, N).
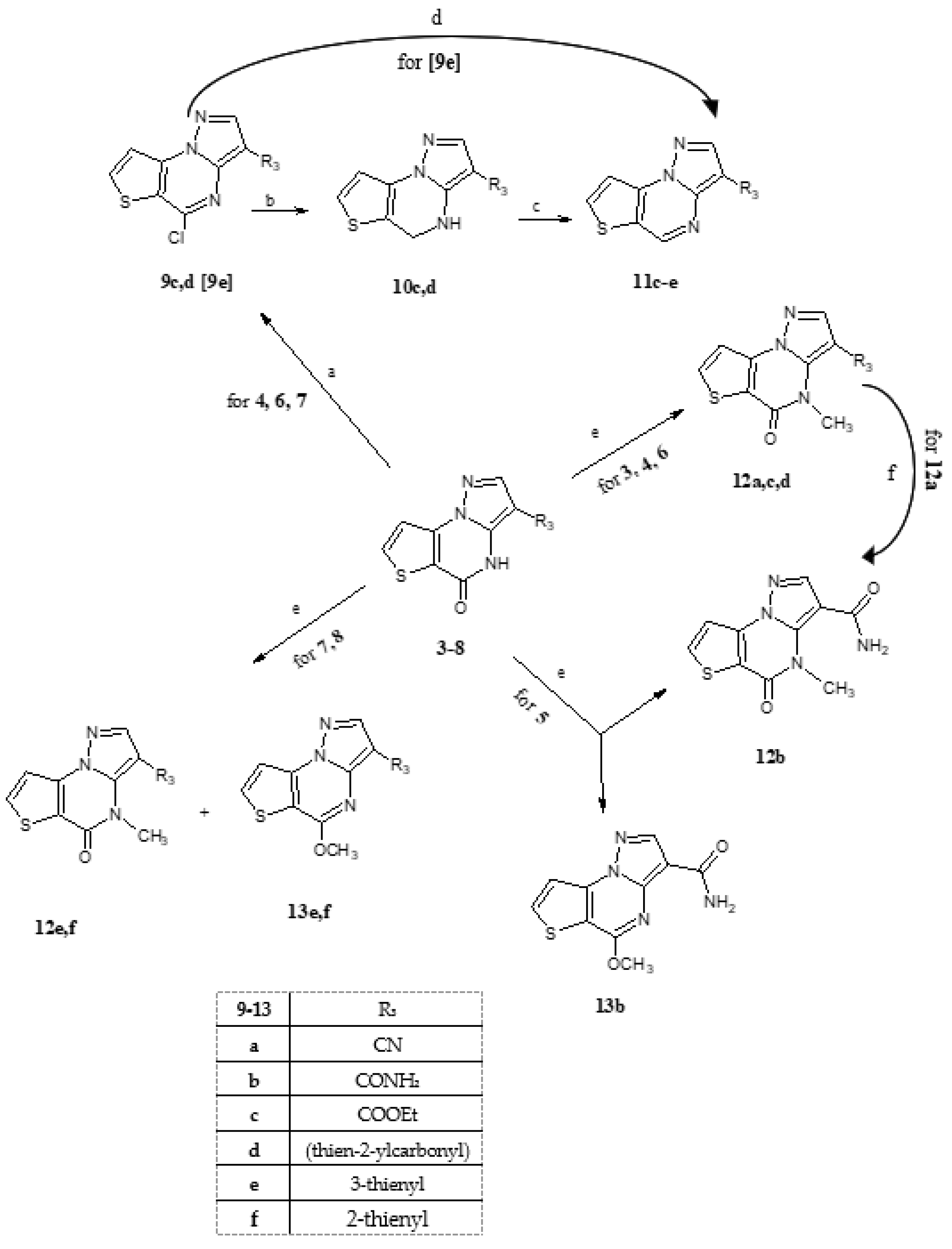
General procedure for the synthesis of compounds 9c,d. Compounds 4, 6 and 7 (0.6 mmol) were suspended in a mixture of POCl3 (5.5 mL) and PCl5 (0.91 mmol, 0.190 g) and refluxed for three hours. The evaporation to dryness to eliminate the excess of POCl3 gave a residue recuperated with ice/water filtered and purified with a suitable solvent, obtaining 9c,d, starting from 4 and 6, respectively. From compound 7, the 5-chloro intermediate 9e was not isolated but used as such, see below.
Ethyl 5-chloropyrazolo[1,5-a]thieno[2,3-e]pyrimidine-3-carboxylate (9c). From 5. Recrystallized by ethanol, yield 90%, cream crystals, mp > 300 °C. TLC: toluene/ethyl acetate/methanol 8:2:1.5 v/v/v (Rf: 0.7); 1H-NMR (400 MHz, DMSO-d6) δ 8.61 (m, 2H, H-2 and H-7); 8.02 (d, 1H, H-8, J = 5.2 Hz); 4.29 (q, 2H, CH2, J = 7.2 Hz); 1.30 (t, 3H, CH3, J = 7.2 Hz). 1H-NMR (400 MHz, CDCl3) δ 8.55 (s, 1H, H-2); 8.08 (d, 1H, H-7, J = 5.6 Hz); 7.95 (d, 1H, H-8, J = 5.6 Hz); 4.45 (q, 2H, CH2, J = 7.2 Hz); 1.44 (t, 3H, CH3, J = 7.2 Hz). 13C-NMR (100 MHz, DMSO-d6) δ 161.80, 155.06, 145.85, 145.55, 130.55, 125.66, 126.70, 118.30, 60.95, 14.13. Anal. C11H8N3O2SCl (C, H, N).
(5-Chloropyrazolo[1,5-a]thieno[2,3-e]pyrimidin-3-yl)(thiophen-2-yl)methanone (9d). From 6. Recrystallized by i-propanol, yield 95%, cream crystals, mp 186–188 °C. TLC: toluene/ethyl acetate/methanol 8:2:1.5 v/v/v (Rf: 0.5); 1H-NMR (400 MHz, DMSO-d6) δ 8.80 (s, 1H, H-2); 8.65 (d, 1H, H-7, J = 5.2 Hz); 8.14 (s, 1H, H-5′); 8.07 (m, 2H, H-8 and H-3′); 7.30 (m, 1H, H-4′). 13C-NMR (100 MHz, DMSO-d6) δ 173.70, 155.06, 145.88, 145.53, 144.35, 135.81, 133.72, 130.50, 127.06, 126.74, 125.66, 126.70, 118.30. Anal. C13H6N3OS2Cl (C, H, N).
General procedure for the synthesis of compounds 10c,d. Compounds 9c,d (0.4 mmol) was dissolved in a mixture of CH2Cl2/EtOH (7.5 mL/15 mL) and NaBH4 (3.6 mmol, 0.136 g) was added in small portions. The reaction was maintained at room temperature for 40 min, and then the evaporation to dryness of the solvent gave a residue which was recovered with water, filtered and purified with a suitable solvent, obtaining 10c,d, respectively.
Ethyl 4,5-dihydropyrazolo[1,5-a]thieno[2,3-e]pyrimidine-3-carboxylate (10c). From 9c. Recrystallized by water, yield 45%, cream crystals, mp 126–128 °C. TLC: toluene/ethyl acetate/methanol 8:2:1.5 v/v/v (Rf: 0.5); 1H-NMR (400 MHz, DMSO-d6) δ 7.57 (m, 2H, H-2 and H-7); 7.18 (d, 1H, H-8, J = 4.4 Hz); 7.05 (s, 1H, NH, exch.); 4.7 (s, 2H, NCH2); 4.17 (q, 2H, CH2, J = 7.2 Hz); 1.25 (t, 3H, CH3, J = 7.2 Hz). 1H-NMR (400 MHz, CDCl3) δ 8.55 (s, 1H, H-2); 8.08 (d, 1H, H-7, J = 5.6 Hz); 7.95 (d, 1H, H-8, J = 5.6 Hz); 4.70 (s, 2H, NCH2); 4.18 (q, 2H, CH2, J = 6.8 Hz); 1.25 (t, 3H, CH3, J = 6.8 Hz). 13C-NMR (100 MHz, DMSO-d6) δ 163.29, 140.97, 125.82, 116.47, 59.18, 41.96, 15.02. Anal. C11H11N3O2S (C, H, N).
4,5-Dihydropyrazolo[1,5-a]thieno[2,3-e]pyrimidin-3-yl(thiophen-2-yl)methanone (10d). From 9d. Recrystallized by ethanol, yield 40%, cream crystals, mp 186–188 °C. TLC: toluene/ethyl acetate/methanol 8:2:1.5 v/v/v (Rf: 0.3); 1H-NMR (400 MHz, DMSO-d6) δ 8.11 (s, 1H, H-2); 7.97 (s, 1H, H-5′); 7.91 (d, 1H, H-7, J = 4.8 Hz); 7.78 (s, 1H, NH, exch.); 7.61 (d, 1H, H-8 J = 4.8 Hz); 7.22 (m, 2H, H-3′ and H-4′); 4.79 (s, 2H, NCH2). 13C-NMR (100 MHz, DMSO-d6) δ 172.98, 158.01, 144.82, 144.35, 137.85, 135.71, 133.57, 129.06, 127.74, 123.66, 119.30, 51.55. Anal. C13H9N3OS2 (C, H, N).
General procedure for the synthesis of compounds 11c,d. Compounds 10c,d (0.4 mmol) was dissolved in toluene (15 mL), and Pd/C as catalyst was added. The reaction was refluxed for 3–5 h and filtered off the catalyst. The evaporation to dryness of the solution yielded a residue recovered with water, filtered and purified with a suitable solvent, obtaining 11c and 11d, respectively.
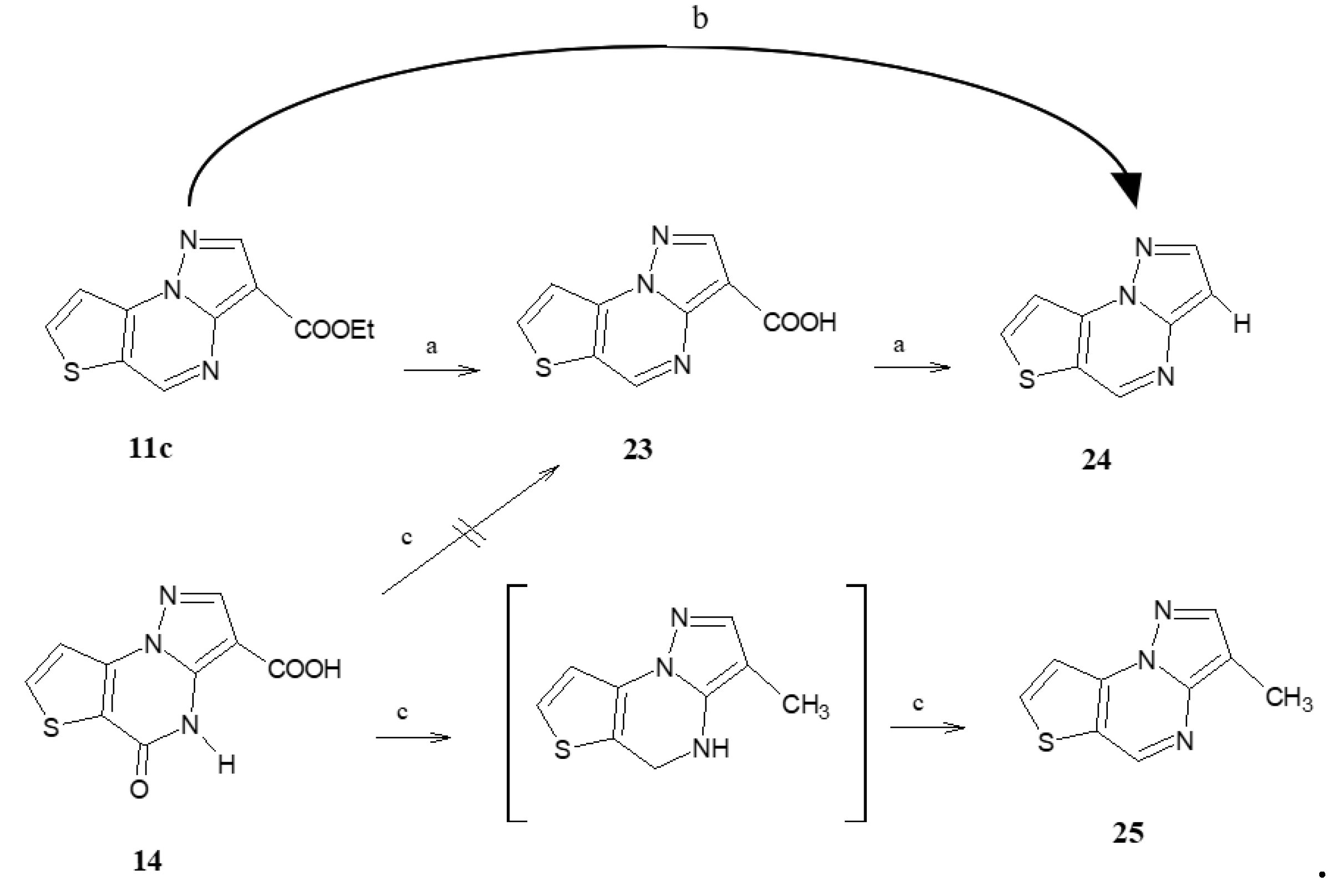
Ethyl pyrazolo[1,5-a]thieno[2,3-e]pyrimidine-3-carboxylate (11c). From 10c. Recrystallized by ethanol yield 50%, white crystals, mp 159–160 °C. TLC: toluene/ethyl acetate 8:2 v/v (Rf: 0.1); 1H-NMR (400 MHz, DMSO-d6) δ 9.42 (s, 1H, H-5); 8.60 (s, 1H, H-2); 8.57 (d, 1H, H-7, J = 5.2 Hz); 7.98 (d, 1H, H-8, J = 5.2 Hz); 4.31 (q, 2H, CH2, J = 6.8 Hz); 1.32 (t, 3H, CH3, J = 6.8 Hz). 13C-NMR (100 MHz, DMSO-d6) δ 162.34, 148.66, 145.99, 145.25, 141.82, 140.02, 123.02, 115.95, 103.38, 60.08, 14.92. Anal. C11H9N3O2S (C, H, N).
Pyrazolo[1,5-a]thieno[2,3-e]pyrimidin-3-yl(thiophen-2-yl)methanone (11d). From 10d. Recrystallized by i-propanol, yield 68%, white crystals, mp 73–75 °C. TLC: toluene/ethyl acetate/methanol 8:2:1.5 v/v/v (Rf: 0.2); 1H-NMR (400 MHz, DMSO-d6) δ 9.42 (s, 1H, H-5); 8.74 (s, 1H, H-2); 8.59 (d, 1H, H-7, J = 4.8 Hz); 8.23 (d, 1H, H-5′ J = 2.4 Hz); 8.03 (m, 2H, H-3′ and H-8); 7.28 (m, 1H, H-4′). 13C-NMR (100 MHz, DMSO-d6) δ 173.08, 149.01, 145.82, 144.85, 144.35, 135.81, 133.43, 130.57, 129.06, 126.74, 125.60, 119.10. Anal. C13H7N3OS2 (C, H, N).
3-(Thiophen-3-yl)pyrazolo[1,5-a]thieno[2,3-e]pyrimidine (11e). Compound 7, 3-(thiophene-3-yl)pyrazolo[1,5-a]thieno[2,3-e]pyrimidin-5(4H)-one (0.6 mmol), was suspended in a mixture of POCl3 (5.5 mL) and PCl5 (0.91 mmol, 0.190 g) and refluxed for three hours. The evaporation to dryness to eliminate the excess of POCl3 gave the corresponding 5-chloro derivative (9e), not isolated but used as such for the next reduction step through a CTH (catalytic transfer hydrogenation). Thus, this intermediate suspended in EtOH (20 mL) was added of ammonium formate (4.08 mmol, 0.275 g) and 10% Pd/C as catalyst. The reaction was maintained at reflux temperature for several hours, during which it was possible to evidence, by TLC, the formation of the 4,5-dihydro derivative in a mixture with the final 4,5-dehydro compound 11e. When the reaction finished, the catalyst was filtered off, the solution evaporated to dryness, and the residue recovered with water. Recrystallized by ethanol 80%, yield 90%, cream crystals, mp 157–160 °C. TLC: toluene/ethyl acetate 8:2 v/v (Rf: 0.5); 1H-NMR (400 MHz, DMSO-d6) δ 9.24 (s, 1H, H-5); 8.65 (s, 1H, H-2); 8.45 (d, 1H, H-7, J = 5.2 Hz); 8.05 (s, 1H, H-2′); 7.95 (d, 1H, H-8, J = 5.2 Hz); 7.86 (d, 1H, H-5′, J = 4.8 Hz); 7.65 (m, 1H, H-4′). 13C-NMR (100 MHz, DMSO-d6) δ 145.56, 141.21, 138.47, 126.77, 119.61, 115.88. Anal. C12H7N3S2 (C, H, N).
General procedure for the synthesis of compounds 12a,c–f and 13b,e,f. A solution of DMF abs. (5 mL), compounds 3–8 (0.40 mmol) and K2CO3 anhydrous (0.80 mmol) was maintained for 15 min at room temperature. After this time, methyl iodide (0.80 mmol) was added and enhanced temperature to 80 °C. After one hour and monitoring the reaction by TLC, adding water gave a precipitate, filtered and purified by a suitable solvent. In the case of compounds 3, 4 and 6, only 4-N-CH3 derivatives were formed (12a,c,d). From compound 4, only the 5-methoxyderivative 13b was recovered, while if the reaction is performed at reflux temperature, only the 4-methyl derivative 12b was obtained. From 7 and 8, a mixture of two products was recovered at the end of alkylation. The chromatographic separation permits isolating the 4-NCH3 (12e,f) and the 5-OCH3 derivatives (13e,f).
4-Methyl-5-oxo-4,5-dihydropyrazolo[1,5-a]thieno[2,3-e]pyrimidine-3-carbonitrile (12a). From 3. Recrystallized by ethanol, yield 89%, cream crystals, mp 224–226 °C. TLC: toluene/ethyl acetate/methanol 8:2:1 v/v/v (Rf: 0.6); 1H-NMR (400 MHz, DMSO-d6) δ 8.44 (s, 1H, H-2); 8.36 (d, 1H, H-7, J = 4.0 Hz); 7.71 29/03/2023 (d, 1H, H-8, J = 4.0 Hz); 3.73 (s, 3H, NCH3). 13C-NMR (100 MHz, DMSO-d6) δ 163.76, 156.51, 148.40, 144.37, 144.30, 135.86, 129.07, 115.41, 106.58, 36.50. Anal. C10H6N4OS (C, H, N).
4-Methyl-5-oxo-4,5-dihydropyrazolo[1,5-a]thieno[2,3-e]pyrimidine-3-carboxamide (12b). From 5 at reflux temperature. Recrystallized by ethanol, yield 93%, cream crystals, mp > 300 °C. TLC: dichlorometane/methanol 9:1 v/v (Rf: 0.3); 1H-NMR (400 MHz, DMSO-d6) δ 8.28 (d, 1H, H-7, J = 4.8 Hz); 8.11 (s, 1H, H-2); 7.86 (bs, 1H, CONH, exch.); 7.65 (d, 1H, H-8, J = 4.8 Hz); 7.32 (bs, 1H, CONH, exch.); 3.73 (s, 3H, NCH3). 13C-NMR (100 MHz, DMSO-d6) δ 165.30, 163.76, 156.51, 150.40, 145.67, 144.31, 135.86, 132.05, 129.08, 36.69. Anal. C10H8N4O2S (C, H, N). The treatment of compound 12a with sulfuric acid at 60 °C and the subsequent addition of ice/water gave the precipitate, 12b recovered by filtration.
Ethyl 4-methyl-5-oxo-4,5-dihydropyrazolo[1,5-a]thieno[2,3-e]pyrimidine-3-carboxylate (12c). From 4. Recrystallized by ethanol, yield 75%, cream crystals, mp 173–174 °C. TLC: toluene/ethyl acetate/methanol 8:2:1.5 v/v/v (Rf: 0.5); 1H-NMR (400 MHz, CDCl3) δ 8.19 (s, 1H, H-2); 7.86 (d, 1H, H-7, J = 5.2 Hz); 7.64 (d, 1H, H-8, J = 5.2 Hz); 4.34 (q, 2H, CH2, J = 7.2 Hz); 4.04 (s, 3H, NCH3); 1.40 (t, 3H, CH3, J = 7.2 Hz). 13C-NMR (100 MHz, DMSO-d6) δ 161.78, 156.10, 145.24, 142.00, 141.69, 138.12, 117.25, 116.95, 98.60, 60.77, 33.57, 14.59. Anal. C12H11N3O3S (C, H, N).
3-(Thiophene-2-carbonyl)-4-methylpyrazolo[1,5-a]thieno[2,3-e]pyrimidin-5(4H)-one (12d). From 6. Recrystallized by ethanol, yield 95%, cream crystals, mp 183–186 °C. TLC: toluene/ethyl acetate/methanol 8:2:1.5 v/v/v (Rf: 0.5); 1H-NMR (400 MHz, DMSO-d6) δ 8.37 (d, 1H, H-7, J = 5.2 Hz); 8.33 (s, 1H, H-2); 8.10 (d, 1H, H-5′ J = 4.8 Hz); 7.82 (d, 1H, H-3′, J = 3.6 Hz); 7.74 (d, 1H, H-8, J = 5.2 Hz); 7.30 (m, 1H, H-4′); 3.56 (s, 3H NCH3). 13C-NMR (100 MHz, DMSO-d6) δ 179.37, 156.18, 145.24, 144.91, 141.71, 138.40, 135.92, 135.62, 129.12, 117.39, 106.50, 33.07. Anal. C14H9N3O2S2 (C, H, N).
3-(Thiophene-3-yl)-4-methylpyrazolo[1,5-a]thieno[2,3-e]pyrimidin-5(4H)-one (12e). From 7, after chromatographic separation, second eluting band (toluene/ethyl acetate/methanol 8:2:1.5 v/v/v as eluent, Rf: 0.5), yield 35%, cream crystals, mp 109–111 °C. 1H-NMR (400 MHz, DMSO-d6) δ 8.29 (d, 1H, H-7, J = 4.8 Hz); 7.85 (s, 1H, H-2); 7.68 (d, 1H, H-8, J = 4.8 Hz); 7.63–7.59 (m, 2H, H-2′ and H-4′); 7.25 (m, 1H, H-5′); 3.40 (s, 3H, NCH3). 1H-NMR (400 MHz, CDCl3) δ 7.83 (d, 1H, H-7, J = 5.2 Hz); 7.71 (s, 1H, H-2); 7.65 (d, 1H, H-8, J = 5.2 Hz); 7.40 (m, 1H, H-2′); 7.26 (m, 1H, H-4′); 7.11 (d, 1H, H-5′, J = 4.4 Hz); 3.41 (s, 3H, NCH3). 13C-NMR (100 MHz, DMSO-d6) δ 157.94, 144.97, 140.00, 138.10, 137.90, 130.86, 125.99, 114.34, 114.23, 106.12, 20.54. Anal. C13H9N3OS2 (C, H, N).
3-(Thiophene-2-yl)-4-methylpyrazolo[1,5-a]thieno[2,3-e]pyrimidin-5(4H)-one (12f). From 8 after chromatographic separation, second eluting band (toluene/ethyl acetate/methanol 8:2:1.5 v/v/v as eluent, Rf: 0.5), yield 36%, yellow light crystals, mp 140–141 °C. 1H-NMR (400 MHz, DMSO-d6) δ 8.30 (d, 1H, H-7, J = 5.2 Hz); 7.90 (s, 1H, H-2); 7.68 (d, 1H, H-8, J = 5.2 Hz); 7.62 (d, 1H, H-5′, J = 5.2 Hz); 7.18 (m, 1H, H-3′); 7.13 (dd, 1H, H-4′, J1 = 4.8 Hz, J2 = 3.6 Hz); 3.31 (s, 3H, NCH3). 13C-NMR (100 MHz, DMSO-d6) δ 165.46, 154.88, 145.71, 137.76, 135.14, 131.79, 129.06, 117.48, 114.01, 56.89. Anal. C13H9N3OS2 (C, H, N).
5-Methoxypyrazolo[1,5-a]thieno[2,3-e]pyrimidine-3-carboxamide (13b). From 5 at room temperature. Recrystallized by ethanol, yield 75%, cream crystals, mp 269–270 °C. TLC: dichlorometane/methanol 8:2 v/v (Rf: 0.6); 1H-NMR (400 MHz, DMSO-d6) δ 8.42 (d, 1H, H-7, J = 4.8 Hz); 8.36 (s, 1H, H-2); 7.90 (d, 1H, H-8, J = 4.8 Hz); 7.49 (bs, 1H, CONH, exch.); 7.34 (bs, 1H, CONH, exch.); 4.22 (s, 3H, OCH3). 13C-NMR (100 MHz, DMSO-d6) δ 164.40, 162.26, 145.87, 145.50, 130.56, 125.68, 114.45, 107.32, 53.79. Anal. C10H8N4O2S (C, H, N).
5-Methoxy-3-(thiophene-3-yl)pyrazolo[1,5-a]thieno[2,3-e]pyrimidine (13e). From 7, after chromatographic separation, first eluting band (toluene/ethyl acetate/methanol 8:2:1.5 v/v/v as eluent, Rf: 0.8), yield 35%, cream crystals, mp 127–130 °C. 1H-NMR (400 MHz, DMSO-d6) δ 8.49 (s, 1H, H-2); 8.38 (d, 1H, H-7, J = 5.2 Hz); 7.98 (m, 1H, H-2′); 7.85 (d, 1H, H-8, J = 5.2 Hz); 7.81 (d, 1H, H-5′, J = 4.8 Hz); 7.62 (m, 1H, H-4′); 4.20 (s, 3H, OCH3). 13C-NMR (100 MHz, DMSO-d6) δ 162.20, 144.86, 139.60, 133.47, 128.37, 128.20, 126.71, 125.60, 124.75, 114.56, 107.90, 53.75. Anal. C13H9N3OS2 (C, H, N).
5-Methoxy-3-(thiophene-2-yl)pyrazolo[1,5-a]thieno[2,3-e]pyrimidine (13f). From 8 after chromatographic separation, first eluting band (toluene/ethyl acetate/methanol 8:2:1.5 v/v/v as eluent, Rf: 0.8), yield 24%, yellow light crystals, mp 118–120 °C. 1H-NMR (400 MHz, DMSO-d6) δ 8.43 (s, 1H, H-2); 8.39 (d, 1H, H-7, J = 5.2 Hz); 7.85 (d, 1H, H-8, J = 5.2 Hz); 7.57 (m, 1H, H-5′); 7.42 (d, 1H, H-3′, J = 4.8 Hz); 7.11 (m, 1H, H-4′); 4.20 (s, 3H, OCH3). 13C-NMR (100 MHz, DMSO-d6) δ 162.20, 144.86, 138.20, 133.65, 130.57, 128.67, 128.00, 126.74, 125.60, 114.56, 107.90, 53.50. Anal. C13H9N3OS2 (C, H, N).
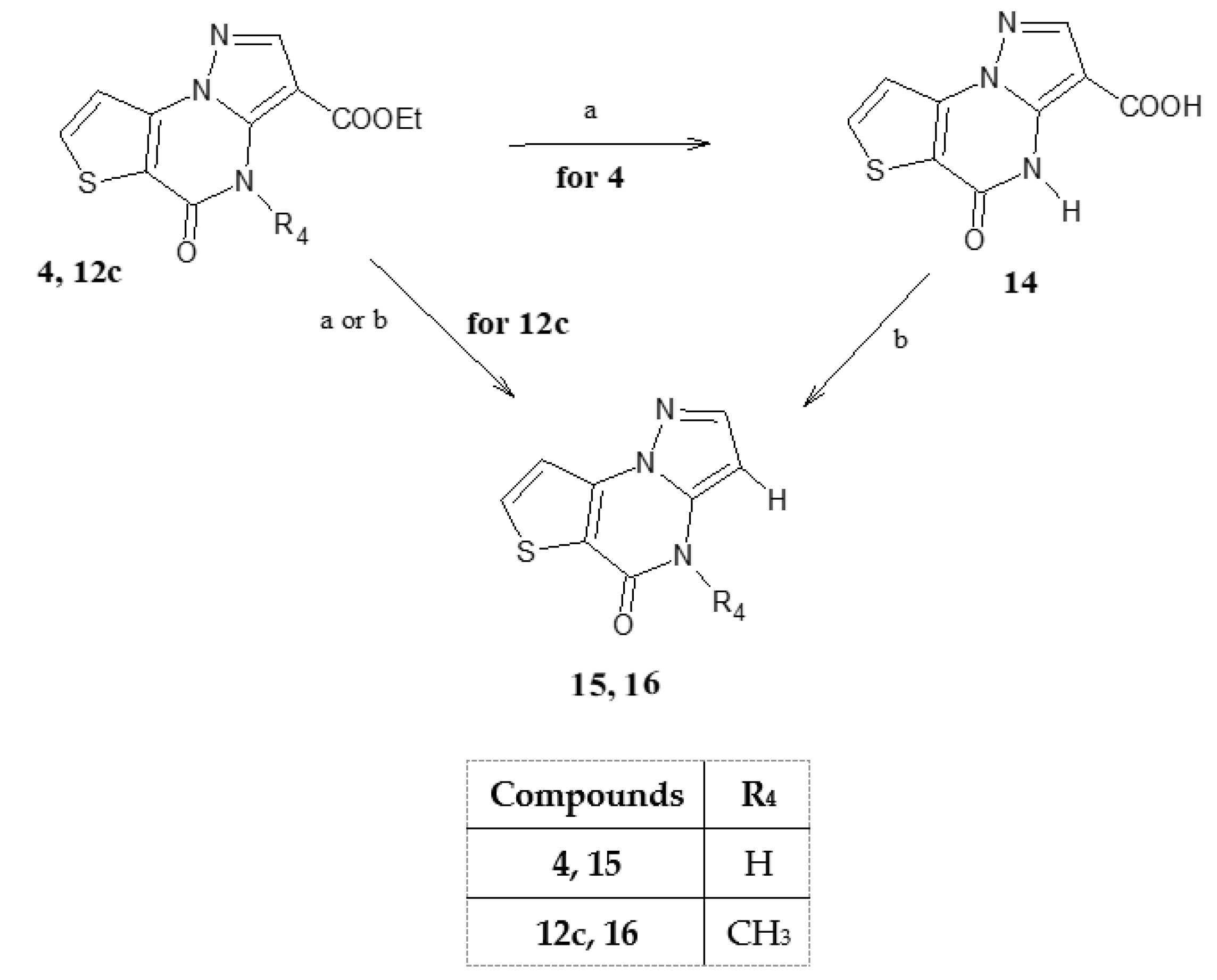
5-Oxo-4,5-dihydropyrazolo[1,5-a]thieno[2,3-e]pyrimidine-3-carboxylic acid (14). To a suspension of 4 (0.40 mmol) in NaOH 10% solution (10 mL), was added 0.5 mL of methoxyethanol to favour the solubilization. The reaction was refluxed for 1.30 h, then ice/water and HCl 6N until pH 1. The precipitate formed was filtered, washed with water and purified by a suitable solvent. Recrystallized by ethanol, yield 78%, cream crystals, mp 282–284 °C. TLC: toluene/ethyl acetate/acetic acid 8:2:2 v/v/v (Rf: 0.5); 1H-NMR (400 MHz, DMSO-d6) δ 12.70 (bs, 1H, OH, exch.); 11.37 (bs, 1H, NH, exch.); 8.34 (d, 1H, H-7, J = 4.4 Hz); 8.13 (s, 1H, H-2); 7.68 (d, 1H, H-8, J = 4.4 Hz). 13C-NMR (100 MHz, DMSO-d6) δ 165.25, 164.96, 156.50, 144.35, 143.97, 135.87, 133.20, 129.04, 117.65. Anal. C9H5N3O3S (C, H, N).
5-Oxo-4,5-dihydropyrazolo[1,5-a]thieno[2,3-e]pyrimidine (15). The acid 14 (0.50 mmol) was suspended in 10 mL of HCl conc. and maintained at reflux temperature for 5 h. Adding ice/water gave a residue filtered and purified by recrystallization with ethanol. Recrystallized by ethanol, yield 65%, cream crystals, mp 290–291 °C. TLC: toluene/ethyl acetate/methanol 8:2:1.5 v/v/v (Rf: 0.7); 1H-NMR (400 MHz, DMSO-d6) δ 12.30 (bs, 1H, NH, exch.); 8.26 (s, 1H, H-7); 7.74 (s, 1H, H-2); 7.64 (s, 1H, H-8); 5.93 (s, 1H, H-3). 13C-NMR (100 MHz, DMSO-d6) δ 164.76, 156.26, 148.45, 144.37, 135.85, 133.21, 129.09, 114.15. Anal. C8H5N3OS (C, H, N).
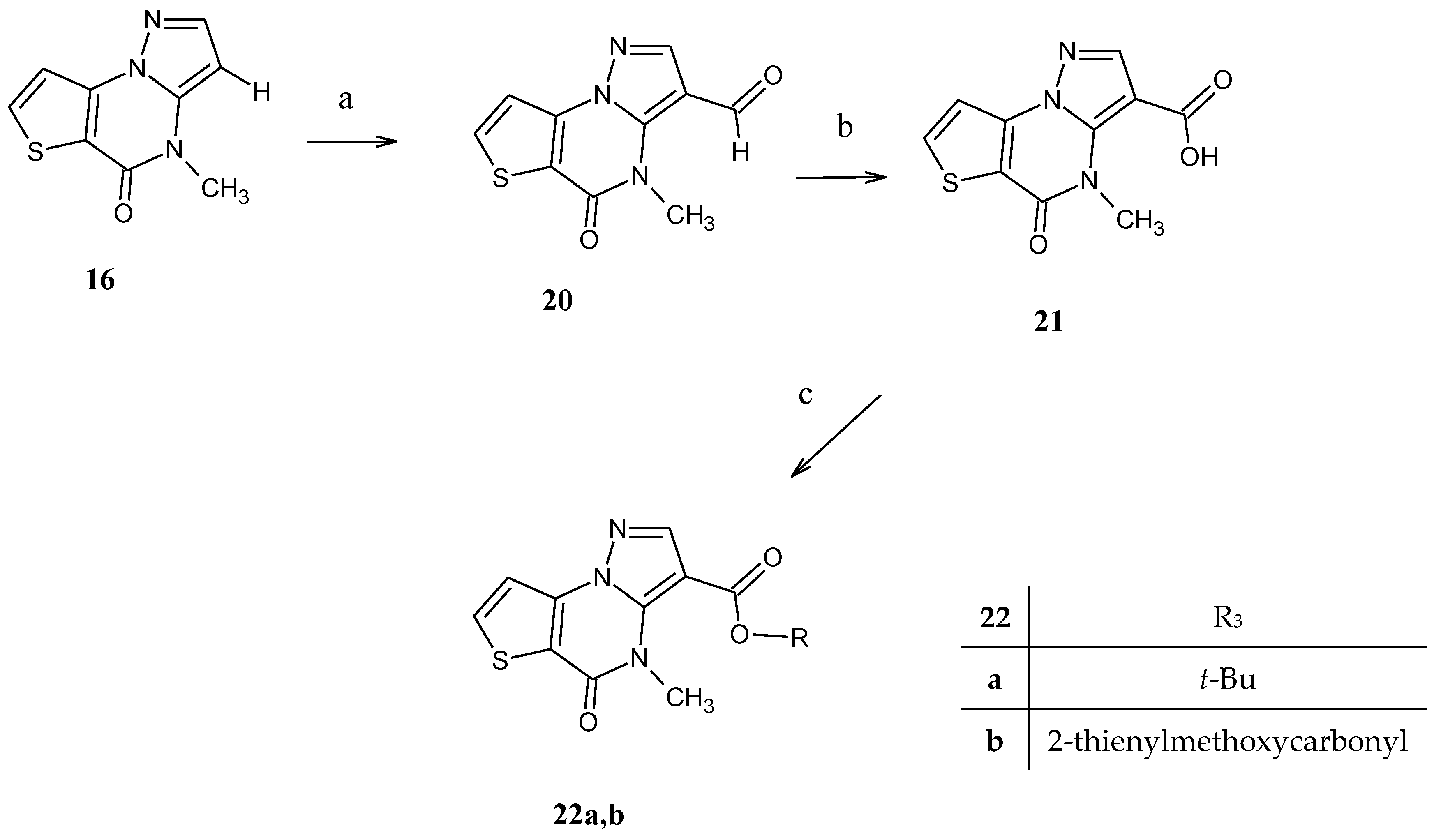
4-Methyl-5-oxo-4,5-dihydropyrazolo[1,5-a]thieno[2,3-e]pyrimidine (16). The ester 12c (0.50 mmol) was suspended in 10 mL of NaOH 10% solution and maintained at 80 °C until the starting material disappeared. The extraction with ethyl acetate and the next usual work up gave a residue which was filtered and purified by recrystallization with ethanol. Yield 71%, cream crystals, mp 157–159 °C. TLC: toluene/ethyl acetate/acetic acid 8:2:1 v/v/v (Rf: 0.8); 1H-NMR (400 MHz, DMSO-d6) δ 8.18 (d, 1H, H-7, J = 5.6 Hz); 7.83 (d, 1H, H-2 J = 2.0 Hz); 7.62 (d, 1H, H-8, J = 5.6 Hz); 6.22 (d, 1H, H-3 J = 2.0 Hz); 3.86 (s, 3H, N-CH3). 1H-NMR (400 MHz, CDCl3) δ 7.82 (d, 1H, H-7, J = 5.6 Hz); 7.87 (d, 1H, H-2 J = 2.0 Hz); 7.64 (d, 1H, H-8, J = 5.6 Hz); 5.96 (d, 1H, H-3 J = 2.0 Hz); 3.63 (s, 3H, N-CH3). 13C-NMR (100 MHz, DMSO-d6) δ 163.71, 156.56, 148.49, 144.30, 135.80, 133.27, 129.09, 114.20, 37.05. Anal. C9H7N3OS (C, H, N).
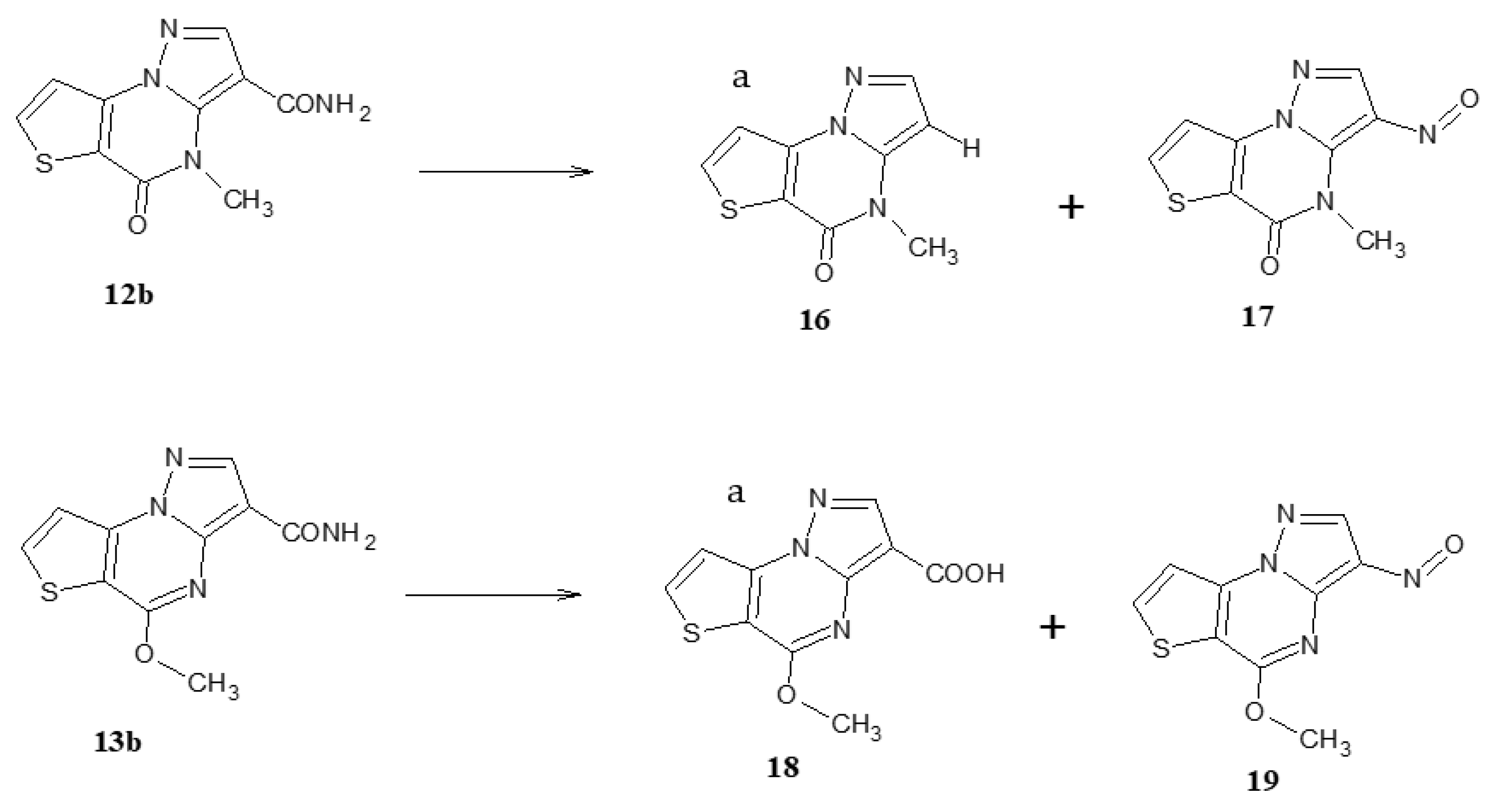
General procedure for the synthesis of compounds 17 and 19. A suspension of 12b or 13b (0.32 mmol) in H2SO4 conc. (8 mL) was stirred until a solution was obtained and then cooled at 0 °C; to this solution, sodium nitrite (0.22g, 3.2 mmol/5 mL of water) was slowly added and the green suspension was maintained for 3 h at 0 °C. The suspension was made alkaline and extracted with ethyl acetate. After the standard work-up, the evaporation of the organic layer gave a green residue that was purified and characterized.
4-Methyl-3-nitrosopyrazolo[1,5-a]thieno[2,3-e]pyrimidin-5(4H)-one (17). From 12b. Recrystallized by ethanol, yield 50%, green crystals, mp 240–242 °C. TLC: toluene/ethyl acetate/acetic acid 8:2:1 v/v/v (Rf: 0.9); 1H-NMR (400 MHz, DMSO-d6) δ 8.44 (d, 1H, H-7, J = 5.6 Hz); 7.74 (d, 1H, H-8, J = 5.6 Hz); 7.72 (s, 1H, H-2); 4.21 (s, 3H, NCH3). 13C-NMR (100 MHz, DMSO-d6) δ 163.70, 156.59, 148.49, 144.30, 134.99, 133.25, 129.01, 104.05, 36.99. ESI-HRMS (m/z) calculated for [M+H]+ ion species C9H6N4O2S = 235,0295; found: 235,0284. Anal. C9H6N4O2S (C, H, N).
5-Methoxy-3-nitrosopyrazolo[1,5-a]thieno[2,3-e]pyrimidine (19). From 13b. Recrystallized by ethanol, yield 48%, green crystals, mp 218–220 °C. TLC: toluene/ethyl acetate/acetic acid 8:2:1 v/v/v (Rf: 0.9); 1H-NMR (400 MHz, DMSO-d6) δ 8.86 (s, 1H, H-2); 8.54 (d, 1H, H-7, J = 5.6 Hz); 7.94 (d, 1H, H-8, J = 5.6 Hz); 4.23 (s, 3H, OCH3). 13C-NMR (100 MHz, DMSO-d6) δ 163.65, 156.60, 148.49, 144.30, 134.99, 133.20, 128.99, 104.10, 55.60. ESI-HRMS (m/z) calculated for [M+H]+ ion species C9H6N4O2S = 235,0294; found: 235,0284. Anal. C9H6N4O2S (C, H, N).
5-Methoxy-3-nitrosopyrazolo[1,5-a]thieno[2,3-e]pyrimidin-3-carboxylic acid (18). From 13b, after acidification of the alkaline solution. The carboxylic acid was recrystallized by ethanol, yield 30%, white crystals, mp 218–220 °C. TLC: toluene/ethyl acetate/acetic acid 8:2:1 v/v/v (Rf: 0.5); 1H-NMR (400 MHz, DMSO-d6) δ 12.24 (bs, 1H, OH, exch.); 8.44 (d, 1H, H-7, J = 4.8 Hz); 8.38 (s, 1H, H-2); 7.88 (d, 1H, H-8, J = 4.8 Hz); 4.17 (s, 3H, OCH3). 13C-NMR (100 MHz, DMSO-d6) δ 169.35, 162.20, 145.80, 145.59, 130.50, 126.70, 125.69, 114.50, 107.10, 53.70. Anal. C10H7N3O3S (C, H, N).
4-Methyl-5-oxo-4,5-dihydropyrazolo[1,5-a]thieno[2,3-e]pyrimidin-3-carbaldehyde (20). A suspension of 16 (150 mg, 0.73 mmol) in glacial acetic acid (6 mL) was added of hexamethylenetetramine (HTMA, 0.36 g) and maintained at reflux temperature for 10 h. After disappearing the starting material, evaluated by TLC (CHX/EtOAc 1:5, v/v as eluent, Rf: 0.8), the addition of ice gave a precipitate that was recovered by filtration. Yield 85%, white crystals, mp 222–224 °C. 1H-NMR (400 MHz, CDCl3) δ 10.02 (s, 1H, CHO); 8.27 (s, 1H, H-2); 7.91 (d, 1H, H-7, J = 5.2 Hz); 7.88 (d, 1H, H-8, J = 5.2 Hz); 4.03 (s, 3H, OCH3). 13C-NMR (100 MHz, DMSO-d6) δ 170.35, 163.20, 156.25, 145.80, 144.59, 135.84, 133.50, 129.50, 107.15, 37.50. Anal. C10H7N3O2S (C, H, N).
4-Methyl-5-oxo-4,5-dihydropyrazolo[1,5-a]thieno[2,3-e]pyrimidin-3-carboxylic acid (21). The aldehyde 20 (150 mg, 0.73 mmol) was suspended in acetone and water (5 mL/5 mL) and a solution of potassium permanganate (1.1 mmol) in water was added after the suspension was made alkaline with sodium hydroxide 10%. The reaction was heated for 8 h, and after cooling and elimination of the manganese dioxide by filtration, the alkaline aqueous phase was extracted to eliminate the starting material not reacting. The next acidification of the aqueous phase gave the corresponding carboxylic acid that was recovered by extraction. Yield 60%, white crystals, mp 220–223 °C. TLC: CHX/EtOAc 1:5, v/v (Rf: 0.2); 1H-NMR (400 MHz, DMSO-d6) δ 12.68 (bs, 1H, OH, exch.); 8.32 (d, 1H, H-7, J = 4.0 Hz); 8.21 (s, 1H, H-2); 7.67 (d, 1H, H-8, J = 4.0 Hz); 3.87 (s, 3H, NCH3). 13C-NMR (100 MHz, DMSO-d6) δ 169.10, 162.40, 145.85, 145.59, 130.50, 126.55, 125.80, 115.50, 107.10, 37.70. Anal. C10H7N3O3S (C, H, N).
General procedure for the synthesis of compounds 22a,b. The carboxylic acid 21 (0.5 mmol) was transformed into the corresponding 3-carbonyl chloride by reaction with excess SOCl2 in anhydrous conditions. After the standard work-up, the residue was suspended in dichloromethane (6 mL), and the suitable alcohol (excess 0.15 mL) was added; TLC monitored the reaction until the disappearance of the starting material. Then, the final solution was evaporated to dryness, and the residue recuperated with isopropyl ether and recrystallized.
tert-Butyl 4-methyl-5-oxo-4,5-dihydropyrazolo[1,5-a]thieno[2,3-e]pyrimidine-3-carboxylate (22a). From 21 and tert-butanol, white crystals recrystallized by 80% ethanol, yield 25%; mp > 300 °C. TLC: toluene/ethyl acetate/methanol 8:2:1.5 v/v/v (Rf: 0.5); 1H-NMR (400 MHz, CDCl3) δ 8.10 (s, 1H, H-2); 7.85 (d, 1H, H-7, J = 4.8 Hz); 7.63 (d, 1H, H-8, J = 4.8 Hz); 4.01 (s, 3H, NCH3); 1.50 (s, 9H, (CH3)3). 13C-NMR (100 MHz, DMSO-d6) δ 161.78, 156.10, 145.24, 142.00, 141.69, 138.12, 117.25, 116.95, 98.60, 33.57, 14.59. Anal. C14H15N3O3S (C, H, N).
Thiophen-2-yl-methyl 4-methyl-5-oxo-4,5-dihydropyrazolo[1,5-a]thieno[2,3-e]pyrimidine-3-carboxylate (22b). From 21 and 2-thiophenmethanol, white crystals recrystallized by 80% ethanol, yield 30%; mp 190–195 °C. TLC: toluene/ethyl acetate/methanol 8:2:1.5 v/v/v (Rf: 0.6); 1H-NMR (400 MHz, CDCl3) δ 8.19 (s, 1H, H-2); 7.85 (d, 1H, H-7, J = 5.2 Hz); 7.63 (d, 1H, H-8, J = 5.2 Hz); 7.35 (d, 1H, H-5 Thiophene, J = 5.2 Hz); 7.18 (s, 1H, H-3 Thiophene); 7.03 (d, 1H, H-4 Thiophene, J1 = 4.8 Hz); 5.47 (s, 2H, OCH2); 4.03 (s, 3H, NCH3). 13C-NMR (100 MHz, CDCl3) δ 145.60, 135.92, 128.25, 126.89, 117.02, 60.81, 33.87. Anal. C15H11N3O3S2 (C, H, N).
Pyrazolo[1,5-a]thieno[2,3-e]pyrimidine-3-carboxylic acid (23). Compound 11c (0.40 mmol) was treated with 10% NaOH solution and maintained at 80 °C until the starting material disappeared in TLC (toluene/ethyl acetate/acetic acid 8:2:2 v/v/v as eluent). However, the reaction gave many compounds, and the fast-eluted band’s fluorescent spot was recovered by purification with a chromatography column (toluene/ethyl acetate/acetic acid 8:2:2 v/v/v as eluent, Rf: 0.5). Yield 15%, white crystals, mp 246–247 °C. 1H-NMR (400 MHz, DMSO-d6) δ 12.05 (bs, 1H, OH, exch.); 9.34 (s, 1H, H-5); 8.55 (s, 1H, H-2); 8.50 (d, 1H, H-7, J = 5.2 Hz); 7.95 (d, 1H, H-8, J = 5.2 Hz). 13C-NMR (100 MHz, CDCl3) δ 145.60, 135.93, 128.25, 126.89, 117.02, 60.81, 33.87. Anal. C9H5N3O2S (C, H, N).
Pyrazolo[1,5-a]thieno[2,3-e]pyrimidine (24). Compound 11c (0.40 mmol) were treated with HCl/CH3COOH solution and maintained at 70 °C until the starting material disappeared in TLC (toluene/ethyl acetate/acetic acid 8:2:2 v/v/v as eluent, Rf: 0.6). The reaction from 11c, monitored by TLC evidenced the formation of 3-carboxylic acid (23) that quickly evolved in the decarboxylated compound 24. The final solution was extracted with ethyl acetate, and after the normal work-up, the residue was purified by recrystallization from ethanol. Yield 25%, white crystals, mp 246–247 °C. 1H-NMR (400 MHz, DMSO-d6) δ 9.14 (s, 1H, H-5); 8.43 (d, 1H, H-7, J = 4.8 Hz); 8.17 (s, 1H, H-2); 7.91 (d, 1H, H-8, J = 4.8 Hz); 6.83 (s, 1H, H-3). 13C-NMR (100 MHz, DMSO-d6) δ 149.90, 146.24, 144.70, 130.52, 126.70, 125.25, 119.15, 101.30. Anal. C8H5N3S (C, H, N).
3-Methylpyrazolo[1,5-a]thieno[2,3-e]pyrimidine (25). Compound 14 (0.34 mmol) was suspended in THF anhydrous (5 mL) and 1.50 mmol of LiAlH4 was added, using a 1M solution of LiAlH4 in THF. After 1 h, the starting material disappeared and adding ice/water quenched the reaction. The extraction with ethyl acetate gave the intermediate 4,5-dihydro derivative not isolated but identified since not fluorescent in TLC, which was treated with toluene and 10% Pd/C at reflux temperature until the dehydrogenation was complete. The final suspension was filtered, toluene was evaporated and the residue was recrystallized by ethanol; yield 35%, white crystals, mp 127–130 °C. TLC: toluene/ethyl acetate/methanol 8:2:1.5 v/v/v (Rf: 0.7); 1H-NMR (400 MHz, DMSO-d6) δ 9.06 (s, 1H, H-5); 8.39 (d, 1H, H-7, J = 5.2 Hz); 8.04 (s, 1H, H-2); 7.87 (d, 1H, H-8, J = 5.2 Hz); 2.33 (s, 1H, CH3). 13C-NMR (100 MHz, DMSO-d6) δ 149.90, 132.74, 132.90, 130.56, 126.70, 125.65, 119.10, 115.63, 14.30. Anal. C9H7N3S (C, H, N).


 ,
, 














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}