

BET Bromodomain Inhibitors: Novel Design Strategies and Therapeutic Applications

Abstract
:1. Introduction
2. BET Family of Bromodomain Proteins Represent Attractive Targets for Cancer Therapy
2.1. Bromodomains (BRDs)
2.2. Bromodomain and Extra-Terminal Domain (BET) Proteins
2.3. Brd4
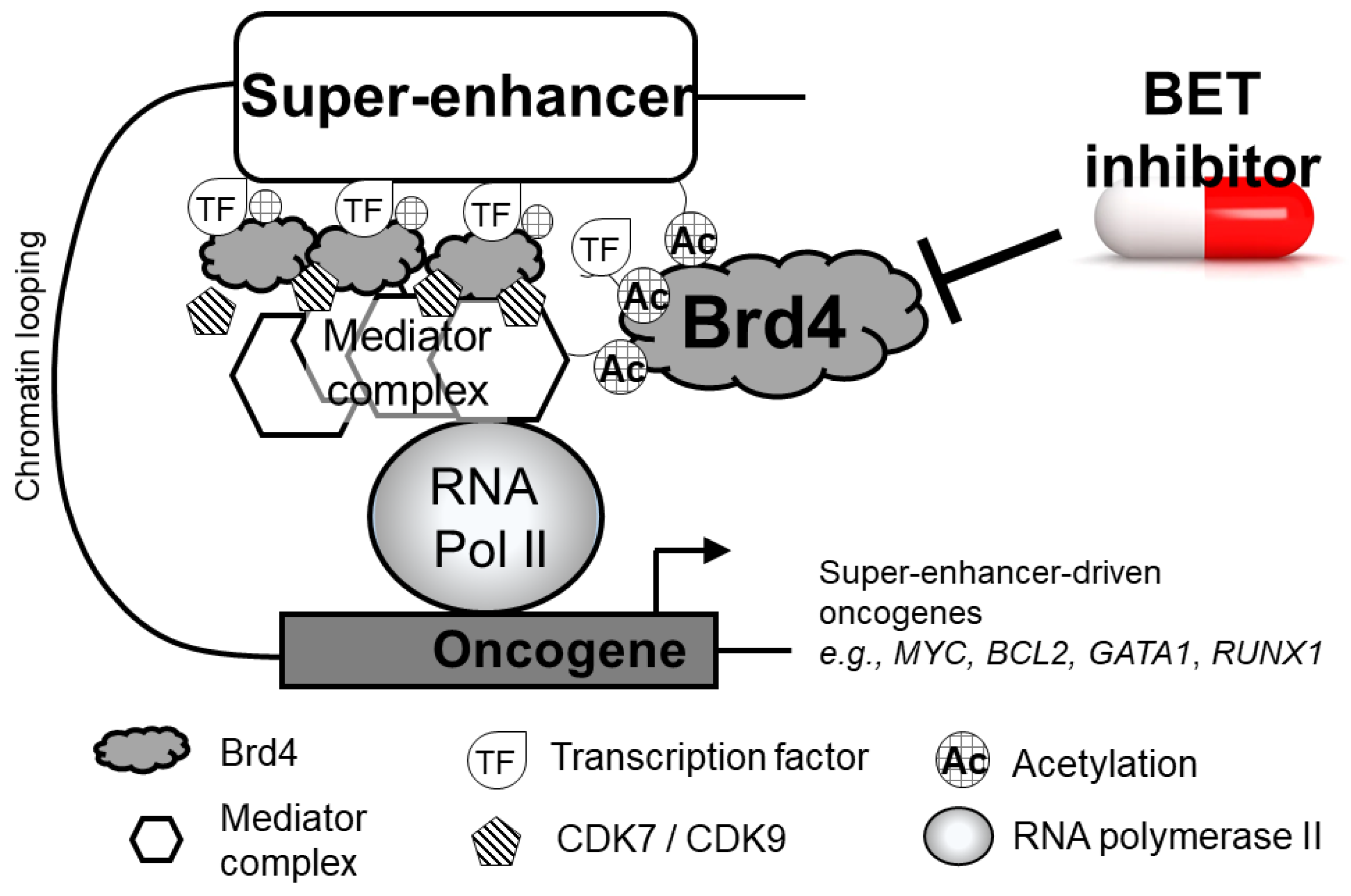
2.4. Brd4 at Super-Enhancers of Actively Transcribed DNA
2.5. BRD4–NUT Fusion
3. Evolution of Small-Molecule Inhibitors of BET Proteins
3.1. Pan BET Inhibitors
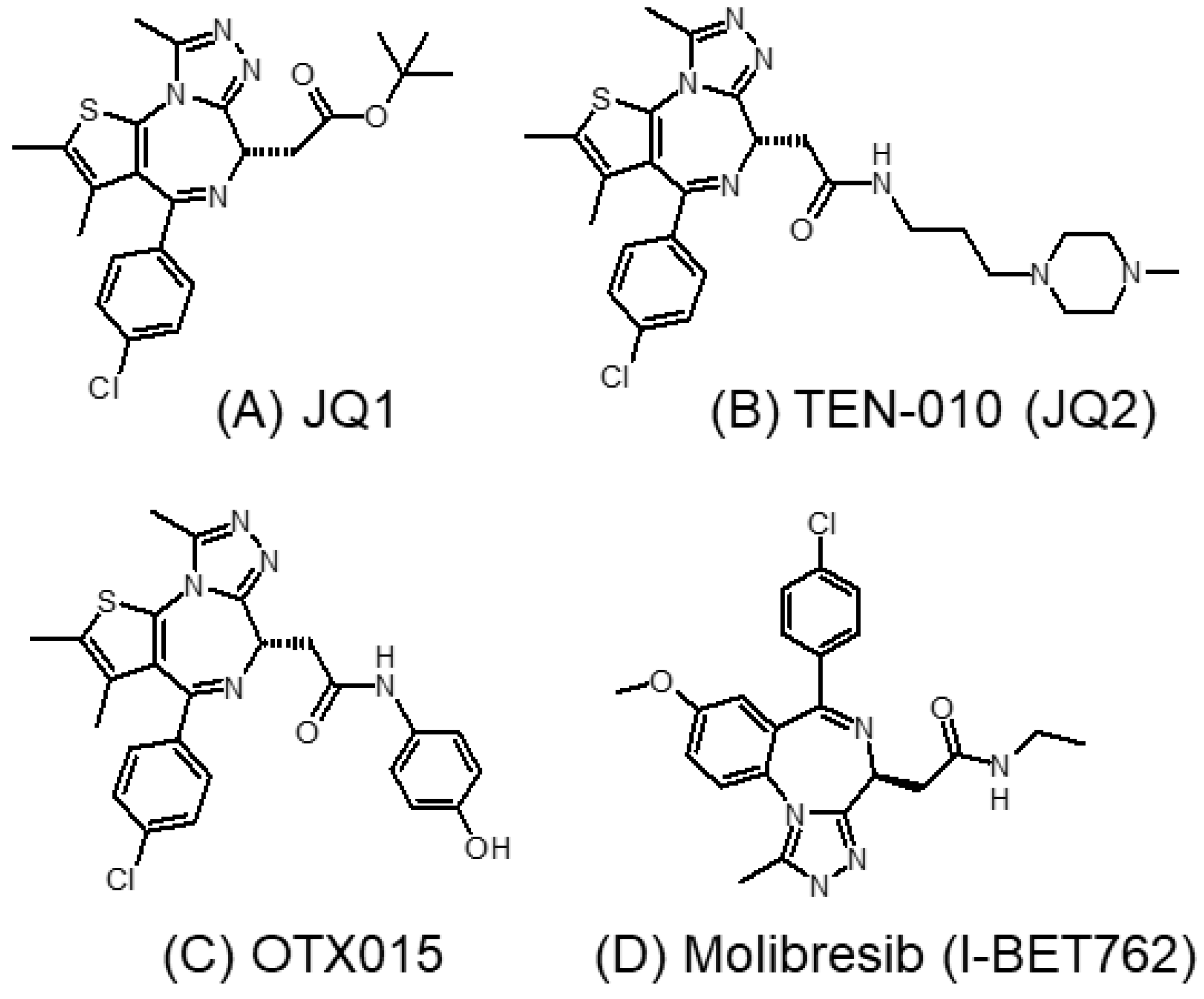
3.1.1. JQ1 and Its Analogs
3.1.2. I-BET762 (GSK525762A, Molibresib)
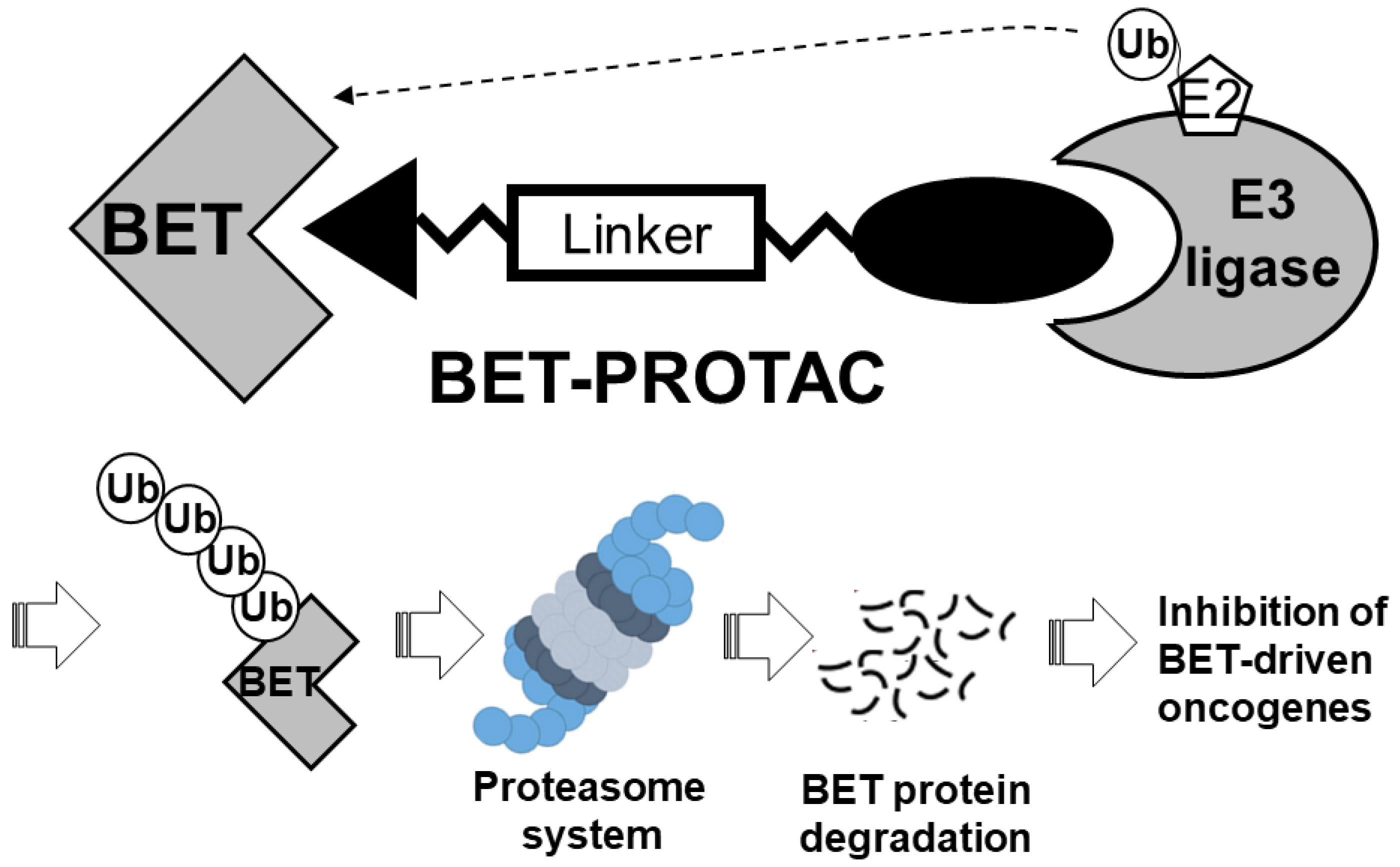
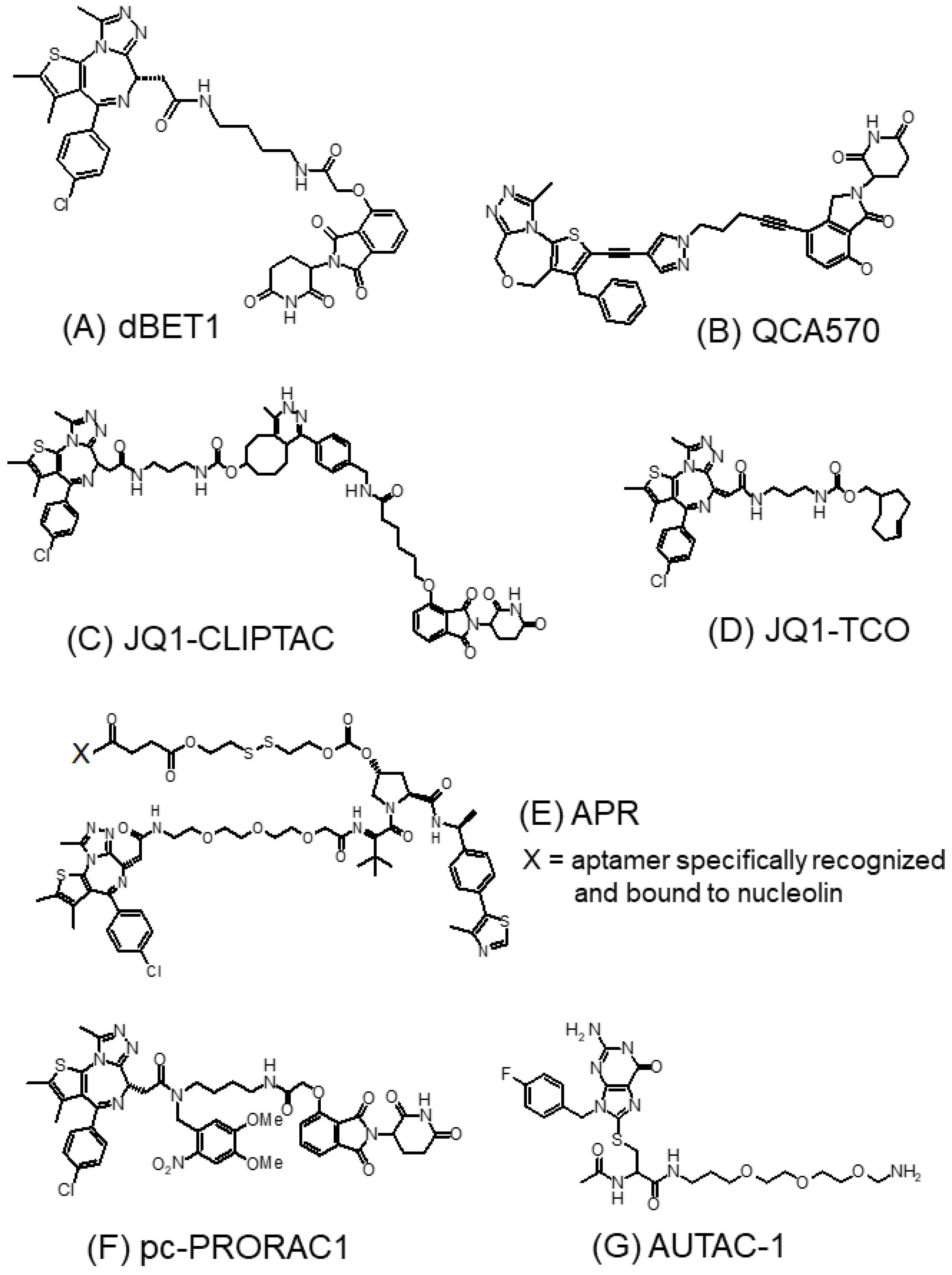
3.1.3. BET-PROTACs
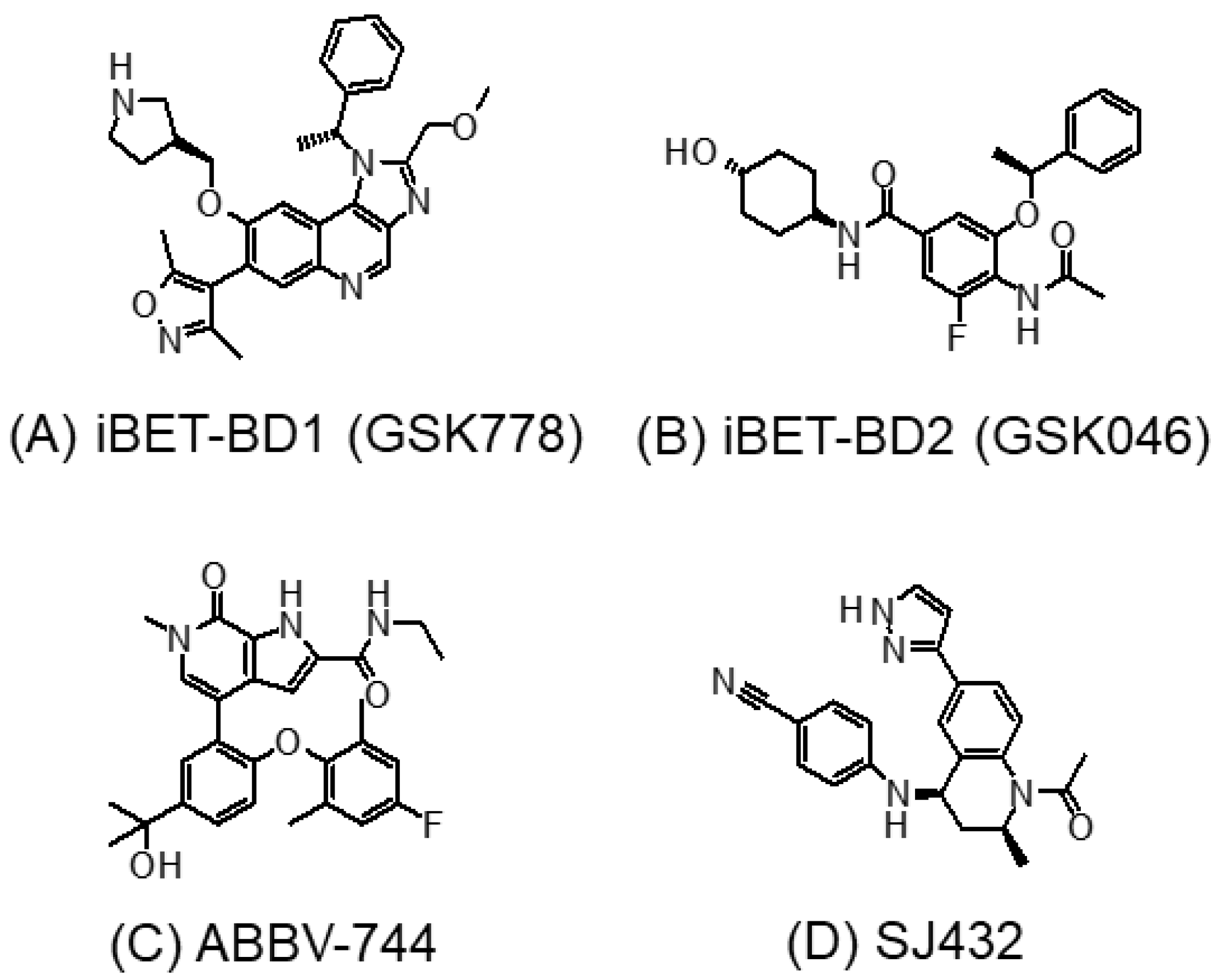
3.2. BD1- or BD2-Selective BET Inhibitors
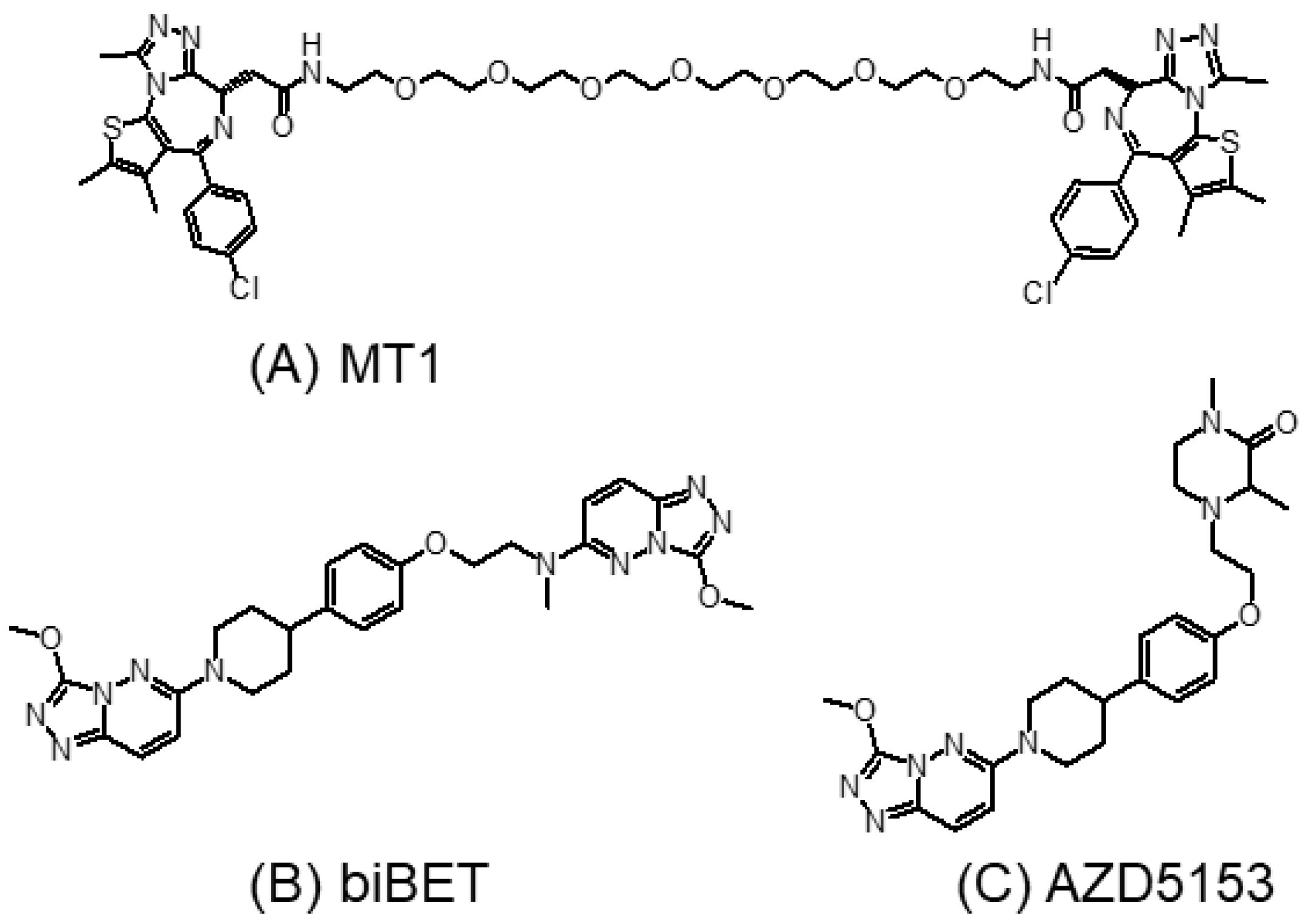
3.3. Bivalent BET Inhibitors
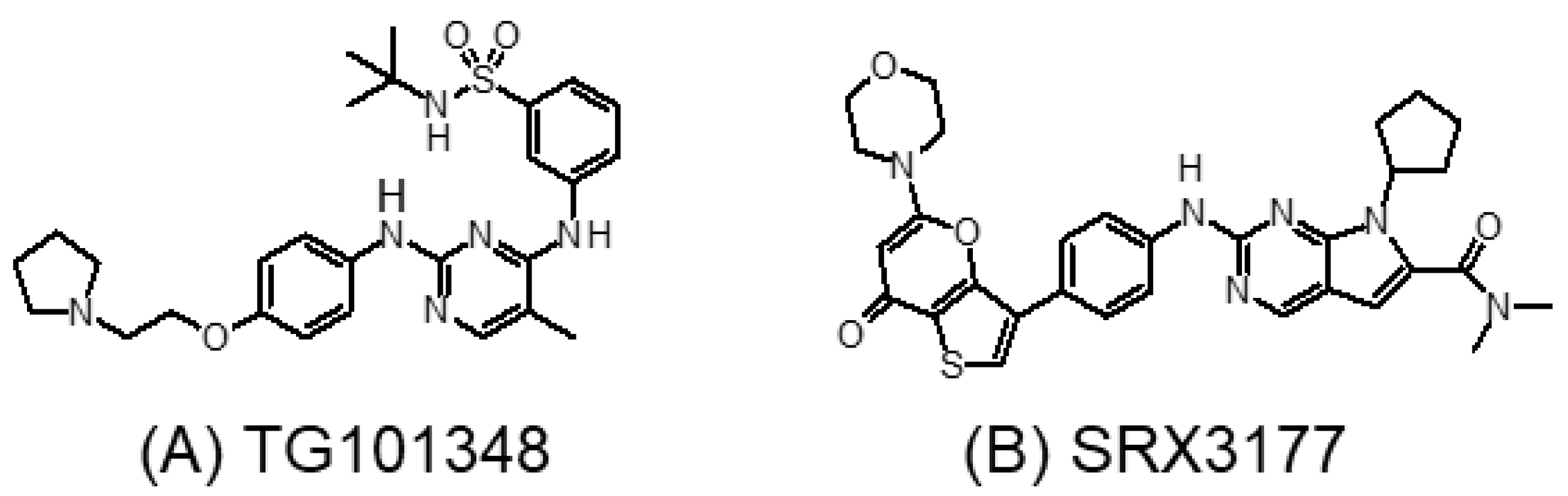
3.4. Tyrosine Kinase and BET Dual Inhibitors
3.5. Brd4-Selective Inhibitors
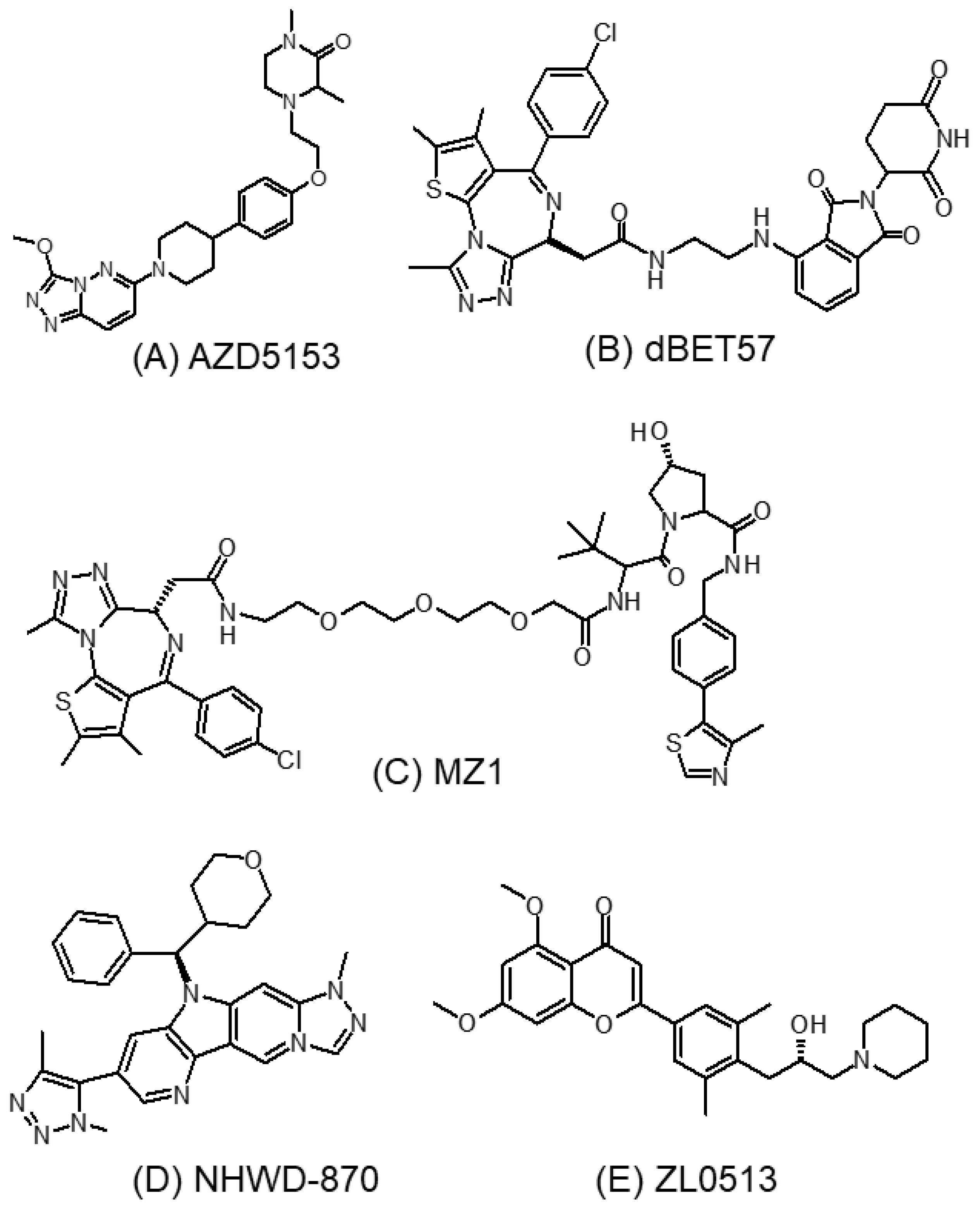
3.5.1. AZD5153
3.5.2. dBET57
3.5.3. MZ1
3.5.4. NHWD-870
3.5.5. ZL0513
4. Toxicities of BET Inhibitors
5. Combination of BET Inhibitor with Other Chemotherapeutic Modalities for Cancer Treatment
5.1. Combination with Conventional Chemotherapeutic Drugs
5.2. Combination with Epigenetic Drugs
5.3. Combination with Molecular-Targeted Agents
5.4. Combination with Cyclin-Dependent Kinase (CDK) Inhibitors
5.5. Combination with Proteasome Inhibitors
5.6. Combination with Immunotherapeutic Drugs
6. Resistance to BET Inhibitors
7. Potential Biomarkers Predicting the Activity and Resistance of BET Inhibitors
7.1. Predictive Biomarkers for the Anticancer Effect of BET Inhibitors
7.1.1. Presence of BRD4-–NUT Fusion
7.1.2. Expression of the BET Family of Bromodomain Proteins
7.1.3. MYC Amplification
7.2. Biomarkers of Resistance
7.2.1. Aberrant Activation of Receptor Tyrosine Kinase (RTK) Signaling Pathways
7.2.2. Aberrant Ubiquitination and Degradation of BET Proteins
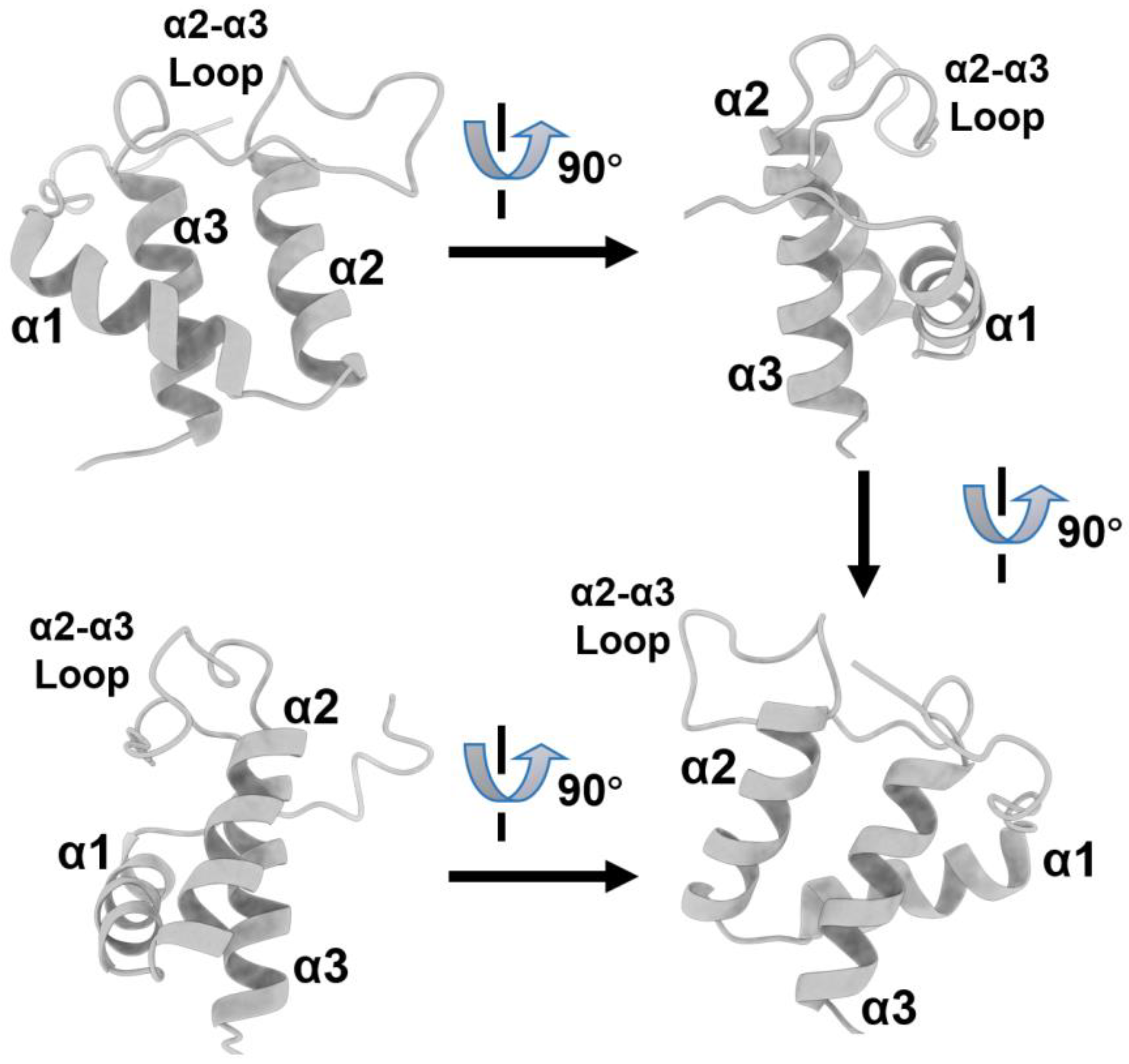
8. Targeting at the Extra-Terminal (ET) Domain of Brd4
8.1. Significance of Targeting the BET/Brd4 Extra-Terminal Domain
8.2. Interaction between Brd4-ET and Other Protein Partners
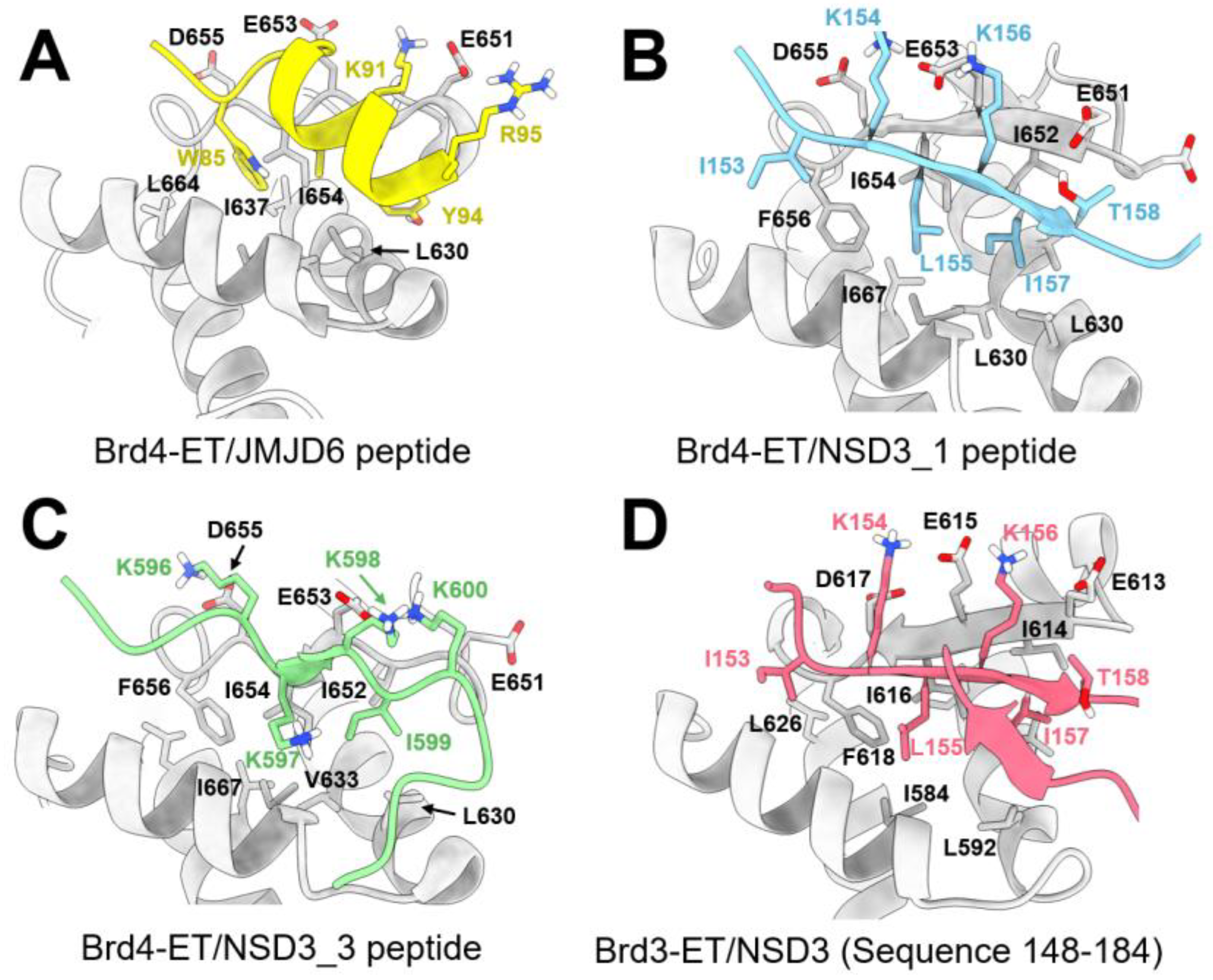
8.2.1. Interaction between the ET Domain and JMJD6
8.2.2. Interaction between the ET Domain and NSD3
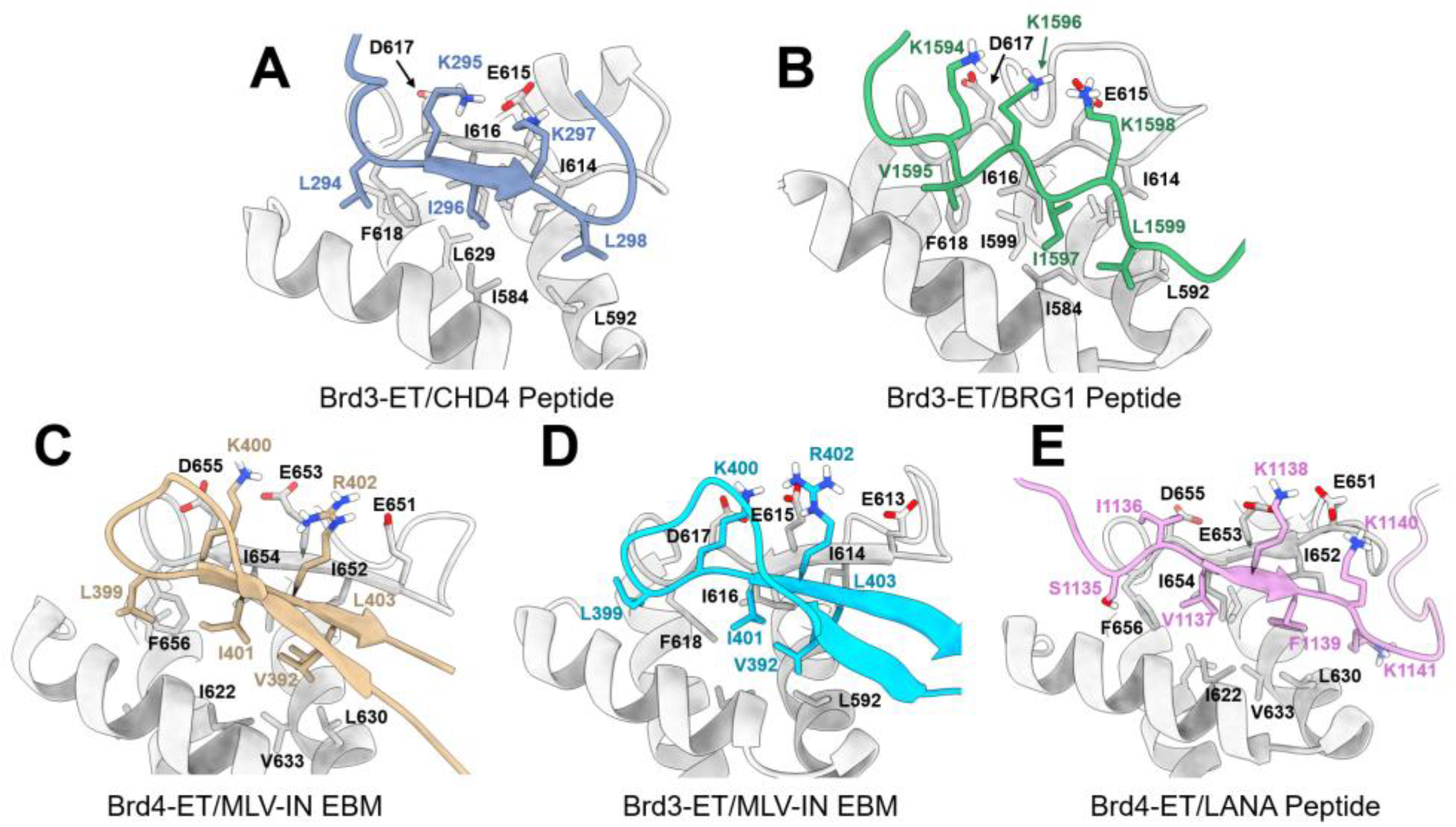
8.2.3. Interaction between the ET Domain and Chromatin-Remodeling Complexes
8.2.4. Interaction between the ET Domain and Viral Protein
9. Further Perspectives
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Millan-Zambrano, G.; Burton, A.; Bannister, A.J.; Schneider, R. Histone post-translational modifications—Cause and consequence of genome function. Nat. Rev. Genet. 2022, 23, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.L.; Guppy, B.J.; Sawchuk, L.; Davie, J.R.; McManus, K.J. Regulation of chromatin structure via histone post-translational modification and the link to carcinogenesis. Cancer Metastasis Rev. 2013, 32, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, F.; Wei, W. Targeting the acetylation signaling pathway in cancer therapy. Semin. Cancer Biol. 2022, 85, 209–218. [Google Scholar] [CrossRef]
- Ali, H.A.; Li, Y.; Bilal, A.H.M.; Qin, T.; Yuan, Z.; Zhao, W. A comprehensive review of BET protein biochemistry, physiology, and pathological roles. Front. Pharmacol. 2022, 13, 818891. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef]
- Chen, Q.; Yang, B.; Liu, X.; Zhang, X.D.; Zhang, L.; Liu, T. Histone acetyltransferases CBP/p300 in tumorigenesis and CBP/p300 inhibitors as promising novel anticancer agents. Theranostics 2022, 12, 4935–4948. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Lin, H. Understanding the function of mammalian sirtuins and protein lysine acylation. Annu. Rev. Biochem. 2021, 90, 245–285. [Google Scholar] [CrossRef]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef]
- Fujisawa, T.; Filippakopoulos, P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 246–262. [Google Scholar] [CrossRef] [PubMed]
- Zaware, N.; Zhou, M.M. Bromodomain biology and drug discovery. Nat. Struct. Mol. 2019, 26, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, R.; Meslamani, J.; Zhou, M.M. The bromodomain: From epigenome reader to druggable target. Biochim. Biophys. Acta Gene Regul. Mech. 2014, 1839, 676–685. [Google Scholar] [CrossRef] [Green Version]
- Prinjha, R.K.; Witherington, J.; Lee, K. Place your BETs: The therapeutic potential of bromodomains. Trends Pharmacol. Sci. 2012, 33, 146–153. [Google Scholar] [CrossRef]
- Alamer, E.; Zhong, C.; Hajnik, R.; Soong, L.; Hu, H. Modulation of BRD4 in HIV epigenetic regulation: Implications for finding an HIV cure. Retrovirology 2021, 18, 3. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yang, C.; Candelario-Jalil, E. Role of BET proteins in inflammation and CNS diseases. Front. Mol. Biosci. 2021, 8, 748449. [Google Scholar] [CrossRef]
- Lin, Y.J.; Umehara, T.; Inoue, M.; Saito, K.; Kigawa, T.; Jang, M.K.; Ozato, K.; Yokoyama, S.; Padmanabhan, B.; Guntert, P. Solution structure of the extraterminal domain of the bromodomain-containing protein BRD4. Protein Sci. 2008, 17, 2174–2179. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S.; Sowa, M.E.; Ottinger, M.; Smith, J.A.; Shi, Y.; Harper, J.W.; Howley, P.M. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell Biol. 2011, 31, 2641–2652. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Ipsaro, J.J.; Shi, J.; Milazzo, J.P.; Wang, E.; Roe, J.-S.; Suzuki, Y.; Pappin, D.J.; Joshua-Tor, L.; Vakoc, C.R. NSD3-short is an adaptor protein that couples BRD4 to the CHD8 chromatin remodeler. Mol. Cell 2015, 60, 847–859. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zeng, L.; Shen, C.; Ju, Y.; Konuma, T.; Zhao, C.; Vakoc, C.R.; Zhou, M.M. Structural mechanism of transcriptional regulator NSD3 recognition by the ET domain of BRD4. Structure 2016, 24, 1201–1208. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.H.; Lin, D.I. Amplification of the NSD3-BRD4-CHD8 pathway in pelvic high-grade serous carcinomas of tubo-ovarian and endometrial origin. Mol. Clin. Oncol. 2017, 7, 301–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Ma, Q.; Wong, K.; Li, W.; Ohgi, K.; Zhang, J.; Aggarwal, A.K.; Rosenfeld, M.G. Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell 2013, 155, 1581–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konuma, T.; Yu, D.; Zhao, C.; Ju, Y.; Sharma, R.; Ren, C.; Zhang, Q.; Zhou, M.-M.; Zeng, L. Structural mechanism of the oxygenase JMJD6 recognition by the extraterminal (ET) domain of BRD4. Sci. Rep. 2017, 7, 16272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, R.; Zhou, M.M. The role of human bromodomains in chromatin biology and gene transcription. Curr. Opin. Drug Discov. Devel. 2009, 12, 659–665. [Google Scholar]
- Fu, L.L.; Tian, M.; Li, X.; Li, J.J.; Huang, J.; Ouyang, L.; Zhang, Y.; Liu, B. Inhibition of BET bromodomains as a therapeutic strategy for cancer drug discovery. Oncotarget 2015, 6, 5501–5516. [Google Scholar] [CrossRef] [Green Version]
- Gilan, O.; Rioja, I.; Knezevic, K.; Bell, M.J.; Yeung, M.M.; Harker, N.R.; Lam, E.Y.; Chung, C.W.; Bamborough, P.; Petrietich, M.; et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science 2020, 368, 387–394. [Google Scholar] [CrossRef]
- Altendorfer, E.; Mochalova, Y.; Mayer, A. BRD4: A general regulator of transcription elongation. Transcription 2022, 13, 70–81. [Google Scholar] [CrossRef]
- Houzelstein, D.; Bullock, S.L.; Lynch, D.E.; Grigorieva, E.F.; Wilson, V.A.; Beddington, R.S.P. Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein Brd4. Mol. Cell Biol. 2002, 22, 3794–3802. [Google Scholar] [CrossRef] [Green Version]
- Devaiah, B.N.; Singer, D.S. Cross-talk among RNA polymerase II kinases modulates C-terminal domain phosphorylation. J. Biol. Chem. 2012, 287, 38755–38766. [Google Scholar] [CrossRef] [Green Version]
- Vollmuth, F.; Bankenfeldt, W.; Geyer, M. Structures of the dual bromodomains of the P-TEFb-activating protein Brd4 at atomic resolution. J. Biol. Chem. 2009, 284, 36547–36556. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Tian, J.; Wu, T. BRD4 in physiology and pathology: “BET” on its partners. Bioessays 2021, 43, e2100180. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Wu, R.; Tang, D.; Kang, R. The BET family in immunity and disease. Signal Transduct. Target Ther. 2021, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Du, L. The therapeutic potential of BRD4 in cardiovascular disease. Hypertens. Res. 2020, 43, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Nishiyama, A.; Karpova, T.; McNally, J.; Ozato, K. Brd4 marks select genes on mitotic chromatin and directs postmitotic transcription. Mol. Biol. Cell 2009, 20, 4899–4909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotekar, A.; Singh, A.K.; Devaiah, B.N. BRD4 and MYC: Power couple in transcription and disease. FEBS J. 2022. [Google Scholar] [CrossRef] [PubMed]
- Pott, S.; Lieb, J.D. What are super-enhancers? Nat. Genet. 2015, 47, 8–12. [Google Scholar] [CrossRef]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 2018, 361, 6400. [Google Scholar] [CrossRef] [Green Version]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [Green Version]
- Jia, Q.; Chen, S.; Tan, Y.; Li, Y.; Tang, F. Oncogenic super-enhancer formation in tumorigenesis and its molecular mechanisms. Exp. Mol. Med. 2020, 52, 713–723. [Google Scholar] [CrossRef]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and cancer: Going beyond transcriptional regulation. Mol. Cancer 2018, 17, 164. [Google Scholar] [CrossRef]
- French, C. NUT midline carcinoma. Nat. Rev. Cancer 2014, 14, 149–150. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; You, J. Mechanistic analysis of the role of bromodomain-containing protein 4 (BRD4) in BRD4-NUT oncoprotein-induced transcription activation. J. Biol. Chem. 2015, 290, 2744–2758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alekseyenko, A.A.; Walsh, E.M.; Wang, X.; Grayson, A.R.; Hsi, P.T.; Kharchenko, P.V.; Kuroda, M.I.; French, C.A. The oncogenic BRD4-NUT chromatin regulator drives aberrant transcription within large topological domains. Genes Dev. 2015, 29, 1507–1523. [Google Scholar] [CrossRef] [Green Version]
- French, C.A. NUT midline carcinoma. Cancer Genet. Cytogenet. 2010, 203, 16–20. [Google Scholar] [CrossRef]
- French, C.A. Pathogenesis of NUT midline carcinoma. Annuv. Rev. Pathol. 2012, 7, 247–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibitors as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [Green Version]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Lauer, U.M.; Hinterleitner, M.; Horger, M.; Ohnesorge, P.V.; Zender, L. NUT carcinoma—An underdiagnosed malignancy. Front. Oncol. 2022, 12, 914031. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [Green Version]
- Beesley, A.H.; Stirnweiss, A.; Ferrari, E.; Endersby, R.; Howlett, M.; Failes, T.W.; Arndt, G.M.; Charles, A.K.; Cole, C.H.; Kees, U.R. Comparative drug screening in NUT midline carcinoma. Br. J. Cancer 2014, 110, 1189–1198. [Google Scholar] [CrossRef] [Green Version]
- Boi, M.; Gaudio, E.; Bonetti, P.; Kwee, I.; Bernasconi, E.; Tarantelli, C.; Rinaldi, A.; Testoni, M.; Cascione, L.; Ponzoni, M.; et al. The BET bromodomain inhibitor OTX015 affects pathogenetic pathways in preclinical B-cell tumor models and synergizes with targeted drugs. Clin. Cancer Res. 2015, 21, 1628–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finley, A.; Copeland, R.A. Small molecule control of chromatin remodeling. Chem. Biol. 2014, 21, 1196–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roboz, G.J.; Desai, P.; Lee, S.; Ritchie, E.K.; Winer, E.S.; DeMario, M.; Brennan, B.; Nuesch, E.; Chesne, E.; Brennan, L.; et al. A dose escalation study of RO6870810/TEN-10 in patients with acute myeloid leukemia and myelodysplastic syndrome. Leuk. Lymphoma 2021, 62, 1740–1748. [Google Scholar] [CrossRef] [PubMed]
- Odore, E.; Lokier, F.; Cvitkovic, E.; Bekradda, M.; Herait, P.; Bourdel, F.; Kahatt, C.; Raffoux, E.; Stathis, A.; Thieblemont, C.; et al. Phase I population pharmacokinetic assessment of the oral bromodomain inhibitor OTX015 in patients with haematological malignancies. Clin. Pharmacokinet. 2016, 55, 397–405. [Google Scholar] [CrossRef]
- Shi, J.; Song, S.; Han, H.; Xu, H.; Huang, M.; Qian, C.; Zhang, X.; Ouyang, L.; Hong, Y.; Zhuang, W.; et al. Potent activity of the bromodomain inhibitor OTX015 in multiple myeloma. Mol. Pharm. 2018, 15, 4139–4147. [Google Scholar] [CrossRef]
- Vazquez, R.; Licandro, S.A.; Astorgues-Xerri, L.; Lettera, E.; Panini, N.; Romano, M.; Erba, E.; Ubezio, P.; Bello, E.; Libener, R.; et al. Promising in vivo efficacy of the BET bromodomain inhibitor OTX015/MK-8628 in malignant pleural mesothelioma xenografts. Int. J. Cancer 2017, 140, 197–207. [Google Scholar] [CrossRef]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef] [Green Version]
- Piha-Paul, S.A.; Hann, C.L.; French, C.A.; Cousin, S.; Brana, I.; Cassier, P.A.; Moreno, V.; de Bono, J.S.; Harward, S.D.; Ferron-Brady, G.; et al. Phase 1 study of molibresib (GSK525762), a bromodomain and extra-terminal domain protein inhibitor, in NUT carcinoma and other solid tumors. JNCI Cancer Spectr. 2020, 4, pkz093. [Google Scholar] [CrossRef]
- Wyce, A.; Matteo, J.J.; Foley, S.W.; Felitsky, D.J.; Rajapurkar, S.R.; Zhang, X.P.; Musso, M.C.; Korenchuk, S.; Karpinich, N.O.; Keenan, K.M.; et al. MEK inhibitors overcome resistance to BET inhibition across a number of solid and hematologic cancers. Oncogenesis 2018, 7, 35. [Google Scholar] [CrossRef] [Green Version]
- Xie, F.; Huang, M.; Lin, X.; Liu, C.; Liu, Z.; Meng, F.; Wang, C.; Huang, Q. The BET inhibitor I-BET762 inhibits pancreatic ductal adenocarcinoma cell proliferation and enhances the therapeutic effect of gemcitabine. Sci. Rep. 2018, 8, 8102. [Google Scholar] [CrossRef] [Green Version]
- Cousin, S.; Blay, J.Y.; Garcia, I.B.; de Bono, J.S.; Le Tourneau, C.; Moreno, V.; Trigo, J.; Hann, C.L.; Azad, A.A.; Im, S.A.; et al. Safety, pharmacokinetic, pharnacodynamic and clinical activity of molibresib for the treatment of nuclear protein in testis carcinoma and other cancers: Results of a Phase I/II open-label, dose escalation study. Int. J. Cancer 2022, 150, 993–1006. [Google Scholar] [CrossRef] [PubMed]
- Guedeney, N.; Cornu, M.; Schwalen, F.; Kieffer, C.; Voisin-Chiret, A.S. PROTAC technology: A new drug design for chemical biology with many challenges in drug discovery. Drug Discov. Today 2022, 28, 103395. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jiang, X.; Feng, F.; Liu, W.; Sun, H. Degradation of proteins by PROTACs and other strategies. Acta Pharm. Sin. B 2020, 10, 207–238. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [Green Version]
- Qin, C.; Hu, Y.; Zhou, B.; Fernandez-Salas, E.; Yang, C.Y.; Liu, L.; McEachern, D.; Przbranowski, S.; Wang, M.; Stuckey, J.; et al. Discovery of QCA570 as an exceptionally potent and efficacious proteolysis targeting chimera (PROTAC) degrader of the bromodomain and extra-terminal (BET) proteins capable of inducing complete and durable tumor regression. J. Med. Chem. 2018, 61, 6685–6704. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, Y.; Yang, S.; Chen, W.; Xing, D. PROTACs for BRDs proteins in cancer therapy: A review. J. Enzyme Inhib. Med. Chem. 2022, 37, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.L.; Zhao, F.; Xu, Y.T.; Guan, Y.Y.; Yu, T.; Zhang, Y.Z.; Duan, Y.C.; Zhao, Y. A comprehensive review of BET-targeting PROTACs for cancer therapy. Bioorg. Med. Chem. 2022, 73, 117033. [Google Scholar] [CrossRef]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [Green Version]
- Asatsuma-Okumura, T.; Ito, T.; Handa, H. Molecular mechanisms of cereblon-based drugs. Pharmacol. Ther. 2019, 202, 132–139. [Google Scholar] [CrossRef]
- Burslem, G.M.; Crews, C.M. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell 2020, 181, 102–114. [Google Scholar] [CrossRef]
- Zhang, K.; Gao, L.; Wang, J.; Chu, X.; Zhang, Z.; Zhang, Y.; Fang, F.; Tao, Y.; Li, X.; Tian, Y.; et al. A novel BRD family PROTAC inhibitor dBET1 exerts great anti-cancer effects by targeting c-MYC in acute myeloid leukemia cells. Pathol. Oncol. Res. 2022, 28, 1610447. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Lv, W.; Rao, Y. Opportunities and challenges of small molecule induced targeted protein degradation. Front. Cell Dev. Biol. 2021, 9, 685106. [Google Scholar] [CrossRef]
- Lebraud, H.; Wright, D.J.; Johnson, C.N.; Heightman, T.D. Protein degradation by in-cell self-assembly of proteolysis targeting chimeras. ACS Central Sci. 2016, 2, 927–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieto-Jimenez, C.; Morafraile, E.C.; Alonso-Moreno, C.; Ocana, A. Clinical considerations for the design of PROTACs in cancer. Mol. Cancer 2022, 21, 67. [Google Scholar] [CrossRef]
- Maneiro, M.A.; Forte, N.; Shchepinova, M.M.; Kounde, C.S.; Chudasama, V.; Baker, J.R.; Tate, E.W. Antibody-PROTAC conjugates enable HER2-dependent targeted protein degradation of BRD4. ACS Chem. Biol. 2020, 15, 1306–1312. [Google Scholar] [CrossRef]
- He, S.; Gao, F.; Ma, J.; Dong, G.; Sheng, C. Aptamer-PROTAC conjugates (APCs) for tumor-specific targeting in breast cancer. Angew. Chem. Int. Ed. Engl. 2021, 60, 23299–23305. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.; Wang, K.; Zhou, D.; Zhong, H.; Pan, Z. Light-induced protein degradation with photocaged PROTACs. J. Am. Chem. Soc. 2019, 141, 18370–18374. [Google Scholar] [CrossRef]
- Pei, J.; Pan, X.; Wang, A.; Shuai, W.; Bu, F.; Tang, P.; Zhang, S.; Zhang, Y.; Wang, G.; Ouyang, L. Developing potent LC3-targeting AUTAC tools for protein degradation with selective autophagy. Chem. Commun. 2021, 57, 13194–13197. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Riley-Gillis, B.; Vijay, P.; Shen, Y. Acquired resistance to BET-PROTACs (proteolysis-targeting chimeras) caused by genomic alterations in core components of E3 ligase complexes. Mol. Cancer Ther. 2019, 18, 1302–1311. [Google Scholar] [CrossRef] [Green Version]
- Bricelj, A.; Steniebach, C.; Kuchta, R.; Gutschow, M.; Sosic, I. E3 ligase ligands in successful PROTACs: An overview of synthesis and linker attachment points. Front. Chem. 2021, 9, 707317. [Google Scholar] [CrossRef]
- Gamsjaeger, R.; Webb, S.R.; Lamonica, J.M.; Billin, A.; Blobel, G.A.; Mackay, J.P. Structural basis and specificity of acetylated transcription factor GATA1 recognition by BET family bromodomain protein Brd3. Mol. Cell Biol. 2011, 31, 2632–2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Shao, X.; Leung, E.L.H.; Chen, Y.; Yao, X. Selectively targeting individual bromodomain: Drug discovery and molecular mechanisms. Pharmacol. Res. 2021, 172, 105804. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.S.; Rioja, I.; Bakr, A.; Veldwijk, M.R.; Sperk, E.; Herskind, C.; Weichenhan, D.; Prinjha, R.K.; Plass, C.; Schmezer, P.; et al. Selective inhibitors of bromodomain BD1 and BD2 of BET proteins modulate radiation-induced profibrotic fibroblast responses. Int. J. Cancer 2022, 151, 275–286. [Google Scholar] [CrossRef]
- Faivre, E.J.; McDaniel, K.F.; Albert, D.H.; Mantena, S.R.; Plotnik, J.P.; Wilcox, D.; Zhang, L.; Bui, M.H.; Sheppard, G.S.; Wang, L.; et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 2020, 578, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, G.S.; Wang, L.; Fidanze, S.D.; Hasvold, L.A.; Liu, D.; Pratt, J.K.; Park, C.H.; Longenecker, K.; Qiu, W.; Torrent, M.; et al. Discovery of N-ethyl-4-[2-(4-fluoro-2,6-dimethyl-phenoxy)-5-(1-hydroxy-1-methyl-ethyl)phenyl]-6-methyl-7-oxo-1H-pyrrolo[2,3-c]pyridine-2-carboxamide (ABBV-744), a BET bromodomain inhibitor with selectivity for the second bromodomain. J. Med. Chem. 2020, 63, 5585–5623. [Google Scholar] [CrossRef]
- Slavish, P.J.; Chi, L.; Yun, M.K.; Tsurkan, L.; Martinez, N.E.; Jonchere, B.; Chai, S.C.; Connelly, M.; Waddell, M.B.; Das, S.; et al. Bromodomain-selective BET inhibitors are potent antitumor agents against MYC-driven pediatric cancer. Cancer Res. 2020, 80, 3507–3518. [Google Scholar] [CrossRef] [PubMed]
- Waring, M.J.; Chen, H.; Rabow, A.A.; Walker, G.; Bobby, R.; Boiko, S.; Bradbury, R.H.; Callis, R.; Clark, E.; Dale, I.; et al. Potent and selective bivalent inhibitors of BET bromodomains. Nat. Chem. Biol. 2016, 12, 1097–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Roberts, J.M.; Seo, H.S.; Souza, A.; Paulk, J.; Scott, T.G.; DeAngelo, S.L.; Dhe-Paganon, S.; Bradner, J.E. Design and characterization of bivalent BET inhibitors. Nat. Chem. Biol. 2016, 12, 1089–1096. [Google Scholar] [CrossRef] [Green Version]
- Rhyasen, G.W.; Hattersley, M.M.; Yao, Y.; Dulak, A.; Wang, W.; Petteruti, P.; Dale, I.L.; Boiko, S.; Cheung, T.; Zhang, J.; et al. AZD5153: A novel bivalent BET bromodomain inhibitor highly active against hematological malignancies. Mol. Cancer Ther. 2016, 15, 2563–2574. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Chen, D.; Qian, D.; Zhang, S.; Zhang, Y.; Guo, S.; Ma, Z.; Wang, S. AZD5153, a novel BRD4 inhibitor, suppresses human thyroid carcinoma cell growth in vitro and in vivo. Biochem. Biophys. Res. Commun. 2018, 499, 531–537. [Google Scholar] [CrossRef]
- Luo, M.; Wu, Q.; Yang, Y.; Sun, L.; Huan, X.; Tian, C.; Xiong, B.; Miao, Z.; Wang, Y.; Chen, D. Design and development of a novel series of oral bivalent BET inhibitors with potent anticancer activities. Eur. J. Med. Chem. 2022, 239, 114519. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Heidenreich, D.; Zhou, S.; Ackloo, S.; Kramer, A.; Nakka, K.; Lima-Fernandes, E.; Deblois, G.; Duan, S.; Vellanki, R.N.; et al. A chemical toolbox for the study of bromodomains and epigenetic signaling. Nat. Commun. 2019, 10, 1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Ku, A.F.; Anglin, J.L.; Sharma, R.; Ucisik, M.N.; Faver, J.C.; Li, F.; Nyshadham, P.; Simmons, N.; Sharma, K.L.; et al. Discovery and characterization of bromodomain 2-specific inhibitors of BRDT. Proc. Natl. Acad. Sci. USA 2021, 118, e2021102118. [Google Scholar] [CrossRef] [PubMed]
- Moreno, V.; Sepulveda, J.M.; Vieito, M.; Hernandez-Guerrero, T.; Doger, B.; Saavedra, O.; Ferrero, O.; Sarmiento, R.; Arias, M.; De Alvaro, J.; et al. Phase I study of CC-90010, a reversible, oral BET inhibitor in patients with advanced solid tumors and relapsed/refractory non-Hodgkin’s lymphoma. Ann. Oncol. 2020, 31, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Matzuk, M.M.; McKeown, M.R.; Filippakopoulos, P.; Li, Q.; Ma, L.; Agno, J.E.; Lemieux, M.E.; Picaud, S.; Yu, R.N.; Qi, J.; et al. Small-molecule inhibition of BRDT for male contraception. Cell 2012, 150, 673–684. [Google Scholar] [CrossRef] [Green Version]
- Shang, E.Y.; Nickerson, H.D.; Wen, D.C.; Wang, X.Y.; Wolgemuth, D.J. The first bromodomain of Brdt, a testis-specific member of the BET sub-family of double-bromodomain-containing proteins, is essential for male germ cell differentiation. Development 2007, 134, 3507–3515. [Google Scholar] [CrossRef] [Green Version]
- Miao, Z.; Guna, X.; Jiang, J.; Georg, G.I. BRDT inhibitors for male contraceptive drug discovery: Current status. In Targeting Protein-Protein Interactions by Small Molecules; Cheng, C., Georg, G.I., Eds.; Springer: Singapore, 2018; pp. 287–315. [Google Scholar]
- Guan, X.; Cheryala, N.; Karim, R.; Chan, A.; Berndt, N.; Qi, J.; Georg, G.I.; Schonbrunn, E. Bivalent BET bromodomain inhibitors confer increased potency and selectivity for BRDT via protein conformational plasticity. J. Med. Chem. 2022, 65, 10441–10458. [Google Scholar] [CrossRef]
- Lee, D.H.; Qi, J.; Bradner, J.E.; Said, J.W.; Doan, N.B.; Forscher, C.; Yang, H.; Koeffler, H.P. Synergistic effect of JQ1 and rapamycin for treatment of human osteosarcoma. Int. J. Cancer 2015, 136, 2055–2064. [Google Scholar] [CrossRef] [Green Version]
- Fiskus, W.; Sharma, S.; Qi, J.; Shah, B.; Devaraj, S.G.; Leveque, C. BET protein antagonists JQ1 is synergistically lethal with FLT3 tyrosine kinase inhibitor (TKI) and overcomes resistance to FL3-TKI in AML cells expressing FLT-ITD. Mol. Cancer Ther. 2014, 13, 2315–2327. [Google Scholar] [CrossRef] [Green Version]
- Stratikopoulos, E.E.; Dendy, M.; Szabolcs, M.; Khaykin, A.J.; Lefebvre, C.; Zhou, M.M. Kinase and BET inhibitors together clamp inhibition of PI3K signaling and overcome resistance to therapy. Cancer Cell 2015, 27, 837–851. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Shah, B.; Fiskus, W.; Qi, J.; Rajapakshe, K.; Coarfa, C.; Li, L.; Devaraj, S.G.; Sharma, S.; Zhang, L.; et al. Synergistic activity of BET protein antagonist-based combinations in mantel cell lymphoma cells sensitive or resistant to ibrutinib. Blood 2015, 126, 1565–1574. [Google Scholar] [CrossRef]
- Baker, E.K.; Taylor, S.; Gupte, A.; Sharp, P.P.; Walia, M.; Walsh, N.C.; Zannettino, A.C.; Chalk, A.M.; Burns, C.J.; Walkley, C.R. BET inhibitors induce apoptosis through a MYC independent mechanism and synergise with CDK inhibitors to kill osteosarcoma cells. Sci. Rep. 2015, 5, 10120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ember, S.W.; Zhu, J.Y.; Olesen, S.H.; Martin, M.P.; Becker, A.; Berndt, N.; Georg, G.I.; Schonbrunn, E. Acetyl-lysine binding site of bromodomain-containing protein 4 (BRD4) interacts with diverse kinase inhibitors. ACS Chem. Biol. 2014, 9, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Cicerci, P.; Muller, S.; O’Mahony, A.; Fedorov, O.; Filippakopoulos, P.; Hunt, J.P.; Lasater, E.A.; Pallares, G.; Picaud, S.; Wells, C.; et al. Dual kinase-bromodoamin inhibitors for rationally designed polypharmacology. Nat. Chem. Biol. 2014, 10, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Wulf, G.M.; Barry, W.T.; Birrer, M.; Westin, S.N.; Farooq, S.; Bell-McGuinn, K.M.; Obermayer, E.; Whalen, C.; Spagnoletti, T.; et al. Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly(ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high grade serous ovarian and breast cancer. Ann. Oncol. 2016, 28, 512–518. [Google Scholar] [CrossRef]
- Ember, S.W.; Lambert, Q.T.; Berndt, N.; Gunawan, S.; Ayaz, M.; Tauro, M.; Zhu, J.Y.; Cranfill, P.J.; Greninger, P.; Lynch, C.C.; et al. Potent dual BET bromodomain-kinase inhibitors as value-added multitargetd chemical probes and cancer therapeutics. Mol. Cancer Ther. 2017, 16, 1054–1067. [Google Scholar] [CrossRef] [Green Version]
- Morales, G.A.; Garlich, J.R.; Su, J.; Peng, X.; Newblom, J.; Weber, K.; Durden, D.L. Synthesis and cancer stem cell-based activity of substituted 5-morpholino-7H-thieno [3,2-b] pyran-7-ones designed as next generation PI3K inhibitors. J. Med. Chem. 2013, 56, 1922–1939. [Google Scholar] [CrossRef]
- Andrews, F.H.; Singh, A.R.; Joshi, S.; Smith, C.A.; Morales, G.A.; Garlich, J.R.; Durden, D.L.; Kutateladze, T.G. Dual-activity PI3K-BRD4 inhibitor for the orthogonal inhibition of MYC to block tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2017, 114, E1072–E1080. [Google Scholar] [CrossRef] [Green Version]
- Carlino, L.; Rastelli, G. Dual kinase-bromodomain inhibitors in anticancer drug discovery: A structural and pharmacological perspective. J. Med. Chem. 2016, 59, 9305–9320. [Google Scholar] [CrossRef]
- Deshaies, R.J. Protein degradation: Prime time for PROTACs. Nat. Chem. Biol. 2015, 11, 634–635. [Google Scholar] [CrossRef]
- Chen, L.; Liu, Z.P.; Li, X. Recent advances in dual BRD4-kinase inhibitors based on polypharmacology. ChemMedChem 2022, 17, e202100731. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, A.M.; Vann, K.R.; Joshi, S.; Morales, G.A.; Vega, F.M.; Singh, A.; Pal, D.; Merati, A.B.; Kutateladze, T.G.; Durden, D.L. A triple action CDK4/6-PI3K-BET inhibitor with augmented cancer cell cytotoxicity. Cell Discov. 2020, 6, 49. [Google Scholar] [CrossRef] [PubMed]
- Andrieu, G.P.; Denis, G.V. BET proteins exhibit transcriptional and functional opposition in the epithelial-to-mesenchymal transition. Mol. Cancer Res. 2018, 16, 580–586. [Google Scholar] [CrossRef] [Green Version]
- Wroblewski, M.; Scheller-Wendorff, M.; Udonta, F.; Bauer, R.; Schlichting, J.; Zhao, L.; Batalla, I.B.; Gensch, V.; Pasler, S.; Wu, L.; et al. BET-inhibition by JQ1 promotes proliferation and self-renewal capacity of hematopoietic stem cells. Haematologica 2018, 103, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Bertoni, F. BET proteins as targets for anticancer treatment. Cancer Discov. 2018, 8, 24–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradbury, R.H.; Callis, R.; Carr, G.R.; Chen, H.; Clark, E.; Feron, L.; Glossop, S.; Graham, M.A.; Hattersley, M.; Jones, C.; et al. Optimization of a series of bivalent triazolopyridazine based bromodomain and extraterminal inhibitors: The discovery of (3R)-4-[2-[4-[1-(3-methoxy-[1,2,4]triazolo[4,3-b]pyridazin-6-yl)-4-piperidyl]phenoxy]ethyl]-1,3-dimethyl-piperazin-2-one (AZD5153). J. Med. Chem. 2016, 59, 7801–7817. [Google Scholar] [CrossRef]
- Zhang, P.; Li, R.; Xiao, H.; Liu, W.; Zeng, X.; Xie, G.; Yang, W.; Shi, L.; Yin, Y.; Tao, K. BRD4 inhibitor AZD5153 suppresses the proliferation of colorectal cancer cells and sensitizes the anticancer effect of PARP inhibitor. Int. J. Biol. Sci. 2019, 15, 1942–1954. [Google Scholar] [CrossRef] [Green Version]
- Nowak, R.P.; DeAngelo, S.L.; Buckley, D.; He, Z.; Donovan, K.A.; An, J.; Safaee, N.; Jedrychowski, M.P.; Ponthier, C.M.; Ishoey, M.; et al. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat. Chem. Biol. 2018, 14, 706–714. [Google Scholar] [CrossRef]
- Jia, S.Q.; Zhuo, R.; Zhang, Z.M.; Yang, Y.; Tao, Y.F.; Wang, J.W.; Li, X.L.; Xie, Y.; Li, G.; Wu, D. The BRD4 inhibitor dBET57 exerts anticancer effects by targeting superenhancer-related genes in neuroblastoma. J. Immunol. Res. 2022, 2022, 7945884. [Google Scholar] [CrossRef]
- Zengerle, M.; Chan, K.H.; Ciulli, A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef] [Green Version]
- Yin, M.; Guo, Y.; Hu, R.; Cai, W.L.; Li, Y.; Pei, S.; Sun, H.; Peng, C.; Li, J.; Ye, R.; et al. Potent BRD4 inhibitor suppresses cancer cell-macrophage interaction. Nat. Commun. 2020, 11, 1833. [Google Scholar] [CrossRef] [Green Version]
- Schupp, J.; Krebs, F.K.; Zimmer, N.; Trzeciak, E.; Schuppan, D.; Tuettenberg, A. Targeting myeloid cells in the tumor sustaining microenvironment. Cell Immunol. 2019, 343, 103713. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Fu, Y.; Day, D.; Sun, Y.; Wang, S.; Liang, X.; Gu, F.; Zhang, F.; Stevens, S.M.; Zhou, P.; et al. VEGF amplifies transcription through ETS1 acetylation to enable angiogenesis. Nat. Commun. 2017, 8, 383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Chen, H.; Wang, P.; Li, Y.; Wold, E.A.; Leonard, P.G.; Joseph, S.; Brasier, A.R.; Tian, B.; Zhou, J. Discovery of orally bioavailable chromosome derivatives as potent and selective BRD4 inhibitors: Scaffold hopping, optimization, and pharmacological evaluation. J. Med. Chem. 2020, 63, 5242–5256. [Google Scholar] [CrossRef]
- Zhou, Z.; Li, X.; Liu, Z.; Huang, L.; Yao, Y.; Li, L.; Chen, J.; Zhang, R.; Zhou, J.; Wang, L.; et al. A bromodomain-containing protein 4 (BRD4) inhibitor suppresses angiogenesis by regulating AP-1 expression. Front. Pharmacol. 2020, 11, 1043. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Han, J.; Wang, Z.; Li, X.; Sun, Y.; Hu, Z. Safety and efficacy of bromodomain and extra-terminal inhibitors for the treatment of hematological malignancies and solid tumors: A systematic study of clinical trials. Front. Pharmacol. 2021, 11, 621093. [Google Scholar] [CrossRef]
- Sarnik, J.; Poplawski, T.; Tokarz, P. BET proteins as attractive targets for cancer therapeutics. Int. J. Mol. Sci. 2021, 22, 11102. [Google Scholar] [CrossRef]
- Dey, A. BRD4 directs hematopietic stem cell development and modulates macrophage inflammatory responses. EMBO J. 2019, 38, e100293. [Google Scholar] [CrossRef]
- Bolden, J.E.; Tasdemir, N.; Dow, L.E.; van Es, J.H.; Wilkinson, J.E.; Zhao, Z.; Clevers, H.; Lowe, S.W. Inducible in vivo silencing of Brd4 identifies potential toxicities of sustained BET protein inhibition. Cell Rep. 2014, 8, 1919–1929. [Google Scholar] [CrossRef] [Green Version]
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer 2021, 124, 1478–1490. [Google Scholar] [CrossRef]
- Zhou, X.; Sun, T.; Meng, Y.; Luo, J.; Chen, J.; Xia, B.; Zhang, Z.; Cheng, Z.; Wang, X. BET inhibitors combined with chemotherapy synergistically inhibit the growth of NSCLC cells. Oncol. Rep. 2021, 45, 70. [Google Scholar] [CrossRef] [PubMed]
- Celano, M.; Gagliardi, A.; Maggisano, V.; Ambrosio, N.; Bulotta, S.; Fresta, M.; Russo, D.; Cosco, D. Co-encapsulation of paclitaxel and JQ1 in Zein nanoparticles as potential innovative nanomedicine. Micromachines 2022, 13, 1580. [Google Scholar] [CrossRef] [PubMed]
- Bhadury, J.; Nilsson, L.M.; Muralidharan, S.V.; Green, L.C.; Li, Z.; Gesner, E.M.; Hansen, H.C.; Keller, U.B.; McLure, K.G.; Nilsson, J.A. BET and HDAC inhibitors induce similar genes and biological effects and synergize to kill in Myc-induced murine lymphoma. Proc. Natl. Acad. Sci. USA 2014, 111, E2721–E2730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jostes, S.; Nettersheim, D.; Fellermeyer, M.; Schneider, S.; Hafezi, F.; Honecker, F.; Schumacher, V.; Geyer, M.; Kristiansen, G.; Schorle, H. The bromodomain inhibitor JQ1 triggers growth arrest and apoptosis in testicular germ cell tumors in vitro and in vivo. J. Cell Mol. Med. 2016, 21, 1300–1314. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, A.; Cullinace, C.; Paoli-Iseppi, R.; Wilmott, J.S.; Gunatilake, D.; Madore, J.; Strbenac, D.; Yang, J.Y.; Gowrishankar, K.; Madore, J.; et al. Combining BET and HDAC inhibitors synergistically induces apoptosis of melanoma and suppresses AKT and YAP signaling. Oncotarget 2015, 6, 21507–21521. [Google Scholar] [CrossRef] [Green Version]
- Mazur, P.K.; Herner, A.; Mello, S.S.; Wirth, M.; Hausmann, S.; Sanchez-Rivera, F.J.; Lofgren, S.M.; Kuschma, T.; Hahn, S.A.; Vangala, D.; et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat. Med. 2015, 21, 1163–1171. [Google Scholar] [CrossRef] [Green Version]
- Shahbazi, J.; Liu, P.Y.; Atmadibrata, B.; Bradner, J.E.; Marshall, G.M.; Lock, R.B.; Liu, T. The bromodomain inhibitor JQ1 and the histone deacetylase inhibitor panobinostat synergistically reduce N-Myc expression and induce anticancer effects. Clin. Cancer Res. 2016, 22, 2534–2544. [Google Scholar] [CrossRef] [Green Version]
- Mishra, V.K.; Wegwitz, F.; Kosinsky, R.L.; Sen, M.; Baumgartner, R.; Wulff, T.; Siveke, J.T.; Schildhaus, H.U.; Najafova, Z.; Kari, V.; et al. Histone deacetylase class-I inhibition promotes epithelial gene expression in pancreatic cancer cells in a BRD4- and MYC-dependent manner. Nucleic Acids Res. 2017, 45, 6334–6349. [Google Scholar] [CrossRef] [Green Version]
- Fiskus, W.; Sharma, S.; Qi, J.; Valenta, J.A.; Schaub, L.J.; Shah, B.; Peth, K.; Portier, B.P.; Rodriguez, M.; Devaraj, S.G.; et al. Highly active combination of BRD4 antagonist and histone deacetylase inhibitor against human acute myelogenous leukemia cells. Mol. Cancer Ther. 2014, 13, 1142–1154. [Google Scholar] [CrossRef] [Green Version]
- Coude, M.M.; Braun, T.; Berrou, J.; Dupont, M.; Bertrand, S.; Masse, A.; Raffoux, E.; Itzykson, R.; Delord, M.; Riveiro, M.E.; et al. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget 2015, 6, 17698–17712. [Google Scholar] [CrossRef] [Green Version]
- Fong, C.Y.; Gilan, O.; Lam, E.Y.; Rubin, A.F.; Ftouni, S.; Tyler, D.; Stanley, K.; Sinha, D.; Yeh, P.; Dawson, M.A. BET inhibitor resistance emerges from leukaemia stem cells. Nature 2015, 525, 538–542. [Google Scholar] [CrossRef]
- Alu, A.; Lei, H.; Han, X.; Wei, Y.; Wei, X. BTK inhibitors in the treatment of hematological malignancies and inflammatory diseases: Mechanisms and clinical studies. J. Hematol. Oncol. 2022, 15, 138. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, V.E.; Smith, C.C. FLT3 mutations in acute myeloid leukemia: Key concepts and emerging controversies. Front. Oncol. 2020, 10, 612880. [Google Scholar] [CrossRef] [PubMed]
- Hydbring, P.; Bahram, F.; Su, Y.; Tronnersjo, S.; Hogstrand, K.; von der Lehr, N.; Sharifi, H.R.; Lilischkis, R.; Hein, N.; Wu, S.; et al. Phosphorylation by Cdk2 is required for Myc to repress Ras-induced senescence in cotransformation. Proc. Natl. Acad. Sci. USA 2010, 107, 58–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolin, S.; Borgenvik, A.; Persson, C.U.; Sundstrom, A.; Qi, J.; Bradner, J.E.; Weiss, W.A.; Cho, Y.J.; Weishaupt, H.; Swartling, F.J. Combined BET bromodomain and CDK2 inhibition in MYC-driven medulloblastoma. Oncogene 2018, 37, 2850–2862. [Google Scholar] [CrossRef] [Green Version]
- Vangala, J.R.; Potluri, A.; Radhakrishnan, S.K. BET inhibitors synergize with carfilzomib to induce cell death in cancer cells via impairing Nrf1 transcriptional activity and exacerbating the unfolded protein response. Biomolecules 2020, 10, 501. [Google Scholar] [CrossRef] [Green Version]
- Perez-Galab, P.; Mora-Jensen, H.; Weniger, M.A.; Shaffer, A.L., 3rd; Rizzatti, E.G.; Chapman, C.M.; Mo, C.C.; Stennett, L.S.; Rader, C.; Liu, P.; et al. Bortezomib resistance in mantle cell lymphoma is associated with plasmacytic differentiation. Blood 2011, 117, 542–552. [Google Scholar] [CrossRef] [Green Version]
- Diaz, T.; Rodriguez, V.; Lozano, E.; Mena, M.P.; Calderon, M.; Rosinol, L.; Martinez, A.; Tovar, N.; Perez-Galan, P.; Blade, J.; et al. The BET bromodomain inhibitor CPI203 improves lenalidomide and dexamethasone activity in in vitro and in vivo models of multiple myeloma by blockade of Ikaros and MYC signaling. Haematologica 2017, 102, 1776–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, M.; Gao, R.; Zhao, R.; Huang, Y.; Chen, C.; Li, K.; Yu, X.; Nie, D.; Chen, Z.; Liu, X.; et al. BET bromodomain inhibition rescues PD-1-mediated T-cell exhaustion in acute myeloid leukemia. Cell Death Dis. 2022, 13, 671. [Google Scholar] [CrossRef]
- Kurimchak, A.M.; Shelton, C.; Duncan, K.E.; Johnson, K.J.; Brown, J.; O’Brien, S.; Gabbasov, R.; Fink, L.S.; Li, Y.; Lounsbury, N.; et al. Resistance to BET bromodomain inhibitors is mediated by kinome reprogramming in ovarian cancer. Cell Rep. 2016, 16, 1273–1286. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Tang, Y.A.; Xiao, Q.; Lee, W.C.; Cheng, B.; Niu, Z.; Oguz, G.; Feng, M.; Lee, P.L.; Li, B.; et al. Stromal induction of BRD4 phosphorylation results in chromatin remodeling and BET inhibitor resistance in colorectal cancer. Nat. Commun. 2021, 12, 4441. [Google Scholar] [CrossRef] [PubMed]
- Engelke, C.G.; Chinnaiyan, A.M. aBETing therapeutic resistance by Wnt signaling. Cell Res. 2015, 25, 1187–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, K.; Raza, S.S.; Knab, L.M.; Chow, C.R.; Kwok, B.; Bentrem, D.J.; Popovic, R.; Ebine, K.; Licht, J.D.; Munshi, H.G. GLI2-dependent c-MYC upregulation mediates resistance of pancreatic cancer cells to the BET bromodomain inhibitor JQ1. Sci. Rep. 2015, 5, 9489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, J.E.; Eom, J.I.; Jeung, H.K.; Cheong, J.-W.; Lee, J.Y.; Kim, J.S.; Min, Y.H. AMPK-ULK1-mediated autophagy confers resistance to BET inhibitor JQ1 in acute myeloid leukemia stem cells. Clin. Cancer Res. 2017, 23, 2781–2794. [Google Scholar] [CrossRef] [Green Version]
- Shu, S.; Lin, C.Y.; He, H.H.; Witwicki, R.M.; Tabassum, D.P.; Roberts, J.M.; Janiszewska, M.; Huh, S.J.; Liang, Y.; Ryan, J.; et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 2016, 529, 413–417. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Gan, W.; Li, X.; Wang, S.; Zhang, W.; Huang, L.; Liu, S.; Zhong, Q.; Guo, J.; Zhang, J.; et al. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat. Med. 2017, 23, 1063–1071. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Wang, D.; Zhao, Y.; Ren, S.; Gao, K.; Ye, Z.; Wang, S.; Pan, C.W.; Zhu, Y.; Yan, Y.; et al. Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. Nat. Med. 2017, 23, 1055–1062. [Google Scholar] [CrossRef]
- Coleman, D.J.; Gao, L.; Schwartzmann, J.; Korkola, J.E.; Sampson, D.; Derrick, D.S.; Urrutia, J.; Balter, A.; Burchard, J.; King, C.J.; et al. Maintenance of MYC expression promotes de novo resistance to BET bromodomain inhibition in castration-resistant prostate cancer. Sci. Rep. 2019, 9, 3823. [Google Scholar] [CrossRef] [Green Version]
- French, C.A. NUT carcinoma: Clinicopathologic features, pathogenesis, and treatment. Pathol. Int. 2018, 68, 583–595. [Google Scholar] [CrossRef]
- Albrecht, T.; Harms, A.; Roessler, S.; Goeppert, B. NUT carcinoma in a nutshell: A diagnosis to be considered more frequently. Pathol. Res. Pract. 2019, 215, 152347. [Google Scholar] [CrossRef]
- Chau, N.G.; Ma, C.; Danga, K.; Al-Sayegh, H.; Nardi, V.; Barrette, R.; Lathan, C.S.; DuBiois, S.G.; Haddad, R.J.; Shapiro, G.I.; et al. An anatomical site and genetic based prognostic model for patients with NUT midline carcinoma: Analysis of 124 patients. JNCI Cancer Spectr. 2019, 4, pkz094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC oncogene—The grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 2022, 19, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J., 3rd. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef] [Green Version]
- Amorim, S.; Stathis, A.; Gleeson, M.; Iyengar, S.; Magarotto, V.; Leleu, X.; Morschhauser, F.; Karlin, L.; Broussais, F.; Rezai, K.; et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: A dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016, 3, e196–e204. [Google Scholar] [CrossRef] [PubMed]
- Berthon, C.; Raffoux, E.; Thomas, C.; Vey, N.; Gomez-Roca, C.; Yee, K.; Taussig, D.C.; Rezai, K.; Rumier, C.; Herait, P.; et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: A dose-escalation, phase 1 study. Lancet Haematol 2016, 3, e186–e195. [Google Scholar] [CrossRef] [PubMed]
- Hann, S.R. Role of post-translational modifications in regulating c-Myc proteolysis, transcriptional activity and biological function. Semin. Cancer Biol. 2006, 16, 288–302. [Google Scholar] [CrossRef]
- Iniguez, A.B.; Alexe, G.; Wang, E.J.; Roti, G.; Patel, S.; Chen, L.; Kitara, S.; Conway, A.; Robichaud, A.L.; Stolte, B.; et al. Resistance to epigenetic-targeted therapy engenders tumor cell vulnerabilities associated with enhancer remodeling. Cancer Cell 2018, 34, 922–938. [Google Scholar] [CrossRef] [Green Version]
- Ostertag, M.S.; Hutwelker, W.; Plettenburg, O.; Sattler, M.; Popowicz, G.M. Structural insights into BET client recognition of endometrial and prostate cancer-associated SPOP mutants. J. Mol. Biol. 2019, 431, 2213–2221. [Google Scholar] [CrossRef]
- Janouskova, H.; El Tekle, G.; Bellini, E.; Udeshi, N.D.; Rinaldi, A.; Ulbricht, A.; Bernasocchi, T.; Civenni, G.; Losa, M.; Svinkina, T.; et al. Opposing effects of cancer-type-specific SPOP mutants on BET protein degradation and sensitivity to BET inhibitors. Nat. Med. 2017, 23, 1046–1054. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [Green Version]
- Stathis, A.; Zucca, E.; Bekradda, M.; Gomez-Roca, C.; Delord, J.-P.; de La Motte Rouge, T.; Uro-Coste, E.; de Braud, F.; Pelosi, G.; French, C.A.J.C.d. Clinical response of carcinomas harboring the BRD4–NUT oncoprotein to the targeted bromodomain inhibitor OTX015/MK-8628. Cancer Discov. 2016, 6, 492–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwalm, M.P.; Knapp, S. BET bromodomain inhibitors. Curr. Opin. Chem. Biol. 2022, 68, 102148. [Google Scholar] [CrossRef] [PubMed]
- Crowe, B.L.; Larue, R.C.; Yuan, C.; Hess, S.; Kvaratskhelia, M.; Foster, M.P. Structure of the Brd4 ET domain bound to a C-terminal motif from γ-retroviral integrases reveals a conserved mechanism of interaction. Proc. Natl. Acad. Sci. USA 2016, 113, 2086–2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.F.; Miller, L.D.; Chan, X.B.; Black, M.A.; Pang, B.; Ong, C.W.; Salto-Tellez, M.; Liu, E.T.; Desai, K.V. JMJD6 is a driver of cellular proliferation and motility and a marker of poor prognosis in breast cancer. Breast Cancer Res. 2012, 14, R85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ni, S.-S.; Zhao, W.-L.; Dong, X.-C.; Wang, J.L. High expression of JMJD6 predicts unfavorable survival in lung adenocarcinoma. Tumor Biol. 2013, 34, 2397–2401. [Google Scholar] [CrossRef]
- Wang, F.; He, L.; Huangyang, P.; Liang, J.; Si, W.; Yan, R.; Han, X.; Liu, S.; Gui, B.; Li, W.; et al. JMJD6 promotes colon carcinogenesis through negative regulation of p53 by hydroxylation. PLoS Biol. 2014, 12, e1001819. [Google Scholar] [CrossRef] [Green Version]
- Poulard, C.; Rambaud, J.; Lavergne, E.; Jacquemetton, J.; Renoir, J.-M.; Tredan, O.; Chabaud, S.; Treilleux, I.; Corbo, L.; Romancer, M.L. Role of JMJD6 in breast tumourigenesis. PLoS ONE 2015, 10, e0126181. [Google Scholar] [CrossRef]
- Lee, C.-R.; Lee, S.H.; Rigas, N.K.; Kim, R.H.; Kang, M.K.; Park, N.-H.; Shin, K.-H. Elevated expression of JMJD6 is associated with oral carcinogenesis and maintains cancer stemness properties. Carcinogenesis 2016, 37, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Aprelikova, O.; Chen, K.; El Touny, L.H.; Brignatz-Guittard, C.; Han, J.; Qiu, T.; Yang, H.H.; Lee, M.P.; Zhu, M.; Green, J.E. The epigenetic modifier JMJD6 is amplified in mammary tumors and cooperates with c-Myc to enhance cellular transformation, tumor progression, and metastasis. Clin. Epigenetics 2016, 8, 38. [Google Scholar] [CrossRef] [Green Version]
- Wong, M.; Sun, Y.; Xi, Z.; Milazzo, G.; Poulos, R.C.; Bartenhagen, C.; Bell, J.L.; Mayoh, C.; Ho, N.; Tee, A.E.; et al. JMJD6 is a tumorigenic factor and therapeutic target in neuroblastoma. Nat. Commun. 2019, 10, 3319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Chen, S.; Yang, Y.; Ma, X.; Shao, B.; Yang, S.; Wei, Y.; Wei, X. Jumonji domain-containing protein 6 protein and its role in cancer. Cell Prolif. 2020, 53, e12747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, C.A.; Rahman, S.; Walsh, E.M.; Kühnle, S.; Grayson, A.R.; Lemieux, M.E.; Grunfeld, N.; Rubin, B.P.; Antonescu, C.R.; Zhang, S.; et al. NSD3–NUT fusion Oncoprotein in NUT midline carcinoma: Implications for a novel oncogenic mechanism. Cancer Discov. 2014, 4, 928–941. [Google Scholar] [CrossRef] [Green Version]
- Shafran, J.S.; Jafari, N.; Casey, A.N.; Gyorffy, B.; Denis, G.V. BRD4 regulates key transcription factors that drive epithelial-mesenchymal transition in castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Corrigan-Curay, J.; Cohen-Haguenauer, O.; O’reilly, M.; Ross, S.R.; Fan, H.; Rosenberg, N.; Somia, N.; King, N.; Friedmann, T.; Dunbar, C.; et al. Challenges in vector and trial design using retroviral vectors for long-term gene correction in hematopoietic stem cell gene therapy. Mol. Ther. 2012, 20, 1084–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, C.; Larochelle, A.; Dunbar, C.E. Hematopoietic stem cell gene therapy: Dead or alive? Trends Biotechnol. 2005, 23, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Hematti, P.; Hong, B.-K.; Ferguson, C.; Adler, R.; Hanawa, H.; Sellers, S.; Holt, I.E.; Eckfeldt, C.E.; Sharma, Y.; Schmidt, M.; et al. Distinct genomic integration of MLV and SIV vectors in primate hematopoietic stem and progenitor cells. PLoS Biol. 2004, 2, e423. [Google Scholar] [CrossRef] [Green Version]
- Haviernik, P.; Bunting, K.D. Safety concerns related to hematopoietic stem cell gene transfer using retroviral vectors. Curr. Gene Ther. 2004, 4, 263–276. [Google Scholar] [CrossRef]
- Yoder, K.E.; Rabe, A.J.; Fishel, R.; Larue, R.C. Strategies for targeting retroviral integration for safer gene therapy: Advances and challenges. Front. Mol. Biosci. 2021, 8, 662331. [Google Scholar] [CrossRef]
- Sharma, A.; Larue, R.C.; Plumb, M.R.; Malani, N.; Male, F.; Slaughter, A.; Kessl, J.J.; Shkriabai, N.; Coward, E.; Aiyer, S.S.; et al. BET proteins promote efficient murine leukemia virus integration at transcription start sites. Proc. Natl. Acad. Sci. USA 2013, 110, 12036–12041. [Google Scholar] [CrossRef] [Green Version]
- Larue, R.C.; Plumb, M.R.; Crowe, B.L.; Shkriabai, N.; Sharma, A.; DiFiore, J.; Malani, N.; Aiyer, S.S.; Roth, M.J.; Bushman, F.D.; et al. Bimodal high-affinity association of Brd4 with murine leukemia virus integrase and mononucleosomes. Nucleic Acids Res. 2014, 42, 4868–4881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiyer, S.; Swapna, G.V.; Ma, L.-C.; Liu, G.; Hao, J.; Chalmers, G.; Jacobs, B.C.; Montelione, G.T.; Roth, M.J. A common binding motif in the ET domain of BRD3 forms polymorphic structural interfaces with host and viral proteins. Structure 2021, 29, 886–898.e6. [Google Scholar] [CrossRef] [PubMed]
- Vignali, M.; Hassan, A.H.; Neely, K.E.; Workman, J.L. ATP-dependent chromatin-remodeling complexes. Mol. Cell Biol. 2000, 20, 1899–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohrmann, L.; Verrijzer, C.P. Composition and functional specificity of SWI2/SNF2 class chromatin remodeling complexes. Biochim. Biophys. Acta 2005, 1681, 59–73. [Google Scholar] [CrossRef]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [CrossRef]
- Biegel, J.A.; Busse, T.M.; Weissman, B.E. SWI/SNF chromatin remodeling complexes and cancer. Proc. Am. J. Med. Genet. Part C: Semin. Med. Genet. 2014, 350–366. [Google Scholar]
- Wai, D.C.; Szyszka, T.N.; Campbell, A.E.; Kwong, C.; Wilkinson-White, L.E.; Silva, A.P.; Low, J.K.; Kwan, A.H.; Gamsjaeger, R.; Chalmers, J.D.; et al. The BRD3 ET domain recognizes a short peptide motif through a mechanism that is conserved across chromatin remodelers and transcriptional regulators. J. Biol. Chem. 2018, 293, 7160–7175. [Google Scholar] [CrossRef] [Green Version]
- Kvaratskhelia, M.; Sharma, A.; Larue, R.C.; Serrao, E.; Engelman, A. Molecular mechanisms of retroviral integration site selection. Nucleic Acids Res. 2014, 42, 10209–10225. [Google Scholar] [CrossRef] [Green Version]
- Schröder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.S.; Maetzig, T.; Maertens, G.N.; Sharif, A.; Rothe, M.; Weidner-Glunde, M.; Galla, M.; Schambach, A.; Cherepanov, P.; Schulz, T.F. Bromo-and extraterminal domain chromatin regulators serve as cofactors for murine leukemia virus integration. J. Virol. 2013, 87, 12721–12736. [Google Scholar] [CrossRef] [Green Version]
- Weissman, J.D.; Singh, A.K.; Devaiah, B.N.; Schuck, P.; Larue, R.C.; Singer, D.S. The intrinsic kinase activity of BRD4 spans its BD2-B-BID domains. J. Biol. Chem. 2021, 297, 101326. [Google Scholar] [CrossRef]
- Xing, E.; Surendranathan, N.; Kong, X.; Cyberski, N.; Garcia, J.D.; Cheng, X.; Sharma, A.; Li, P.-K.; Larue, R.C. Development of murine leukemia virus integrase-derived peptides that bind Brd4 extra-terminal domain as candidates for suppression of acute myeloid leukemia. ACS Pharmacol. Transl. Sci. 2021, 4, 1628–1638. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, Y.; Tian, G.; Jiang, Y.; Liu, Z.; Meng, X.; Bao, X.; Feng, L.; Sun, H.; Deng, H.; et al. Chemical proteomic profiling of bromodomains enables the wide-spectrum evaluation of bromodomain inhibitors in living cells. J. Am. Chem. Soc. 2019, 141, 11497–11505. [Google Scholar] [CrossRef] [PubMed]
- Borthakur, G.; Odenike, O.; Aldoss, I.; Rizzieri, D.A.; Prebet, T.; Chen, C.; Popovic, R.; Modi, D.A.; Joshi, R.H.; Wolff, J.E.; et al. A phase 1 study of the pan-bromodomain and extraterminal inhibitor mivebresib (ABBV-075) alone or in combination with venetoclax in patients with relapsed/refractory acute myeloid leukemia. Cancer 2021, 127, 2943–2953. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Starodub, A.N.; Koh, B.D.; Xing, G.; Armstrong, A.J.; Carducci, M.A. Phase Ib study of the BET inhibitor GS-5829 as monotherapy and combined with enzalutamide in patients with metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2022, 28, 3979–3989. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I.; LoRusso, P.; Dowlati, A.; Do, K.T.; Jacobson, C.A.; Vaishampayan, U.; Weise, A.; Caimi, P.F.; Eder, J.P.; French, C.A.; et al. A phase 1 study of RO6870810, a novel bromodomain and extra-terminal protein inhibitor, in patients with NUT carcinoma, other solid tumors, or diffuse large B-cell lymphoma. Br. J. Cancer 2021, 124, 744–753. [Google Scholar] [CrossRef]
- Albert, D.H.; Goodwin, N.C.; Davies, A.M.; Rowe, J.; Feuer, G.; Boyiadzis, M.; Dorritie, K.A.; Mancini, M.; Gandour-Edwards, R.; Jonas, B.A.; et al. Co-clinical modeling of the activity of the BET inhibitor mivebresib (ABBV-075) in AML. In Vivo 2022, 36, 1615–1627. [Google Scholar] [CrossRef]
- Dawson, M.A.; Borthakur, G.; Huntly, B.J.P.; Karadimitris, A.; Alegre, A.; Charidos, A.; Vogl, D.T.; Pollyea, D.A.; Davies, F.E.; Morgan, G.J.; et al. A phase I/II open-label study of molibresib for the treatment of relapsed/refractory hematologic malignancies. Clin. Cancer Res. 2023, 29, 711–722. [Google Scholar] [CrossRef]
- Riddell, K.; Patel, A.; Collins, G.; Zhou, Y.; Schramek, D.; Kremer, B.E.; Ferron-Brady, G. An adaptive physiologically based pharmacokinetic-driven design to investigate the effect of itraconazole and rifampicin on the pharmacokinetics of molibresib (GSK525762) in healthy female volunteers. J. Clin. Pharmacol. 2021, 61, 125–137. [Google Scholar] [CrossRef]
- Lewin, J.; Soria, J.C.; Stathis, A.; Delord, J.P.; Peters, S.; Awarda, A.; Aftimos, P.G.; Bekradda, M.; Rezai, K.; Zeng, Z.; et al. Phase Ib trial with birabresib, a small-molecule inhibitor of bromodomain and extraterminal proteins, in patients with selected advanced solid tumors. J. Clin. Oncol. 2018, 36, 3007–3014. [Google Scholar] [CrossRef]
- Blum, K.A.; Supko, J.G.; Maris, M.B.; Flinn, I.W.; Goy, A.; Younes, A.; Bobba, S.; Senderowicz, A.M.; Efuni, S.; Rippley, R.; et al. A phase I study of pelabresib (CPI-0610), a small-molecule inhibitor of BET proteins, in patients with relapsed or refractory lymphoma. Cancer Res. Commun. 2022, 2, 795–805. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subfamily Number | Representative Members | Cellular Function(s) |
|---|---|---|
| I | PCAF | Histone acetyltransferase |
| GCN5L | Histone acetyltransferase | |
| FALZ | Chromatin remodeling factor | |
| CECR2 | Chromatin remodeling factor | |
| II (the BET BRDs) | BRD2 | Transcriptional regulator |
| BRD3 | Transcriptional regulator | |
| BRD4 | Transcriptional regulator | |
| BRDT | Chromatin remodeling factor | |
| III | BRD8B | Transcriptional regulator |
| CREBBP | Histone acetyltransferase | |
| EP300 | Histone acetyltransferase | |
| BAZ1B | Tyrosine-protein kinase; transcriptional regulator | |
| BRWD3 domain 2 | JAK/STAT signaling | |
| PHIP domain 2 | Insulin signaling | |
| IV | BRD7 | Transcriptional regulator |
| BDR1 | Transcriptional regulator | |
| BDPF1 | Transcriptional regulator | |
| ATAD2 | Transcriptional regulator | |
| BRD9 | Unknown | |
| BRPF3 | Transcriptional regulator | |
| V | TRIM66 | Transcriptional repressor |
| TRIM33 | Ubiquitin E3 ligase; transcriptional regulator | |
| TIF1α | Ubiquitin E3 ligase; transcriptional regulator | |
| SP100 | Transcriptional regulator | |
| SP110 | Transcriptional regulator | |
| VI | MLL | Histone methyltransferase |
| TRIM28 | SUMO E3 ligase; transcriptional regulator | |
| VII | TAF1 | Transcriptional initiation |
| TAF1L | Transcriptional initiation | |
| BRWD3 domain 1 | JAK/STAT signaling | |
| PHIP domain 1 | Insulin signaling | |
| VIII | ASH1L | Histone-lysine methyltransferase |
| SMARCA2A | Chromatin remodeling factor | |
| SMARCA4 | Chromatin remodeling factor | |
| PB1 | Transcriptional regulator |
| Targeting Isoform | BET Inhibitor | Study Phase/ Patient Population | ClinicalTrials.gov Numbera (Current Status) |
|---|---|---|---|
| pan-BET | ABBV-075 (Mivebresib) | Phase I/1st-in-human, dose escalation study/ Breast, NSCLC, AML, MM, prostate, SCLC, non-Hodgkin lymphoma | NCT02391480 (completed; findings reported in [205]) |
| EP31670 (dual BET and CBP/p300 inhibitor) | Phase I/dose escalation study/ Castration-resistant prostate cancer and NUT carcinoma | NCT05488548 (not yet recruiting) | |
| FT-1101 | Phase I/Ib/Safety, pharmacokinetics and pharmacodynamics study/ AML, myelodysplastic syndrome, non-Hodgkin lymphoma | NCT02543879 (completed; findings reported in [127]) | |
| GS-5829 (Alobresib) | Phase Ib/II/ Part I—Safety, tolerability, pharmacokinetics and pharmacodynamics study of GS-5829 as a single agent Part II—Combination of GS-5829 and enzalutamide in patients with metastatic castration-resistant prostate cancer | NCT02607228 (completed; findings reported in [206]) | |
| GSK525762 (Molibresib) | Phase I/II Safety, pharmacokinetics and pharmacodynamics study/ Patients with relapsed, refractory hematologic malignancies | NCT01943851 (completed; findings reported in [58]) | |
| INCB054329 (BD2-selective) | Phase I/II Dose escalation, safety and tolerability study Solid tumors and hematological malignancy | NCT02431260 (terminated by sponsor in Jan 2018 due to pharmacokinetics variability) | |
| RO680810 | Phase I/dose escalation study/ NUT carcinoma, advanced solid tumors, or DLBCL | NCT01987362 (completed; findings reported in [207]) | |
| TEN-010 | Phase I/dose escalation study/ AML and myelodysplastic syndrome | NCT02308761 (completed; findings reported in [53]) | |
| TEN-010 | Phase I/Part 1—dose escalation; Part 2—Expansion cohort in patients with selected malignancies/ Advanced solid tumors | NCT01987362 (completed; findings reported in [207]) | |
| ZEN003694 | Phase I/Safety and tolerability study/ Metastatic CRPC | NCT02705469 (completed) | |
| ZEN003694 | Phase II/Single group assignment Squamous cell lung cancer patients harboring NSD3 amplification | NCT05607108 (recruiting) | |
| Brd2/3/4 | ABBV-744 (BD2-selective) | Phase I/Safety and pharmacokinetics study/ Breast, NSCLC, AML, MM, prostate, SCLC, non-Hodgkin lymphoma | NCT02391480 (completed; findings reported in [208]) |
| I-BET151 (GSK2820151) | Phase I/dose escalation study/ Advanced or recurrent solid tumors | NCT02630251 (terminated in 2017 due to development of another BET inhibitor (GSK525762) with a better understanding of the risk–benefit profile | |
| I-BET762 (GSK525762; Molibresib) | Phase I/II/dose escalation study/ Relapsed or refractory hematologic malignancies | NCT01943851 (completed; findings reported in [209]) | |
| I-BET762 (GSK525762; Molibresib) | Phase I/Open label cross-over study to evaluate the effect of itraconazole and rifampicin on the pharmacokinetics of I-BET0762) | NCT02706535 (completed; findings reported in [210]) | |
| OTX-015 (Birabresib) | Phase I/dose finding study/ AML, DLBCL, ALL, MM | NCT01713582 (completed; findings reported in [165]) | |
| OTX-015 (Birabresib) | Phase IIa/dose optimization/ Recurrent GBM | NCT02296476 (terminated due to lack of clinical activity and not due to safety reasons) | |
| OTX-015 (Birabresib) | Phase IB/dose exploration trial/ NMC, TNBC, NSCLC, CRPC | NCT02698176 (terminated due to limited efficacy and not due to safety reasons) | |
| OTX-015 (Birabresib) | Phase IB/ NMC, TNBC, NSCLC with rearranged ALK gene/fusion protein or KRAS mutation, CRPC, PDAC | NCT02259114 (completed; findings reported in [211]) | |
| Brd2/4 | CC-90010 (Trotabresib) | Phase I/1st-in-human/dose escalation and expansion/ Advanced solid tumors and relapsed/refractory non-Hodgkin lymphoma | NCT03220347 (completed; findings reported in [94]) |
| Brd4 | AZD5153 | Phase I/dose escalation study/ Relapsed or refractory malignant solid tumors, lymphomas | NCT03205176 (completed) |
| AZD5153 | Phase I/Platform protocol for the treatment of relapsed/refractory/aggressive non-Hodgkin lymphoma | NCT03527147 (completed) | |
| CPI-0610 (Pelabresib) | Phase I/1st-in-human Patients with relapsed/refractory lymphomas | NCT01949883 (completed; findings reported in [212]) | |
| CPI-0610 | Phase I/ Patients with previously treated multiple myeloma | NCT02157636 (completed) | |
| CPI-0610 | Phase 1 (dose escalation of CPI-0610 in patients with hematological malignancies) Phase 2 (dose expansion of CPI-0610 with and without ruxolitinib in patients with myelofibrosis and essential thrombocytopenia) | NCT02158858 (active, not recruiting) | |
| NUV-868 (BD2-selective) | Phase 1/1st-in-human dose escalation and expansion study/ Advanced solid tumors Phase 2/combination with olaparib or enzalutamide/ Advanced solid tumors | NCT05252390 (recruiting) | |
| PLX51107 | Phase I/combination of PLX51107 and azacytidine/ Myelodysplastic syndrome and AML | NCT04022785 (completed in October 2022) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
To, K.K.W.; Xing, E.; Larue, R.C.; Li, P.-K. BET Bromodomain Inhibitors: Novel Design Strategies and Therapeutic Applications. Molecules 2023, 28, 3043. https://doi.org/10.3390/molecules28073043
To KKW, Xing E, Larue RC, Li P-K. BET Bromodomain Inhibitors: Novel Design Strategies and Therapeutic Applications. Molecules. 2023; 28(7):3043. https://doi.org/10.3390/molecules28073043
Chicago/Turabian StyleTo, Kenneth K. W., Enming Xing, Ross C. Larue, and Pui-Kai Li. 2023. "BET Bromodomain Inhibitors: Novel Design Strategies and Therapeutic Applications" Molecules 28, no. 7: 3043. https://doi.org/10.3390/molecules28073043