2. Results and Discussion
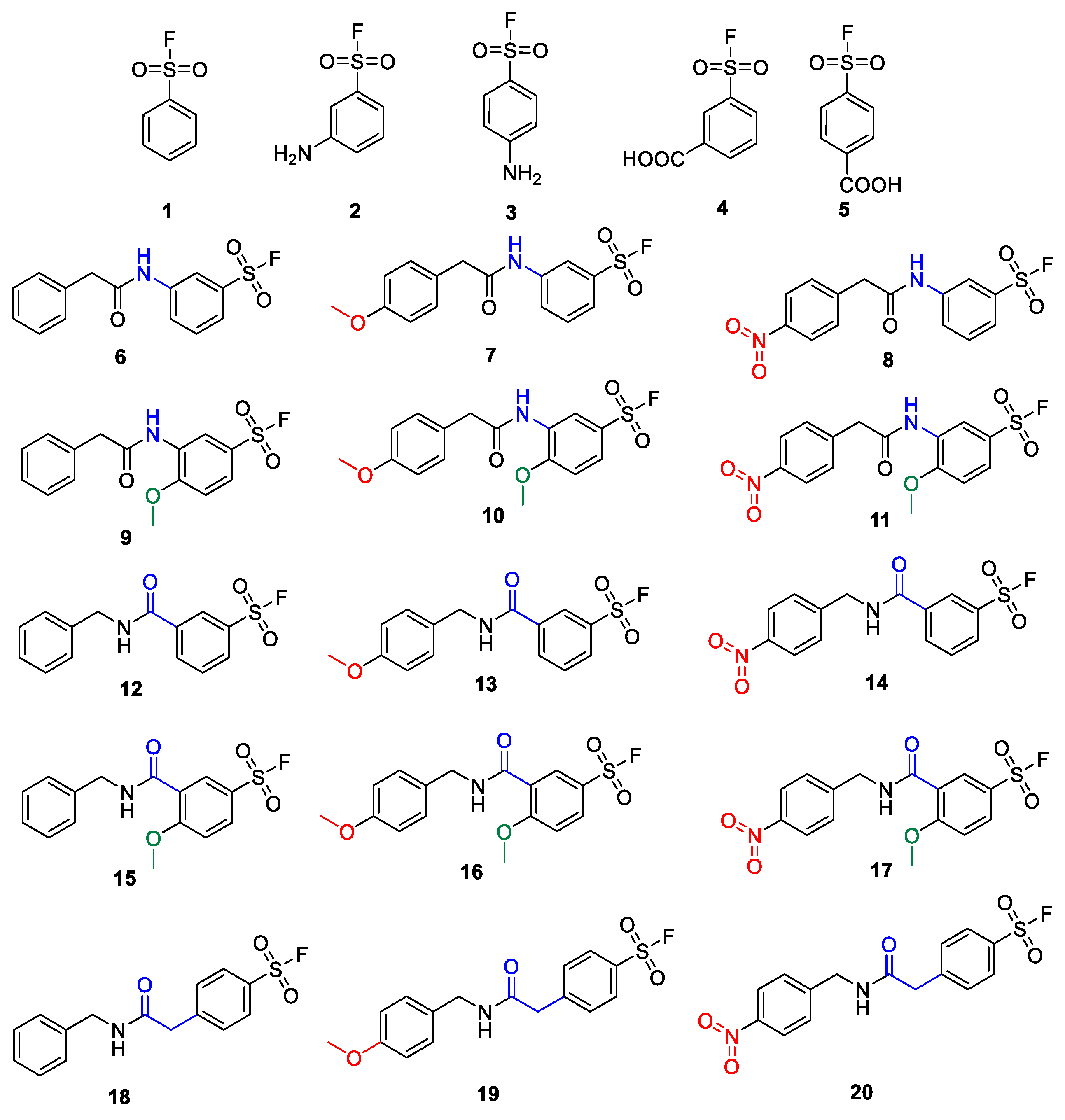
Developing the SuFBits fragment screening platform, we first required an appropriate tag that contains the sulfonyl fluoride warhead and a suitable attachment point/functionality to enable general application in library synthesis. We imagined that this functional group should be easily available for robust reactions, e.g., acylation or well-established multicomponent reactions. We have taken commercially available aryl sulfonyl fluorides that are modified in the
para or
meta position with an amino or carboxylic acid functional group (
1–
5) as the variable fragment binding element (
Figure 1). We have synthesized derivatives of these with or without additional substituents on the phenyl ring or more distant from the warhead (
6–
17), and with or without a methylene spacer as well (
18–
20), separating the amide group from the aromatic core. The additional substituents attached to the phenylsulfonyl fluoride were benzyl, 4′-methoxybenzyl and 4′-nitrobenzyl groups in order to check the possible effect of a distant electron donating or electron withdrawing group on the reactivity.
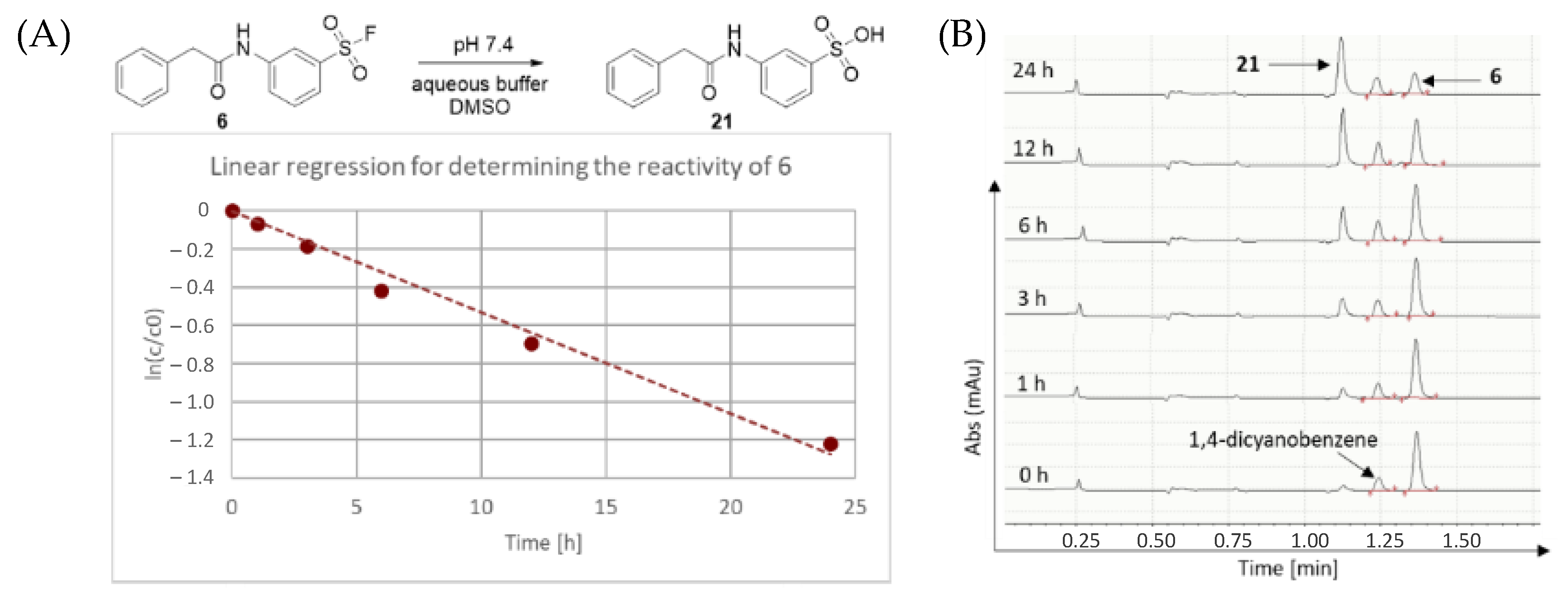
First, we characterized the intrinsic reactivity of the library members (
Table 1). Compounds were added in DMSO to PBS buffer (pH 7.4) together with the internal standard 1,4-dicyanobenzene. The vial was mixed, then analysed by HPLC-MS in 1, 2, 3, 6, 12 and 24 h time points (
Figure 2). In all cases, hydrolysis of the corresponding sulfonic acid could be observed (
Figure 2). These data provided the pseudo first order kinetic rate constant (k) and aqueous stability half-lives (t
1/2) after linear regression (
Table 1 and
Figure 2).
In comparison to benzenesulfonyl fluoride (
1), the amino (
2,
3) and the carboxylic acid substituents (
4,
5) caused an increased half-life, suggesting higher stability and lower aqueous reactivity (
Table 1). The less reactive one was 4-aminobenzenesulfonyl fluoride (
3), followed by the 3-amino derivative (
2), while among the carboxylic acids, the meta-substituted compound (
4) was more stable. These differences might be explained by the electron donating character of the amino group contrary to the electron withdrawing effect of the acids. Based on these results, 3-aminobenzenesulfonyl fluoride (
2) seemed to be the better choice for further library design than the less reactive
para analogue (
3). Among the carboxylic acids, both seemed advantageous, but the
meta substitution (
4) had moderate reactivity that might be considered a better choice. Acylation on the
meta amino group with phenylacetic acids and the addition of a methoxy to the benzenesulfonyl core (
6–
8 and
9–
11, respectively) increased the reactivity to t
1/2 = 10–19 h, which was generally not effected significantly by the substituents. This might also be an advantage during library design if the reactivity of the covalent warhead is stable regardless of the non-covalent part of the molecule. On the contrary, when acylating benzylamines with 3-carboxybenzenesulfonyl fluoride (
12–
14), the reactivity increased to t
1/2 = 4–5 h regardless of the substituents.
Interestingly, the additional methoxy group led to higher half-lives for compounds
15 and
16, while for the nitro-substituted sulfonyl fluoride (
17), no change could be seen. With these experiments, we show that to achieve constant or flexible reactivity, either the amino (
2) or the carboxy warhead (
4) can be chosen during the design of a fragment library. In order to detach the effect of the substituents and check other types of warhead connection through a longer linker, we have used 2-(4-(fluorosulfonyl)phenyl)acetic acid for acylating benzylamines. Compounds
18–
20 provided the lowest half-life values (t
1/2 = 1–4 h), suggesting the highest reactivity in parallel the lowest aqueous stability, which might limit the application of these compounds to rapid labelling approaches. We also presume that the large range of reactivity and stability of compounds
6–
20 (t
1/2 = 1–38 h) might enable fine tuning of the reaction with the target of interest. Notably, these results were in accordance with similar research published recently naming similar compounds to
meta-carboxy derivatives such as
12–
20, which exhibit desirable properties for applications in biological systems [
25].
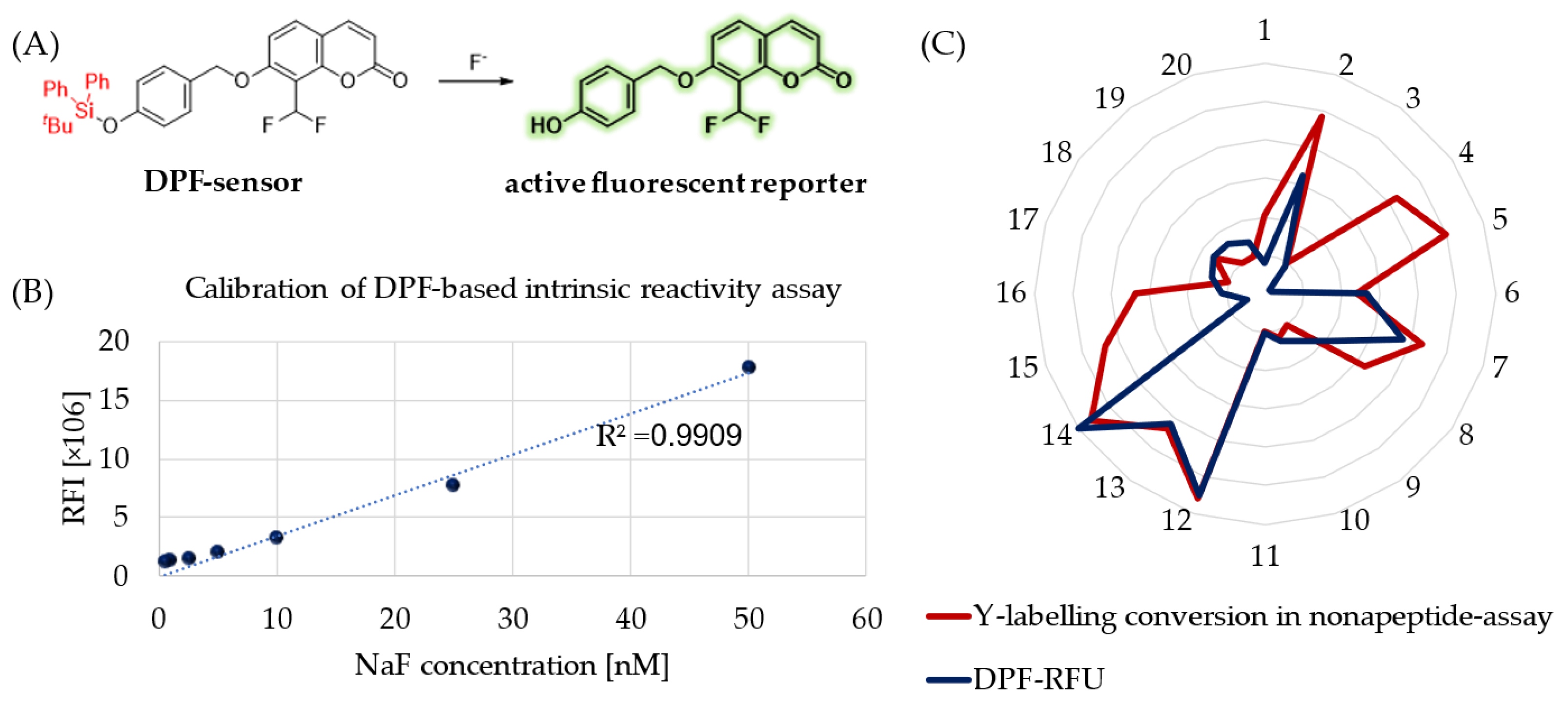
Besides hydrolytic characterization, we aimed to measure the intrinsic reactivity of sulfonyl fluoride probes against a tyrosine surrogate in physiological buffer (PBS, pH = 7.4). Therefore, we developed a 96-well-plate-based spectroscopic analysis applying DPF as a sensitive fluoride sensor molecule (
Figure 3A) [
31]. This molecule is a fluoride-dependent on–off fluorogenic probe, where a 4-(
tert-butyldiphenylsilyloxy) trigger group is attached to the latent reporter coumarin. Fluoride ions induce the removal of a silyl protecting trigger group on DPF (
Figure 3A). After calibration of the chromogenic assay with a NaF concentration series (
Figure 3B), we measured the amount of the released fluoride in the reaction of SuFBits with
N-acetyl tyrosine as a protein surrogate. After 15 minutes of incubation at room temperature, the release of the 7-hydroxy-8- formylcoumarin was monitored by measuring the fluorescence endpoint at 𝜆
ex = 360 nm and 𝜆
em = 445 nm. This measured-reactivity-related fluorescence intensity (
Supplementary Table S1) was then normalized and used as relative fluorescence units (RFU) to describe the reactivity of the library members.
To compare the assay outcome to a more complex surrogate, we performed a nonapeptide assay (KGDYHFPIC) developed in our group [
32] and took the direct conversion of the tyrosine-specific labelling events (
Figure 3C). We observed here a significant correlation between these orthogonal methods describing the tyrosine reactivity of the SuFBit probes. Interestingly, compounds
4,
5,
15 and
16 resulted in significant tyrosine labelling of the nonapeptide; however, they were completely unreactive based on the DPF assay. With all the other probes, the two parallel methods corresponded very well, resulting in probes
12 and
14 as the two strongest consensus hits (
Figure 3C).
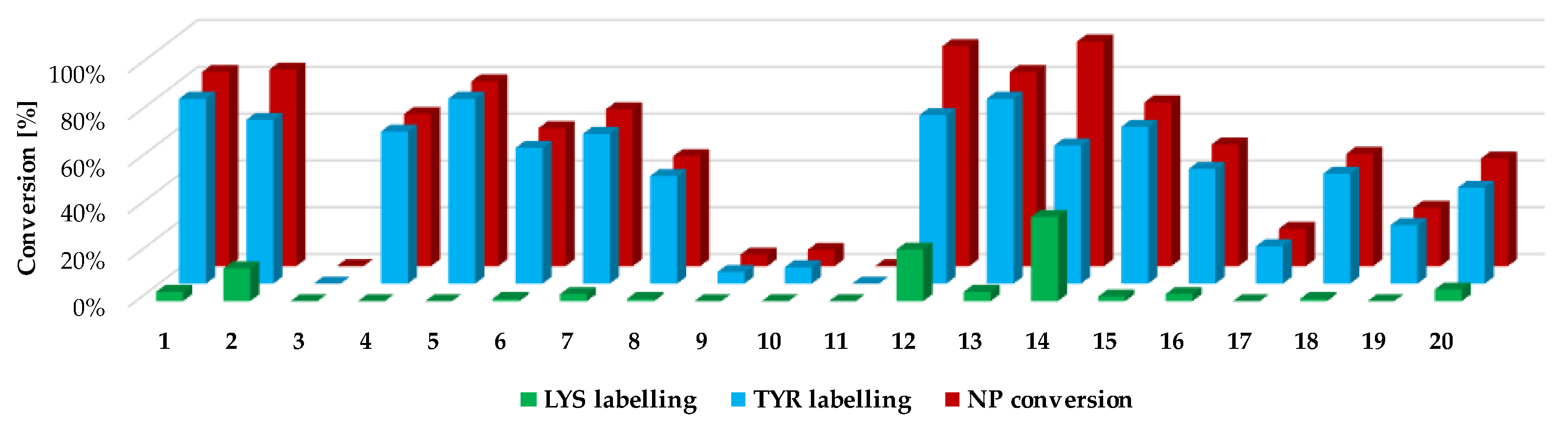
Next, we investigated the residue specificity of the labelling reaction. The nonapeptide contains several nucleophilic amino acid residues (Lys, His, Tyr and Cys); therefore, it is a useful surrogate for the estimation of labelling selectivity/promiscuity (number and proportion of labelled amino acid residues). In this assay, we detected labelling only on Tyr and Lys side chains, and in all cases the cysteines dimerized to disulphide bridges, forming nonapeptide dimers. This phenomenon is unique among the electrophilic chemotypes we have investigated up to now, although the assay conditions were the same as before [
32]. It should be noted that single lysine labelling was not observed; lysines were labelled only together the Tyr residues, suggesting that at first Tyr is labelled and lysine is the secondary target. Considering the reported promiscuity of sulfonyl fluorides, labelling multiple residues, including Cys, His, Thr and Ser, is expected in a protein environment. In the nonapeptide reactivity screening (
Figure 4 and
Supplementary Table S1), phenylsulfonyl fluoride (
1) showed 83% conversion and 4-aminophenylsulfonyl fluoride (
3) did not label the nonapeptide, while conversion was 84% in the case of 3-aminophenylsulfonyl fluoride (
2). The carboxylic acid substituents led to similar trends as that of the intrinsic reactivity assay, as sulfonyl fluoride
4 was less reactive than
5 (65% and 79%, respectively). Moving forward to the larger analogues, the lowest labelling efficacy was observed for
18–
20 (25–48% conversion), where the phenylsulfonyl fluoride is attached through an acetyl linker in the
para position. This might be explained by the high rate of the aqueous hydrolysis side reaction. Comparing this group to
12–
14, where the carboxyl group is directly attached to the phenylsulfonyl core in the
meta position, a significant increase in reactivity could be observed (83–96% conversion), resulting in the most reactive compounds from the whole library. Sulfonyl fluorides
12–
14 also had higher reactivity than that of the free carboxylic acid
4 (65%). Thus, it could be concluded that direct attachment in the
meta position is advantageous, and an aqueous half-life of 4–5 h is still tolerated in labelling reactions. Notably, the addition of a methoxy group to the phenylsulfonyl fluoride warhead significantly decreased the reactivity (
15–
17, 16–70%) compared to
12–
14. When the warhead was attached using 3-aminophenylsulfonyl fluoride (
6–
8), the conversion was moderate (47–67%), and adding the methoxy group similarly resulted in lower values; in particular, the compounds almost lost their ability for labelling (
9–
12, 0–7%). Comparing
6–
11 to
2 shows that increasing the size of
2 decreases the nonapeptide reactivity and thus the Tyr affinity. The nitro substituents distant from the sulfonyl fluoride (
8,
11,
14,
17 and
20) generally showed a decrease in the reactivity, while there was no regularity in the case of the methoxy-substituted analogues (
7,
10,
13,
16 and
19). It could be concluded that coupling through the carboxyl group results in more efficient labelling (average nonapeptide conversion 69 ± 30%) than coupling through the amine (31 ± 30%). The carboxyl modification keeps the originally observed reactivity and amino acid affinity of the phenylsulfonyl warhead. This might be rationalized by the similar electronic properties of amides and carboxylic acids, and the very different ones of amines and amide nitrogens. A recent work reporting a multicomponent synthesis of sulfonyl fluorides using 3-carboxyphenylsulfonyl fluoride gives further support to our observation [
15].
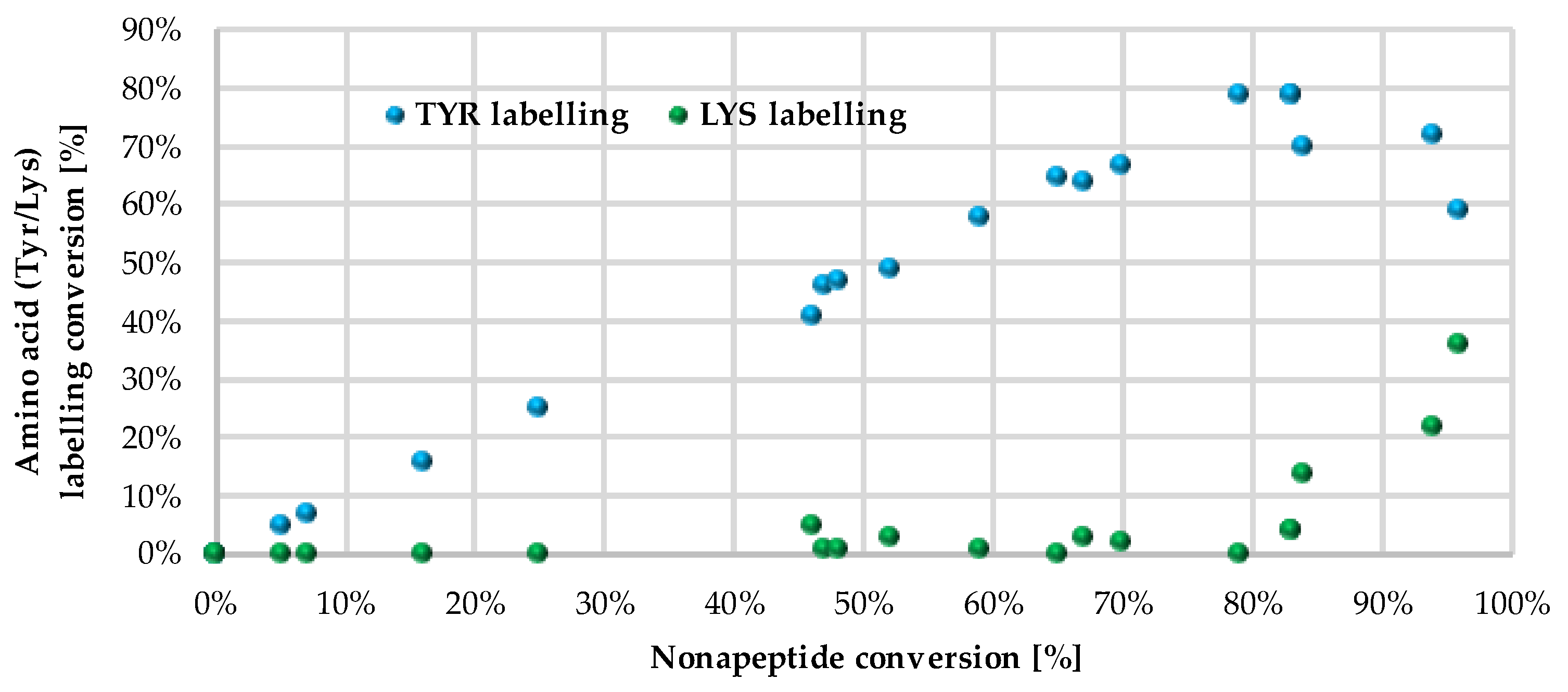
We followed up the oligopeptide assay by analysing the promiscuity of sulfonyl fluorides. Probe
14 labelled both nucleophilic residues (double labelling on the nonapeptide for Tyr and Lys) with significant yield (
Figure 4, blue and green columns,
Supplementary Table S1 and Supplementary Figure S2). The compounds with the highest reactivity usually showed 60–80% Tyr labelling, in addition to 10–40% Lys labelling with single and double adducts. Only
12 and
14 provided a considerate conversion of lysine labelling (22% and 36%, respectively). In general, sulfonyl fluorides reacted preferentially with Tyr and only the most reactive ones could label Lys. Comparing the nonapeptide conversions to the amino acid labelling percentage, the increase in Tyr labelling percentage changed linearly with the increase in the nonapeptide conversion, which suggests that nonapeptide labelling is more selective for tyrosine (
Figure 5).
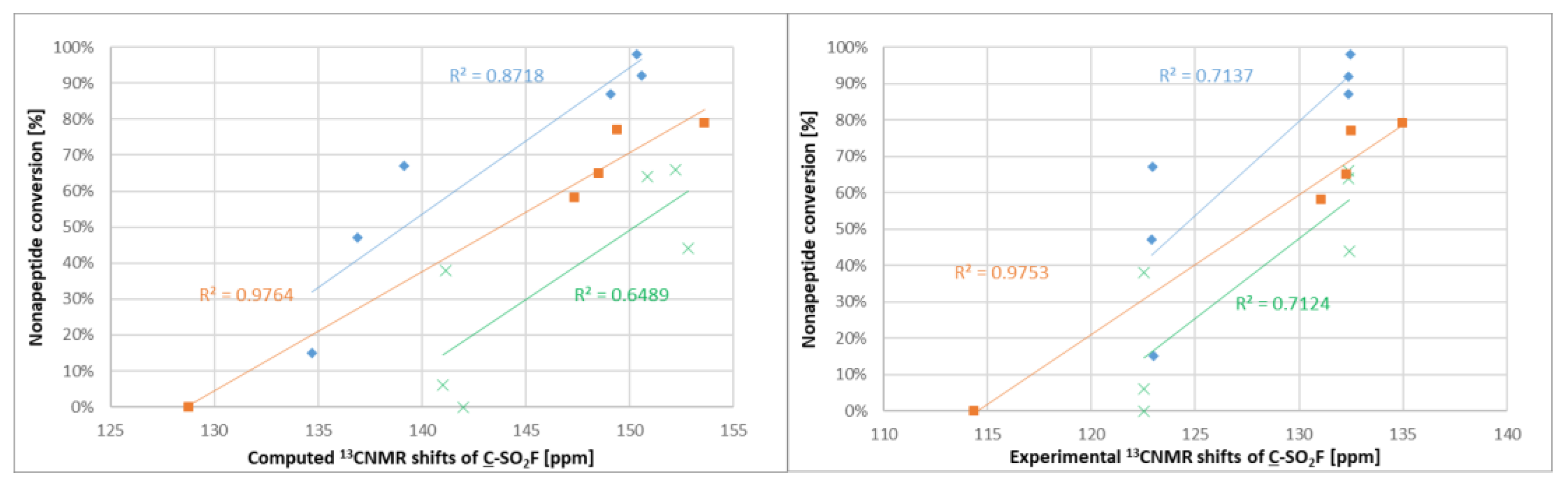
After experimental characterization of the library members, we aimed to find a descriptor that might be useful for reactivity estimation. Therefore,
19F NMR shifts were calculated for the sulfonyl fluorides, but there was no correlation observed between the
19F NMR shifts and the intrinsic reactivity or the nonapeptide conversion. However, when computing and measuring the
13C NMR shifts (
Figure S1) of the sulfonyl-attached aromatic carbon and comparing to the nonapeptide conversions, one could see that among the structurally similar compounds
1–
5 and
12–
17, there was a correlation between the NMR shifts and the reactivity; the more reactive compounds had a higher chemical shift (
Figure 6). A similar observation was found for compounds
6–
11. The methoxyphenylsulfonyl fluoride derivatives (
9–
11 and
15–
17) had systematically lower chemical shifts than the unsubstituted analogues
6–
8 and
12–
14.
Finally, we turned to confirm the biological application of the most potent SuFBit probes. To demonstrate efficient protein labelling, we selected the probes
12 and
14 and applied them to label Kirsten rat sarcoma oncogene mutant (KRas
G12D). KRas is part of the Ras protein family of membrane-bound GTPases, which act as molecular switches. Somatic KRas mutations are found in several cancers [
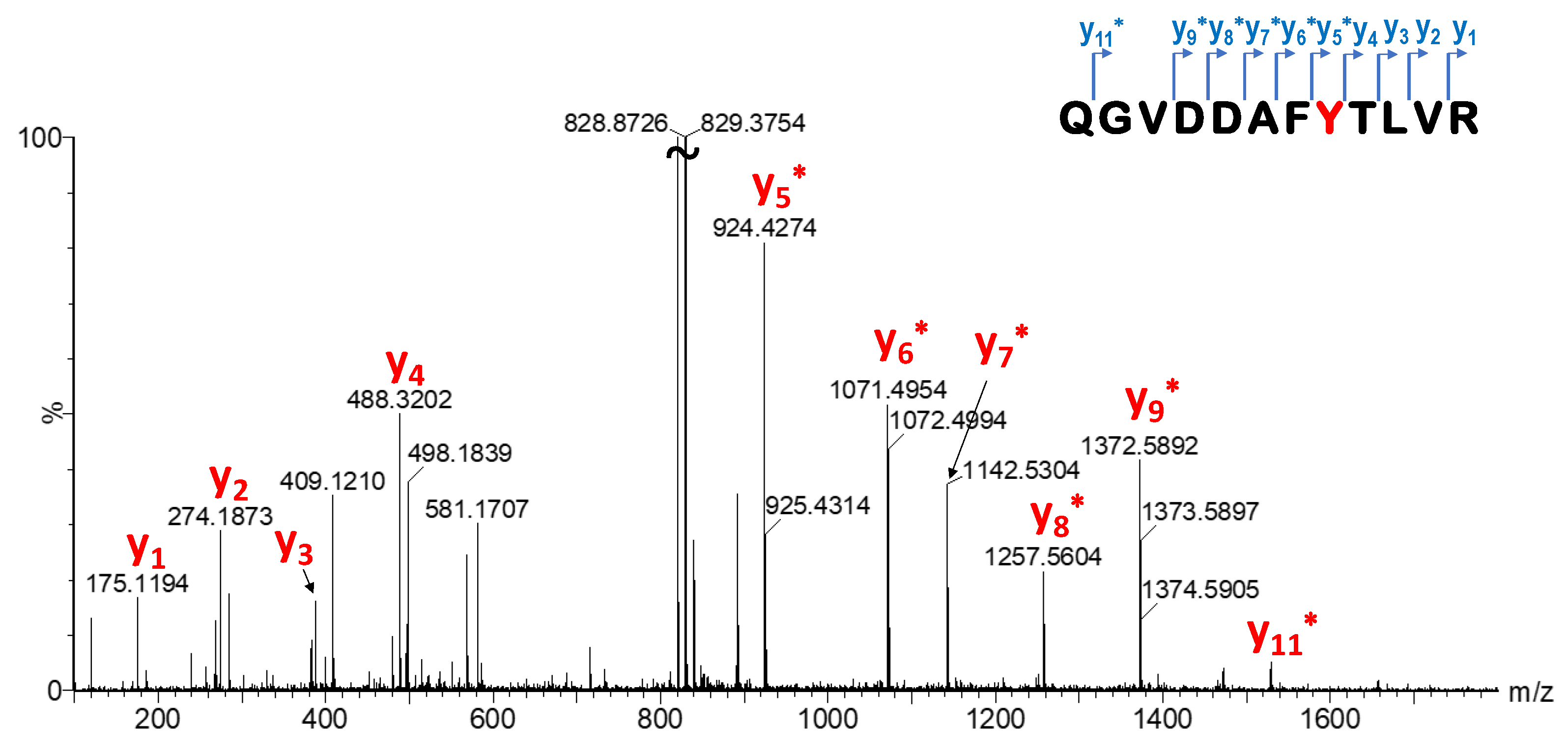
33]. The labelling efficiency of library members was investigated by MS measurements. After enzymatic digestion, single labelling located on Y157 was confirmed by LC-MS/MS peptide mapping (
Figure 7,
Supplementary Table S2, Supplementary Figures S3 and S4). This particular tyrosine is located on the edge of the SOS–KRas protein–protein interaction surface area, and there has not been confirmed covalent labelling at this residue until now. However, we should also note that Y157 is not a mutation-dependent residue.
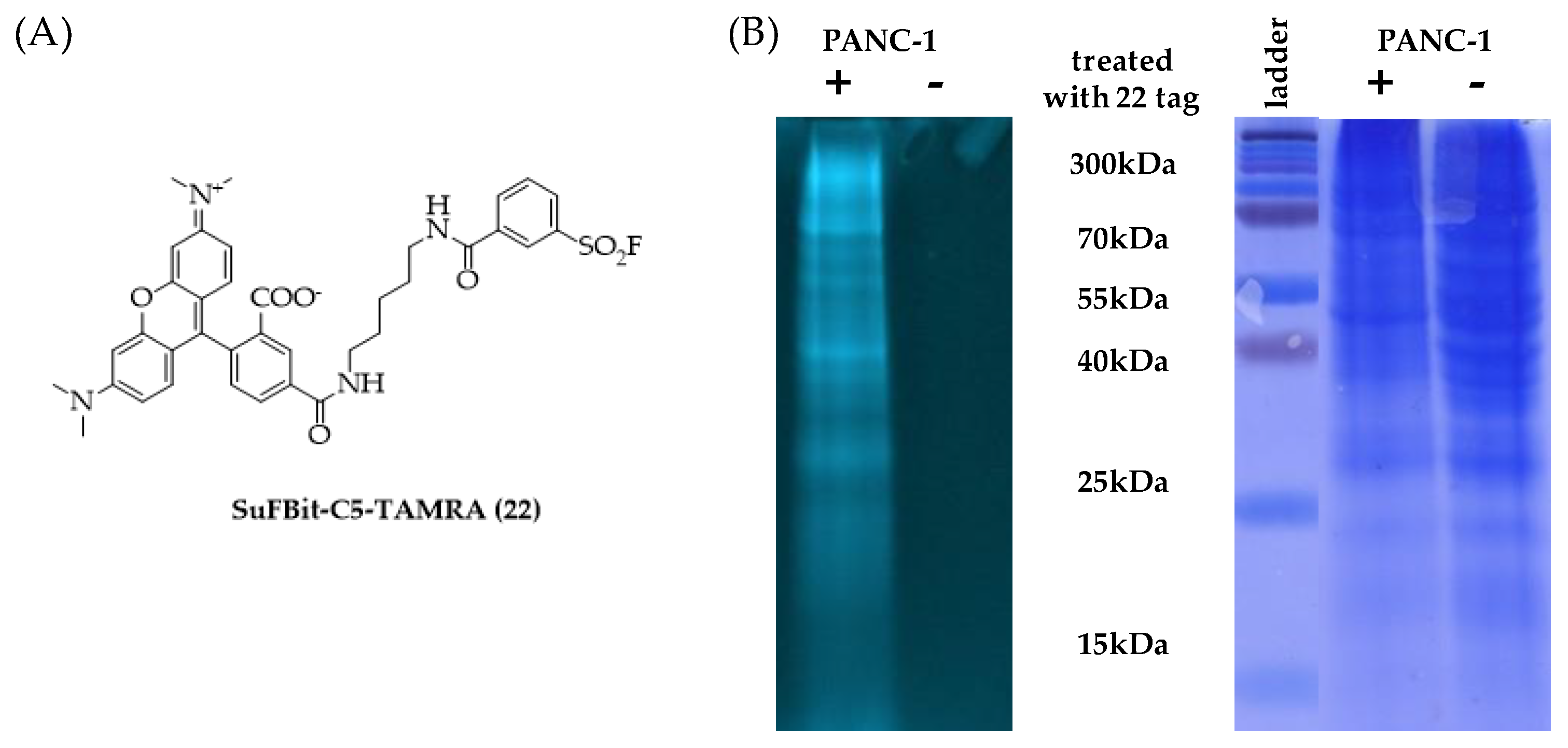
Finally, we moved forward to proteome labelling experiments to demonstrate the SuFBit approach. Therefore, we equipped the warhead moiety of probe
12 to a fluorescence reporter backbone and used this specific fluorescent tag to label PANC-1 cell lysate. After we synthesized the SuFBit-C5-TAMRA (
22) (
Figure 8A), we conducted labelling of PANC-1 cell lysate membrane fractions in the presence of 7 mM of probe
22, applying 2 h of incubation at 37 °C. Next, we applied a general gel electrophoresis protocol to achieve successful confirmation of the lysate labelling by SDS-PAGE analysis (
Figure 8B). To identify the exact modification sites, a MS proteomic study was performed. Finally, it was revealed that the SuFBit-derived fluorescent tag (
22) successfully labelled 18 proteins out of the identified 1529 in the cell lysate. Interestingly, most of the modified peptides were ribosomal proteins; however, some other targets could be identified. Vimentin was also labelled successfully, which was discovered recently to be an attachment factor for SARS-CoV-2 in the process of ACE2-dependent viral entry [
34]. Another identified target was the X-ray repair cross-complementing protein 6 (XRCC6), which is generally is required for DNA repair biochemical processes; furthermore, XRCC6 mutations have been described recently as a potential factor in the development of autism [
35]. However, we identified a moderate number of labelled proteins and we were able to covalently modify several proteins of PANC-1 cells, which might be valuable starting points for further developments in the field of therapeutics and diagnostics.
The SuFBit methodology and the results we demonstrated herein, identifying the possibly most prevalent targets of these electrophilic fragments to capture proteins in cells (full list of labelled proteins is shown in
Supplementary Table S3), might contribute to target validation efforts of early phase oncology programs.
3. Materials and Methods
NMR spectra were recorded in DMSO-d6 or CDCl3 solution at room temperature on a Varian Unity Inova 500 spectrometer (500 and 125 MHz for 1H, 13C and APT NMR spectra, respectively), with the residual solvent signal as the lock and TMS as the internal standard, or on a Bruker AVII600 (1H = 400 and 600 MHz, 19F = 376 MHz) referenced to residual nondeuterated solvent. Chemical shifts (δ) and coupling constants (J) are given in ppm and Hz, respectively. HPLC–MS measurements were performed using a Shimadzu LCMS-2020 device equipped with a Reprospher 100 C18 (5 µm; 100 × 3 mm) column and a positive-negative double ion source (DUIS±) with a quadrupole MS analyser in the range of 50–1000 m/z. The samples were eluted with gradient elution using eluent A (0.1% formic acid in water) and eluent B (0.1% formic acid in acetonitrile). The flow rate was set to 1.5 mL/min. The initial condition was 5% eluent B, followed by a linear gradient to 100% eluent B by 1.5 min, then from 1.5 to 4.0 min, 100% eluent B was retained and from 4.0 to 4.5 min, we reverted back to the initial condition of 5% eluent B which was retained until 5 min. The column temperature was kept at room temperature, and the injection volume was 1–10 µL. HPLC–MS/MS measurements were performed using an ExionLC AC instrument coupled with a Sciex 3500 triple quadrupole mass spectrometer with a Turbo V ion source in electrospray ionization mode. Chromatographic separation was achieved using a Phenomenex 2.6 μm, 100 × 4.6 mm column. Eluents were ultrapure water (A) and acetonitrile (B), both with 0.1% formic acid. The flow rate was 0.5 mL/min. Gradient elution was used, starting at 5% B. The initial ratio was kept for 2 min, then within 8 min, the ratio was raised to 95% B. It was kept there for another 3 min, then dropped back in 0.5 min to 5% and kept there for another 6.5 min for equilibration. The column temperature was kept at room temperature and the injection volume was 10 μL. The MS parameters were as follows: 450 °C source temperature, 5000 V ion spray voltage and 120 V declustering potential. Compressed air was used as the nebulizer gas (GS1) and heater gas (GS2), and nitrogen was used as curtain gas with values set at 35, 45 and 45, respectively. Gasses were provided by a Claind NiGen LCMS gas generator. Compounds 1–5 were purchased from Enamine, while 6–20 were synthesized. The purity of the compounds was assessed by HPLC with UV detection at 215 and 254 nm; all starting compounds were known, purchased or synthesized and >95% pure. LC-MS grade solvents were purchased from Merck (Darmstadt, Germany). RapiGest SF was obtained from Waters (Milford, MA, USA). Mass spectrometry grade trypsin and trypsin/Lys-C mix digestion enzymes were obtained from Promega (Promega Corporation, Madison, WI, USA). Reagents used for enzymatic digestion (1,4-Dithiothreitol (DTT) and iodoacetamide (IAA)) were purchased from Roche Diagnostics (Roche Diagnostics GmbH, Mannheim, DE) and Fluka Chemie GmbH (Buchs, CH). Other consumables (formic acid—FA, trifluoroacetic acid—TFA, heptafluorobutyric acid—HFBA and NH4HCO3) were from Merck Life Science Kft. (Merck KGaA., Darmstadt, Germany).
DFT computations at the M062X/6-311++G(3df,2p) level of theory were performed considering the solvent effect of the corresponding NMR solvent using the CPCM solvent model with the Gaussian 09 and Gaussian 16 program packages. The geometries of the molecules were optimized in all cases, and frequency calculations were also performed to assure that the structures were in a local minimum. The NMR spectra were computed using the GIAO method considering the built-in internal standard, TMS [
36,
37,
38,
39,
40,
41].
General method for the synthesis of sulfonyl fluorides
HATU (1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate, 0.209 g, 0.550 mmol) was added to a solution of the appropriate acid (0.458 mmol), amine (0.458 mmol, 1 equiv.) and DIPEA (0.160 mL, 0.917 mmol) in N,N-dimethylformamide (DMF) (4 mL). The reaction mixture was stirred at room temperature followed by LCMS. After 48 h, the mixture was concentrated under reduced pressure. The resulting crude material was partitioned between dichloromethane (10 mL) and water (10 mL) and the organic layers were passed through a phase separator. The solvent was removed by vacuum, and the crude product was purified by preparative HPLC.
3-(2-Phenylacetamido)benzenesulfonyl fluoride (6)
Yield: 98%, 1H NMR (400 MHz, DMSO-d6) δ 3.71 (s, 2 H) 7.25–7.42 (m, 6 H) 7.71–7.83 (m, 2 H) 8.52 (t, J = 2.0 Hz, 1 H) 10.74 (s, 1 H) ppm; 19F NMR (376 MHz, DMSO-d6) δ 66.03 (s, 1 F) ppm; 13C NMR (126 MHz, DMSO-d6) δ 170.55, 141.11, 135.74, 132.40 (d, J = 23.4 Hz), 131.60, 129.63, 128.81, 127.16, 126.70, 123.03, 117.95, 43.71 ppm. HRMS (ESI): (M + H)+ calcd. for C14H13NO3FS, 294.0600; found 294.0606.
3-(2-(4-Methoxyphenyl)acetamido)benzenesulfonyl fluoride (7)
Yield: 76%, 1H NMR (400 MHz, DMSO-d6) δ 3.63 (s, 2 H) 3.74 (s, 3 H) 6.87–6.94 (m, 2 H) 7.24–7.30 (m, 2 H) 7.71–7.83 (m, 3 H) 8.52 (t, J = 2.0 Hz, 1 H) 10.68 (s, 1 H) ppm; 19F NMR (376 MHz, DMSO-d6) δ 66.02 (s, 1 F) ppm; 13C NMR (126 MHz, DMSO-d6) δ 170.90, 158.62, 141.17, 132.39 (d, J = 23.4 Hz), 131.58, 130.64, 127.63, 126.67, 122.96, 117.93, 114.27, 55.51, 42.85 ppm. HRMS (ESI): (M + H)+ calcd. for C15H15NO4FS, 324.0705; found 324.0711.
3-(2-(4-Nitrophenyl)acetamido)benzenesulfonyl fluoride (8)
Yield: 52%, 1H NMR (400 MHz, DMSO-d6) δ 3.92 (s, 2 H) 7.63 (m, 4 H) 8.22 (m, 3 H) 8.51 (s, 1 H) 10.84 (s, 1 H) ppm; 19F NMR (376 MHz, DMSO-d6) δ 66.03 (s, 1 F) ppm; 13C NMR (126 MHz, DMSO-d6) δ 169.45, 146.96, 143.67, 140.90, 132.42 (d, J = 23.4 Hz), 131.66, 131.19, 126.79, 123.82, 123.65, 123.21, 118.06, 43.15 ppm. HRMS (ESI): (M + H)+ calcd. for C14H12N2O5FS, 339.0450; found 339.0467.
4-Methoxy-3-(2-phenylacetamido)benzenesulfonyl fluoride (9)
Yield: 98%, 1H NMR (400 MHz, DMSO-d6) δ 3.84 (s, 2 H) 4.03 (s, 3 H) 7.23–7.43 (m, 6 H) 7.84–7.89 (m, 1 H) 8.80–8.83 (m, 1 H) 9.79 (s, 1 H) ppm; 19F NMR (376 MHz, DMSO-d6) δ ppm 67.75 (s, 1 F) ppm; 13C NMR (126 MHz, DMSO-d6) δ 170.88, 155.38, 136.07, 129.69, 129.20, 128.78, 127.07, 126.01, 122.55 (d, J = 23.4 Hz), 120.03, 112.70, 57.29, 43.17 ppm. HRMS (ESI): (M + H)+ calcd. for C15H15NO4FS, 324.0705; found 324.0711.
4-Methoxy-3-(2-(4-methoxyphenyl)acetamido)benzenesulfonyl fluoride (10)
Yield: 98%, 1H NMR (400 MHz, DMSO-d6) δ 3.71–3.79 (m, 6 H) 4.02 (s, 2 H) 6.85–6.99 (m, 2 H) 7.19–7.31 (m, 2 H) 7.40 (d, J = 8.8 Hz, 1 H) 7.86 (dd, J = 8.7, 2.6 Hz, 1 H) 8.80 (d, J = 2.5 Hz, 1 H) 9.70 (s, 1 H) ppm; 19F NMR (376 MHz, DMSO-d6) δ 67.75 (s, 1 F) ppm; 13C NMR (126 MHz, DMSO-d6) δ 171.22, 158.58, 155.32; 130.70, 129.24, 127.93, 125.94, 122.55 (d, J = 24.0 Hz), 119.94, 114.25, 112.67, 57.29, 55.51, 42.31 ppm. HRMS (ESI): (M + H)+ calcd. for C16H17NO5FS, 354.0811; found 354.0819.
4-Methoxy-3-(2-(4-nitrophenyl)acetamido)benzenesulfonyl fluoride (11)
Yield: 20%, 1H NMR (400 MHz, DMSO-d6) δ 4.00–4.15 (m, 5 H) 7.40–7.45 (m, 1 H) 7.63 (d, J = 8.9 Hz, 2 H) 7.86–7.90 (m, 1 H) 8.19–8.24 (d, J = 8.9 Hz, 2 H) 8.79 (d, J = 2.5 Hz, 1 H) 9.99 (s, 1 H) ppm; 19F NMR (376 MHz, DMSO-d6) δ 67.76 (s, 1 F) ppm; 13C NMR (126 MHz, DMSO-d6) δ 169.81, 155.53, 146.89, 144.11, 131.13, 129.01, 126.23, 123.80, 122.55 (d, J = 24.0 Hz), 120.35, 112.80, 57.32, 42.74 ppm. HRMS (ESI): (M + H)+ calcd. for C15H14N2O6FS, 369.0556; found 369.0569.
3-(Benzylcarbamoyl)benzenesulfonyl fluoride (12)
Yield: 54%, 1H NMR (400 MHz, CDCl3-d) δ 4.70 (d, J = 5.9 Hz, 2 H) 6.50 (br s, 1 H) 7.40 (m, 4 H) 7.76 (t, J = 7.9 Hz, 1 H) 8.15–8.20 (m, 1 H) 8.26 (d, J = 7.9 Hz, 1 H) 8.40 (t, J = 1.7 Hz, 1 H) ppm; 19F NMR (376 MHz, CDCl3) δ 66.14 (s, 1 F) ppm; 13C NMR (126 MHz, DMSO-d6) δ 164.22, 139.46, 136.48, 135.74, 132.39 (d, J = 24.1 Hz), 131.34, 131.29, 128.80, 127.87, 127.39, 127.29, 43.43 ppm. HRMS (ESI): (M + H)+ calcd. for C14H13NO3FS, 294.0600; found 294.0606.
3-((4-Methoxybenzyl)carbamoyl)benzenesulfonyl fluoride (13)
Yield: 70%, 1H NMR (400 MHz, DMSO-d6) δ 3.74 (s, 3 H) 4.45 (d, J = 5.4 Hz, 2 H) 6.91 (d, J = 8.9 Hz, 2 H) 7.28 (d, J = 8.9 Hz, 2 H) 7.89–7.95 (m, 1 H) 8.31 (d, J = 8.4 Hz, 1 H) 8.43 (d, J = 7.9 Hz, 1 H) 8.58 (s, 1 H) 9.38 (br t, J = 5.9 Hz, 1 H) ppm; 19F NMR (376 MHz, DMSO-d6) δ 66.37 (s, 1 F); 13C NMR (126 MHz, DMSO-d6) δ 164.07, 158.81, 136.56, 135.72, 132.36 (d, J = 24.1 Hz), 131.41, 131.31, 131.24, 129.29, 127.25, 114.23, 55.54, 42.90 ppm. HRMS (ESI): (M + H)+ calcd. for C15H15NO4FS, 324.0705; found 324.0713.
3-((4-Nitrobenzyl)carbamoyl)benzenesulfonyl fluoride (14)
Yield: 34%, 1H NMR (400 MHz, CDCl3) δ ppm 4.79 (d, J = 5.9 Hz, 2 H) 6.93 (br s, 1 H) 7.52–7.58 (m, 2 H) 7.79 (t, J = 7.9 Hz, 1 H) 8.06–8.27 (m, 4 H) 8.29 (d, J = 7.9 Hz, 1 H) ppm; 19F NMR (376 MHz, CDCl3) δ 66.17 (s, 1 F) ppm; 13C NMR (126 MHz, DMSO-d6) δ 164.50, 147.52, 147.03, 136.16, 135.77, 132.45 (d, J = 24.1 Hz), 131.48, 131.42, 128.86, 127.32, 123.99, 43.00 ppm. HRMS (ESI): (M + H)+ calcd. for C14H12N2O5FS, 339.0450; found 339.0461.
3-(Benzylcarbamoyl)-4-methoxybenzenesulfonyl fluoride (15)
Yield: 91%, 1H NMR (400 MHz, DMSO-d6) δ 4.04 (s, 3 H) 4.52 (d, J = 6.4 Hz, 2 H) 7.26 (d, J = 3.9 Hz, 1 H) 7.35 (d, J = 4.4 Hz, 4 H) 7.52 (d, J = 8.9 Hz, 1 H) 8.22–8.29 (m, 2 H) 8.87–8.96 (m, 1H) ppm; 19F NMR (376 MHz, DMSO-d6) δ 67.83 (s, 1 F) ppm; 13C NMR (126 MHz, DMSO-d6) δ 163.55, 163.15, 139.57, 133.17, 130.93, 128.75, 127.58, 127.24, 125.88, 122.92 (d, J = 24.4 Hz), 114.53, 57.56, 43.24 ppm. HRMS (ESI): (M + H)+ calcd. for C15H15NO4FS, 324.0705; found 324.0714.
4-Methoxy-3-((4-methoxybenzyl)carbamoyl)benzenesulfonyl fluoride (16)
Yield: 56%, 1H NMR (400 MHz, DMSO-d6) δ 3.74 (s, 3 H) 4.03 (s, 3 H) 4.44 (d, J = 5.9 Hz, 2 H) 6.88–6.95 (m, 2 H) 7.25–7.31 (m, 2 H) 7.50 (d, J = 8.4 Hz, 1 H) 8.21–8.27 (m, 2 H) 8.80–8.88 (m, 1 H) ppm; 19F NMR (376 MHz, DMSO-d6) δ 67.83 (s, 1 F) ppm; 13C NMR (126 MHz, DMSO-d6) δ 163.41, 163.12, 158.70, 133.12, 131.50, 130.88, 128.97, 125.97, 122.90 (d, J = 24.6 Hz), 114.50, 114.19, 57.55, 55.53, 42.70 ppm. HRMS (ESI): (M + H)+ calcd. for C16H17NO5FS, 354.0811; found 354.0820.
4-Nitro-3-((4-methoxybenzyl)carbamoyl)benzenesulfonyl fluoride (17)
Yield: 28%, 1H NMR (400 MHz, DMSO-d6) δ 4.07 (s, 3 H) 4.64 (d, J = 5.9 Hz, 2 H) 7.54 (d, J = 8.9 Hz, 1 H) 7.62 (d, J = 8.9 Hz, 2 H) 8.23 (d, J = 8.9 Hz, 4 H) 9.09 (br t, J = 5.9 Hz, 1 H) ppm; 19F NMR (376 MHz, DMSO-d6) δ 67.81 (s, 1 F) ppm; 13C NMR (126 MHz, DMSO-d6) δ 163.81, 163.20, 147.75, 146.93, 133.35, 131.03, 128.60, 125.48, 123.95, 123.00 (d, J = 24.6 Hz), 114.59, 57.63, 42.95 ppm. HRMS (ESI): (M + H)+ calcd. for C15H14N2O6FS, 369.0556; found 369.0566.
4-(2-(Benzylamino)-2-oxoethyl)benzenesulfonyl fluoride (18)
Yield: 41%, 1H NMR (400 MHz, CDCl3) δ 3.70 (s, 2 H) 4.47 (d, J = 5.4 Hz, 2 H) 5.85 (br s, 1 H) 7.24–7.38 (m, 5 H) 7.58 (d, J = 8.4 Hz, 2 H) 7.95–8.04 (m, 2 H) ppm; 19F NMR (376 MHz, CDCl3) δ 66.10 (1 F) ppm; 13C NMR (101 MHz, CDCl3) 43.32, 44.05, 127.82, 128.84, 130.04 (d, J = 24.6 Hz), 130.55, 131.80, 132.04, 137.65, 143.33, 168.58 ppm. HRMS (ESI): (M + H)+ calcd. for C15H15NO3FS, 308.0756; found 308.0766.
4-(2-((4-Methoxybenzyl)amino)-2-oxoethyl)benzenesulfonyl fluoride (19)
Yield: 22%, 1H NMR (400 MHz, DMSO-d6) δ 2.51 (t, J = 2.0 Hz, 2 H) 3.73 (s, 3 H) 4.22 (d, J = 5.4 Hz, 2 H) 6.85–6.91 (m, 2 H) 7.14–7.22 (m, 2 H) 7.68 (d, J = 8.4 Hz, 2 H) 8.06–8.12 (m, 2 H) 8.60 (br t, J = 5.7 Hz, 1 H) ppm; 19F NMR (376 MHz, DMSO-d6) δ 66.62 (1 F) ppm; 13C NMR (101 MHz, DMSO-d6) δ 42.32, 42.52, 55.55, 114.20, 128.79, 129.12, 130.01 (d, J = 23.4 Hz), 131.50, 131.53, 146.46, 158.75, 169.05 ppm. HRMS (ESI): (M + H)+ calcd. for C16H17NO4FS, 338.0862; found 338.0873.
4-(2-((4-Nitrobenzyl)amino)-2-oxoethyl)benzenesulfonyl fluoride (20)
Yield: 28%, 1H NMR (400 MHz, CDCl3) δ 3.76 (s, 2 H) 4.57 (d, J = 5.9 Hz, 2 H) 5.95 (br s, 1 H) 7.36–7.51 (m, 2 H) 7.60 (d, J = 7.9 Hz, 2 H) 7.96 -8.09 (m, 2 H) 8.13–8.28 (m, 2 H) ppm; 19F NMR (376 MHz, CDCl3) δ 66.05 (1 F) ppm; 13C NMR (125 MHz, DMSO-d6) 169.58, 147.81, 146.95, 146.12, 131.56, 130.11 (d, J = 24.6 Hz), 128.83, 128.67, 123.94, 42.38, 42.37 ppm. HRMS (ESI): (M + H)+ calcd. for C15H14N2O5FS, 353.0607; found 353.0620.
2-(6-(Dimethylamino)-3-(dimethyliminio)-3H-xanthen-9-yl)-5-((5-(3-(fluorosulfonyl)benzamido)pentyl)carbamoyl)benzoate (22)
1H NMR (500 MHz, DMSO-d6) δ 8.83 (s, 1H), 8.73–8.66 (m, 2H) 8.54 (t, J = 5.6 Hz, 2H), 8.27 (d, J = 7.9 Hz, 1H), 8.11 (d, J = 1.8 Hz, 1H), 7.77 (dt, J = 7.9, 1.4 Hz, 2H), 7.71 (dt, J = 7.8, 1.5 Hz, 1H), 7.37 (t, J = 7.7 Hz, 2H), 7.03 (broaden s, 2H), 3.06 (s, 6H), 2.64 (t, J = 2.0 Hz, 2H), 2.37 (t, J = 1.5, 2H), 1.66–1.57 (m, 4H), 1.44–1.38 (m, 2H), 1.25 (s, 6H) ppm. 13C NMR (125 MHz, DMSO-d6) 171.0; 167.5; 154.6; 150.8; 142.4; 139.8; 135.3; 134.9; 134.1; 134.0; 133.2; 132.9; 130.9; 130.1; 128.9; 128.5; 127.2; 123.3; 113.9; 105.1; 100.2; 96.7; 54.5; 42.4; 17.3; 15.9; 11.7 ppm. HRMS (ESI): (M + H)+ calcd. for C37H38N4O7SF+, 701.2439; found, 701.2432.
Intrinsic reactivity assay
In an LC-MS vial, 800 uL PBS (pH 7.4) buffer, 188 uL DMSO, 6 uL 100 mM 1,4-dicyanobenzene as internal standard in DMSO and 6 uL 100 mM sulfonyl fluoride in DMSO was mixed. The final concentration of the sulfonyl fluoride was 600 uM. The final mixture was analysed by HPLC-MS after 0, 1, 2, 3, 6, 12 and 24 h time intervals. The AUC (area under the curve) values were determined via integration of HPLC spectra, then corrected with internal standard. The fragment AUC values were applied for ordinary least squares (OLS) linear regression and for computing the important parameters (kinetic rate constant and half-life time), a programmed excel (Visual Basic for Applications) was utilized. The data are expressed as means of duplicate determinations, and the standard deviations were within 15% of the given values.
Nonapeptide reactivity and promiscuity assay
For the nonapeptide assay, two initial solutions were prepared: a 1 mM solution of the fragment in dimethyl sulfoxide (DMSO) (A) and a 100 μM solution of the nonapeptide in PBS buffer (pH 7.4) (B). The reaction mixture contained 10 μL A, 50 μL B and 40 μL PBS. The samples were incubated at room temperature for 16 hours. An information-dependent acquisition (IDA) LC-MS/MS experiment was used to identify the number of bindings and their sites. MS scanning was applied as a survey scan and product ion was the dependent scan. The collision energy in the product ion scan was set to 30 eV. The reference data for identification were provided by the GPMAW 12.20 software. The relative quantitation of the nonapeptide and its fragment covalent conjugates was calculated from the total ion chromatograms. For every component, three corresponding ions were selected and their sums of area under the curve were used.
Chromogenic intrinsic reactivity assay with DPF sensor
Stock solutions of the DPF sensor and the compounds were prepared in DMSO. In the assay, N-acetyl-tyrosine (1 mM) and DBU (1 mM) were incubated for 10 min at 25 °C. The SuFex compounds (50 μM) were added to the plate, followed by the DPF sensor (50 μM). After 15 minutes incubation at room temperature, the release of 7-hydroxy-8-formylcoumarin was monitored by measuring the fluorescence endpoint at 𝜆ex = 360 nm and 𝜆em = 445 nm. The fluorescent reactivity assay was measured by a SpectraMax iD5 Multi-Mode Microplate Reader (Molecular Devices, San Jose, CA, USA).
LC-MS/MS peptide mapping analysis
Mass spectrometric experiments were performed on a high-resolution hybrid quadrupole-time-of-flight mass spectrometer (Waters Select Series Cyclic IMS, Waters Corp., Wilmslow, U.K.). The mass spectrometer operated in positive V mode. Leucine enkephalin was used as the Lock Mass standard. Chromatographic separations were performed on a Waters Acquity I-Class UPLC system coupled directly to the mass spectrometer. Modification sites on the KRasG12D protein were determined by RPLC-MS/MS peptide mapping after proteolysis using trypsin. Briefly, proteins were enzymatically digested after buffer exchange using Amicon Ultra-0.5 mL Centrifugal Filter units (10 KDa, Merck Millipore). Protein samples were reduced by dithiothreitol at 37 °C for 30 min. Tryptic cleavage was performed in 50 mM NH4HCO3 solution. A trypsin–LysC mixture (Promega Corporation, Madison, WI, USA) was used for enzymatic digestion. Proteins were digested for 4 h using a 1:50 enzyme:protein ratio at 37 °C. Digestion was stopped by adding formic acid in a final concentration of 0.2% (V/V). Gradient elution was performed on a Waters Acquity CSH Peptide C18 UPLC column (2.1 × 150 mm, 1.7 µm) under the following parameters: mobile phase “A”: 0.1% formic acid in water, mobile phase “B”: 0.1% formic acid in acetonitrile; flow rate: 300 µL/min; column temperature: 60 °C; gradient: 2 min: 2% B, 80 min: 45% B, 81 min: 85% B. HDMSE experiments were performed using collision voltage ramping in the transfer cell under the following parameters: m/z 50–2000; scan time: 0.3 sec; single Lock Mass: leucine enkephalin; low energy: 6 V; high energy: ramping 19–45 V. The cyclic ion mobility cell was operated in single pass mode. BiopharmaLynx 1.3.5 software (Waters Corp., Wilmslow, U.K.) was used for data analysis.
Production of PANC-1 cell lysates
Cells were grown in RPMI 1640 medium at 37 °C in a humidified atmosphere with 5% CO2, as described. When the confluency of pancreatic cancer cells, PANC-1, bearing a KRasG12D mutation reached 70%, the growth medium was discarded and cells were washed twice with phosphate-buffered saline (PBS; Biosera), harvested in 5 mL PBS (Biosera) using a cell scraper (Sarstedt) and collected in a 15 mL conical tube (Sarstedt). Then, cells were centrifuged for 5 min at 1000× g at 4 °C (Eppendorf 5804R centrifuge, Eppendorf AG, Hamburg, Germany), the supernatant was removed, and to the pellet was added a protease and phosphatase inhibitor cocktail mix (Thermo Scientific, Rockford, IL, USA). The cell pellet was resuspended in a 4× volume of PBS (Biosera) on ice, transferred into a 1.5ml tube (Corning, Reynosa, Mexico) and homogenized using a probe sonicator (model Q55, QSonica, Newtown, CT, USA), 10× for 1 sec at 20% power, on ice. After this, centrifugation was applied for 5 min at 12,000× g at 4 °C (Heraeus Biofuge Fresca centrifuge, Kendro Laboratory Products, Osterode, Germany) to pellet nuclei and unbroken cells. The supernatant was transferred to an ultracentrifuge sealing tube (OptiSeal, Beckman Coulter, Brea, CA, United States) and centrifuged for 1 h at 100,000× g at 4 °C (Optima MAX-XP Ultracentrifuge, Beckman Coulter) to separate the membrane (pellet) and soluble (supernatant) protein fractions. The supernatant fraction was transferred to a clean 1.5ml tube (Corning), while the pellet was washed with cold PBS and resuspend in cold PBS (Biosera). The protein concentrations in the membrane and the soluble fractions were quantified by a Pierce BCA protein assay kit (Thermo Scientific) according to the manufacturer’s protocol, and the protein concentration was adjusted to the desired concentration (1–2 mg/mL) with cold PBS (Biosera). Samples were aliquoted in a volume of 50 µL and stored at −80 °C. Prior to ABPP probe labelling, samples were thawed on ice and briefly allowed to warm up to room temperature.
Fluorogenic labelling of PANC-1 cell lysates
Membrane fractions of pancreatic cancer cells (PANC-1) lysates, bearing the KRasG12D mutation, were applied for labelling with the SuFBit-C5-TAMRA (22) fluorescent tag as follows. An amount of 75 µL of 1 mg/mL lysate solutions in PBS buffer were treated with 5.6 µL of 100 mM SF-C5-TAMRA (22) in DMSO. The reaction mixture was incubated at 37 °C for 2 h. Next, 25 µL was submitted for SDS-PAGE analysis and 50 µL of the resulting mixture was submitted for MS analysis.
Cell lysate analysis by SDS-PAGE
Polyacrylamide gel electrophoresis was performed with an mPAGE®TurboMix Bis-Tris Gel Casting Kit (TMKIT, Merck & Co., Rahway, NJ, USA), following standard lab procedures. An 8% resolving gel and a 4% stacking gel were used and a broad-range MW marker (4.6–300 kDa, ProSieve QuadColor Protein Marker, Lonza) was co-run to estimate protein weights. Samples were mixed with the loading buffer in a 3:1 ratio (composition for 6 × SDS: 1 g SDS, 3 mL glycerol, 6 mL 0.5 M Tris buffer pH = 6.8 and 2 mg Coomassie blue R250 in 10 mL) and heated at 65 °C for 5 minutes. Samples were subsequently loaded into the wells. All gels were run at a constant 200 mA for 60 minutes. Fluorescent gel images were recorded with a Bio-Rad Gel Doc XR+ (Bio-Rad Laboratories, Hercules, CA, USA), applying 𝜆ex = 542 nm and 𝜆em = 568 nm. Gels were then stained using a Coomassie stain (0.12 g Coomassie blue G-250, 0.10 g Coomassie blue R-250, 500 mL MeOH, 400 mL distilled water and 100 mL acetic acid), and after washing, it was rested at room temperature for 16 h in a water–ethanol mixture.
Cell lysate analysis by mass spectrometry
The sample solvent was exchanged to 50 mM NH
4HCO
3 solution using a Millipore Amicon Ultra 10 kDa centrifugation filter. The filters were rinsed with LC-MS water for 10 min (13,500 rpm at 4 °C). Then, the samples were added and their volume was made up to 200 μL with 200 mM NH
4HCO
3 solution and centrifuged for 10 min (13,500 rpm at 4 °C). Two additional cycles (10 min, 13,500 rpm, 4 °C) were performed, the first one by adding 200 μL 200 mM NH
4HCO
3 solution and the second one using 200 μL 50 mM NH
4HCO
3 solution. The samples were digested in solution using trypsin, as previously described with minor modifications [
42]. In brief, portions of each sample, each containing approximately 10 µg of protein, were diluted with water and methanol to a final volume of 25 µL, with a final methanol concentration of 5 %. Denaturation of the proteins was performed by adding 0.5% RapiGest SF (5 µL). For S-S bridge reduction, DTT was added at a final concentration of 12.5 mM and incubated at 60 °C for 30 min. For alkylation, IAA was added at a final concentration of 12.5 mM and incubated in the dark at room temperature for 30 min. Enzymatic digestion was performed with the addition of 2 µL of 0.25 µg/µL trypsin/Lys-C mix, with an incubation time of 1 hour at 37 °C, followed by the addition of 2 µL of 1 µg/µL trypsin, incubated for 2 h at 37 °C. The reaction was quenched by the addition of 1.5 μL formic acid and the sample was dried in a SpeedVac at 50 °C and dissolved in 50 µL 0.1% TFA prior to cleaning. Peptide clean-up and desalting were performed on Pierce C18 spin columns (Thermo Fisher Scientific, Waltham, MA, USA). Briefly, column conditioning and equilibration was performed using 2 × 200 µL 50% MeOH and 2 × 200 µL 0.5% TFA and 5% acetonitrile (AcN), respectively. The column was washed with 2 × 200 µL 0.1% HFBA. Then, the sample, reconstituted in 50 µL 0.1% HFBA, was applied and the flowthrough was reapplied to the column. HFBA (0.1%, 2 × 100 µL) was used for sample washing. Finally, sample elution was performed using 2 × 50 µL 0.1% TFA and 70% AcN and 50 µL 0.1% FA and 70% AcN. In each step, centrifugation was carried out for 2 min at 2000 rpm. Then, the sample was dried in a SpeedVac at 50 °C.
Mass spectrometry measurements were performed on a Maxis II ETD Q-TOF (Bruker Daltonics, Bremen, Germany) equipped with a CaptiveSpray nanoBooster ion source coupled to an Ultimate 3000 nanoRSLC system (Dionex, Sunnyvale, CA, USA). Samples were dissolved in 2% can and 0.1% FA, and in each run, 2 µg protein was injected onto an Acclaim PepMap100 C-18 trap column (100 Å, 5 µm, 100 µm × 20 mm, Thermo Fisher Scientific, Sunnyvale, CA, USA) for sample desalting. Peptides were separated on an ACQUITY UPLC M-Class Peptide BEH C18 column (130 Å, 1.7 µm, 75 µm × 250 mm, Waters, Milford, MA, USA) at 48 °C, applying gradient elution (2% B from 0 to 11 min, followed by a 120 min gradient to 50% B). Eluent A consisted of water + 0.1% formic acid, while eluent B was acetonitrile + 0.1% formic acid. MS spectra were recorded over a mass range of 150–2200 m/z at 3 Hz, while the CID was performed at 16 Hz for abundant precursor ions and at 4 Hz for low abundance ones. Sodium formate was used as an internal standard and raw data were recalibrated by the Compass Data Analysis software 4.3 (Bruker Daltonik GmbH, Bremen, Germany).
Proteins were identified by searching against the SwissProt Human (downloaded: 12/08/2022) database with addition of a mutated sequence of the RASK_HUMAN (P01116) protein (G12 → D12) using the Byonic (v3.5.0, Protein Metrics Inc., Cupertino, CA, USA) software search engine. The LC-MS/MS results were searched in Byonic with the following parameters: 4 ppm peptide mass tolerance, 20 ppm fragment mass tolerance, 2 missed cleavages, trypsin as the enzyme, carbamidomethylation of cysteines as the fixed modification, ammonia loss (N-term C), oxidation (M), acetyl (Protein N-term), Glu->Pyro-Glu (N-term E), Gln->Pyro-Glu (N-term Q) and carbamidomethylation (C) as the variable modification. SuFBit-C5-TAMRA (22) was added as a custom modification of +680.230473 Da mass shift, at the following possible sites: N-term, K, R, Q, S, T, Y. The requirements for protein identification were set in Scaffold 4.10 (Proteome Software, USA) with the following parameters: LFDR-rescoring on 95% peptide confidence threshold, minimum of two unique peptides and a protein-level False Discovery Rate of 1%.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}