
[3+3]-Annulation of Cyclic Nitronates with Vinyl Diazoacetates: Diastereoselective Synthesis of Partially Saturated [1,2]Oxazino[2,3-b][1,2]oxazines and Their Base-Promoted Ring Contraction to Pyrrolo[1,2-b][1,2]oxazine Derivatives

Abstract
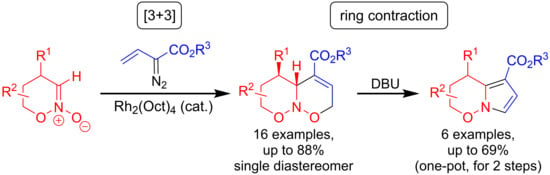
:
1. Introduction
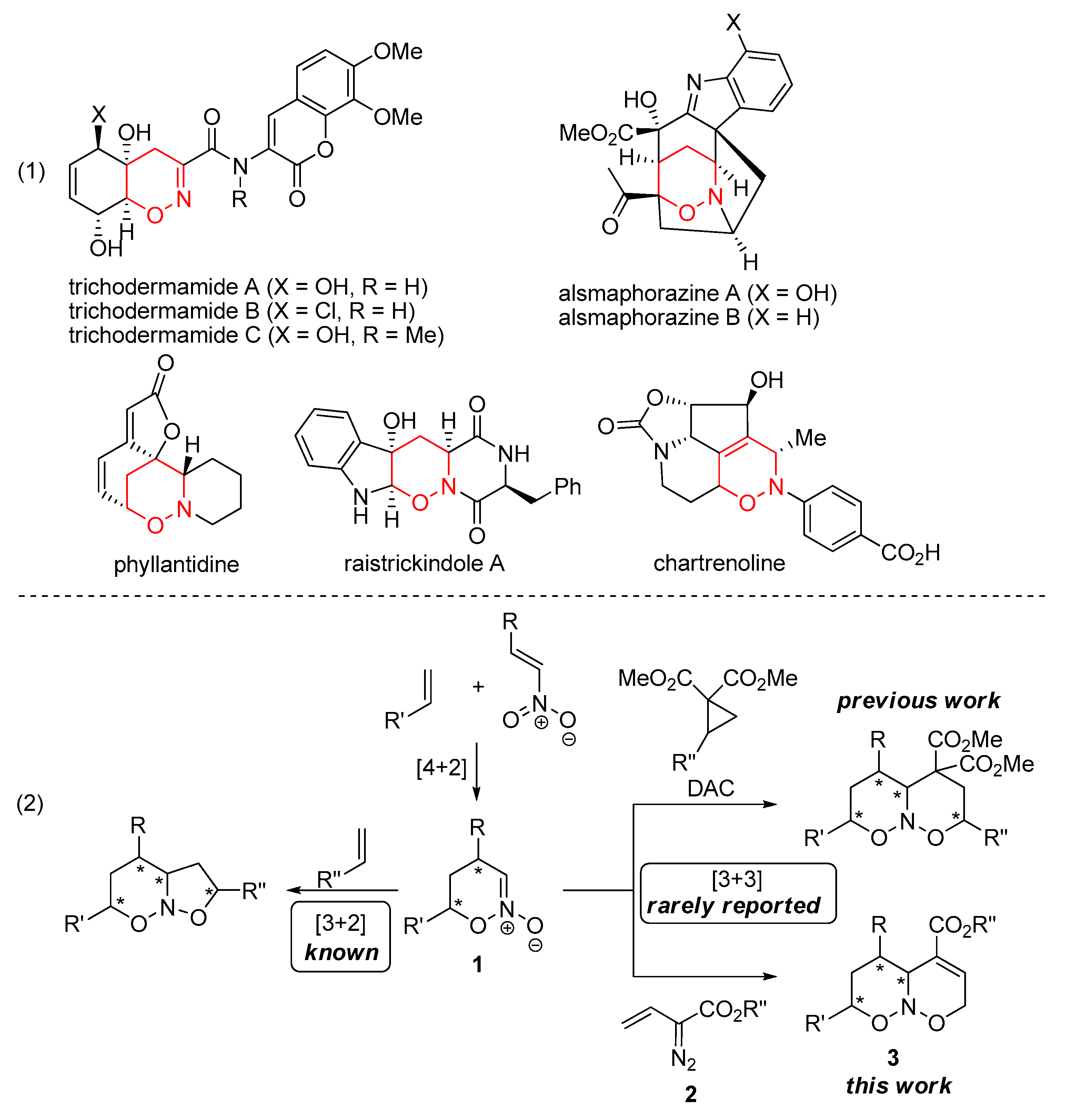
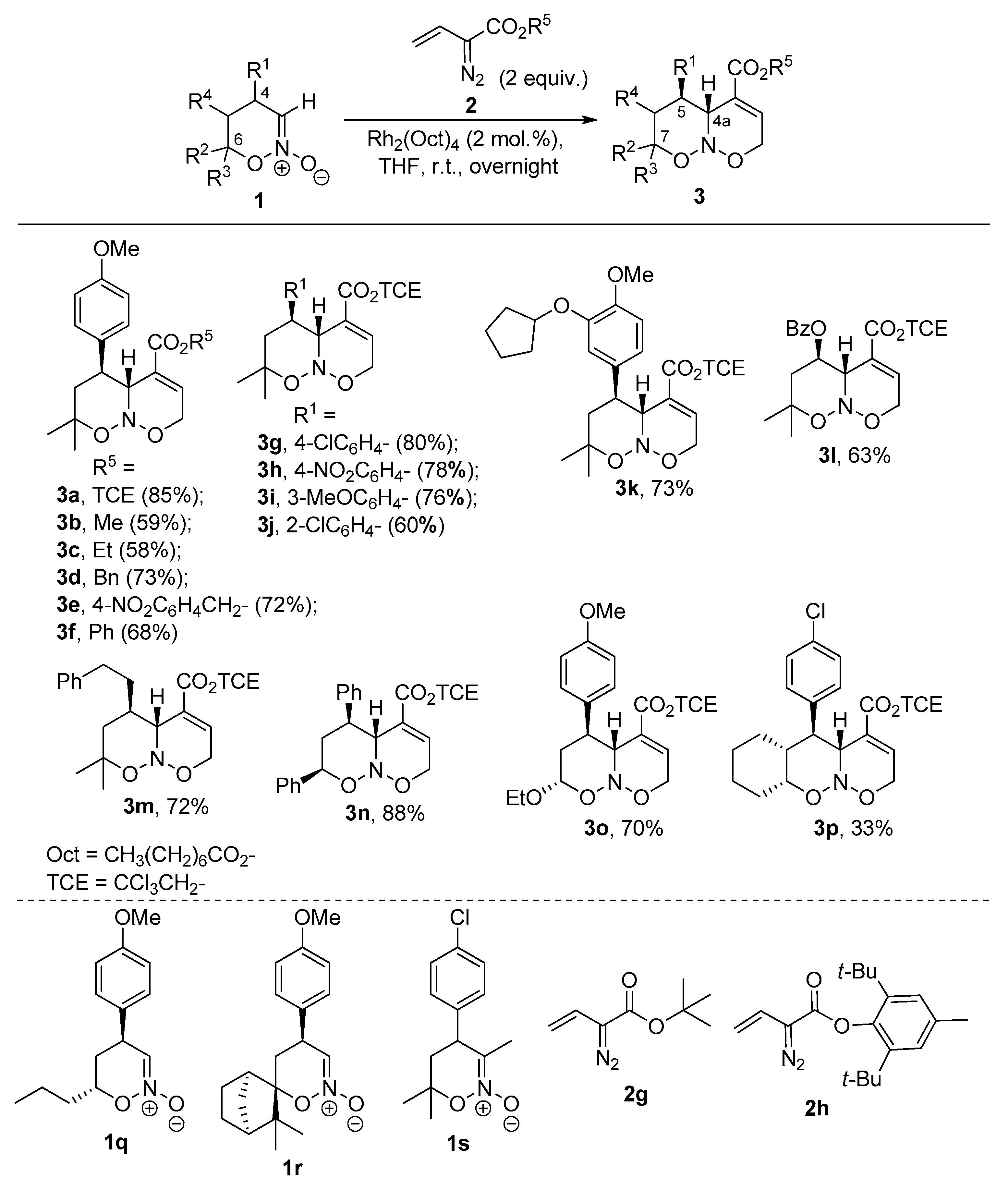
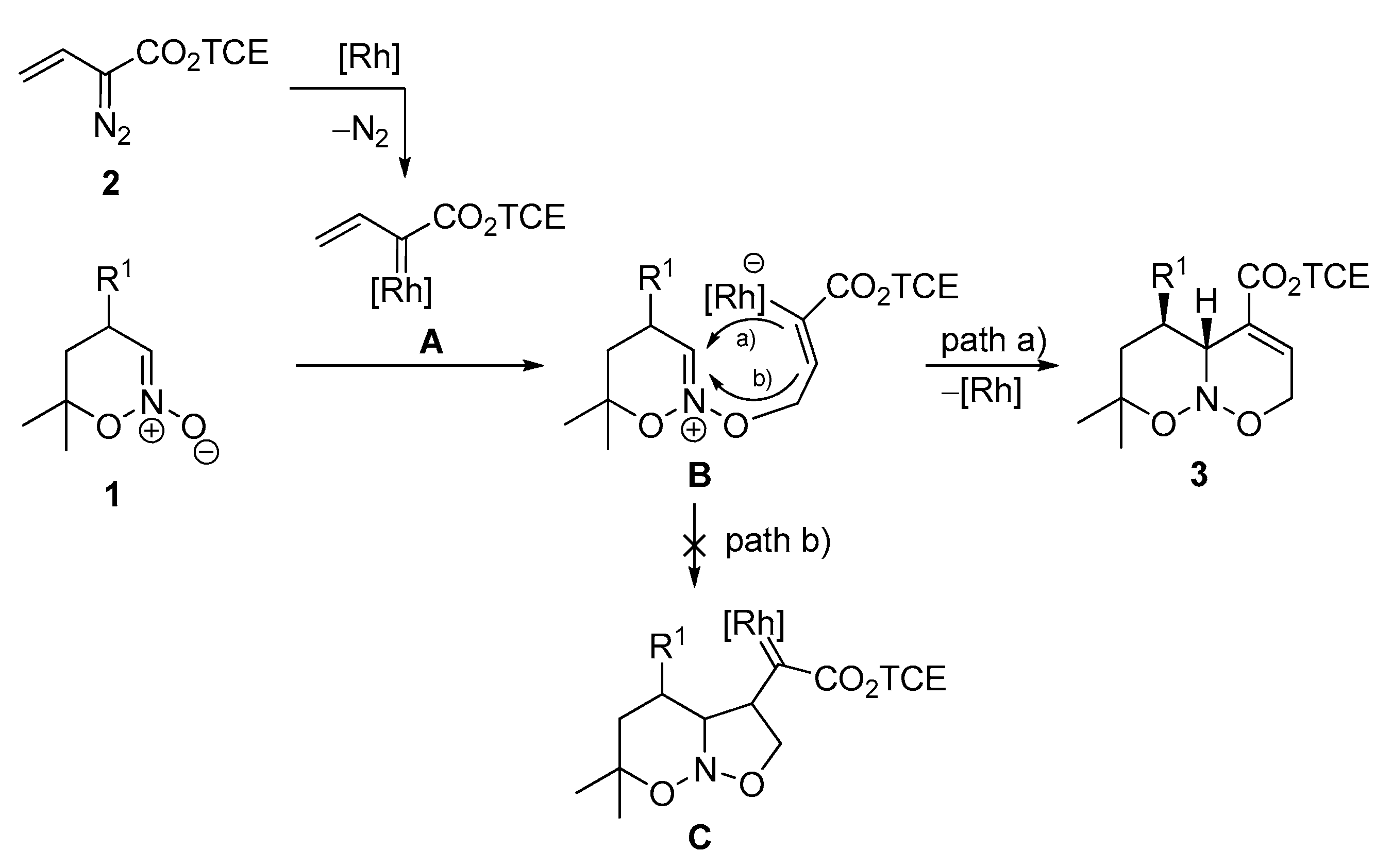
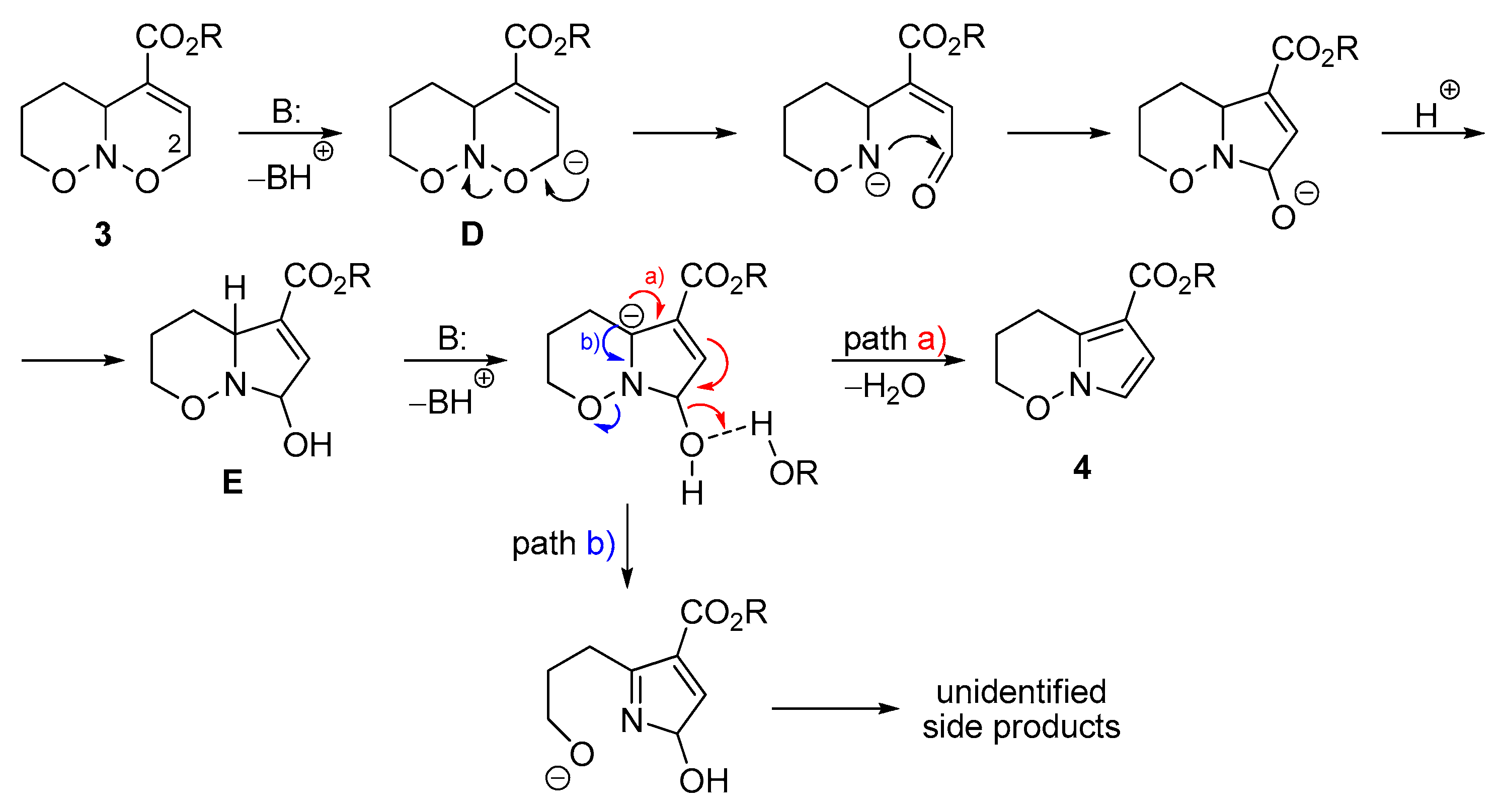
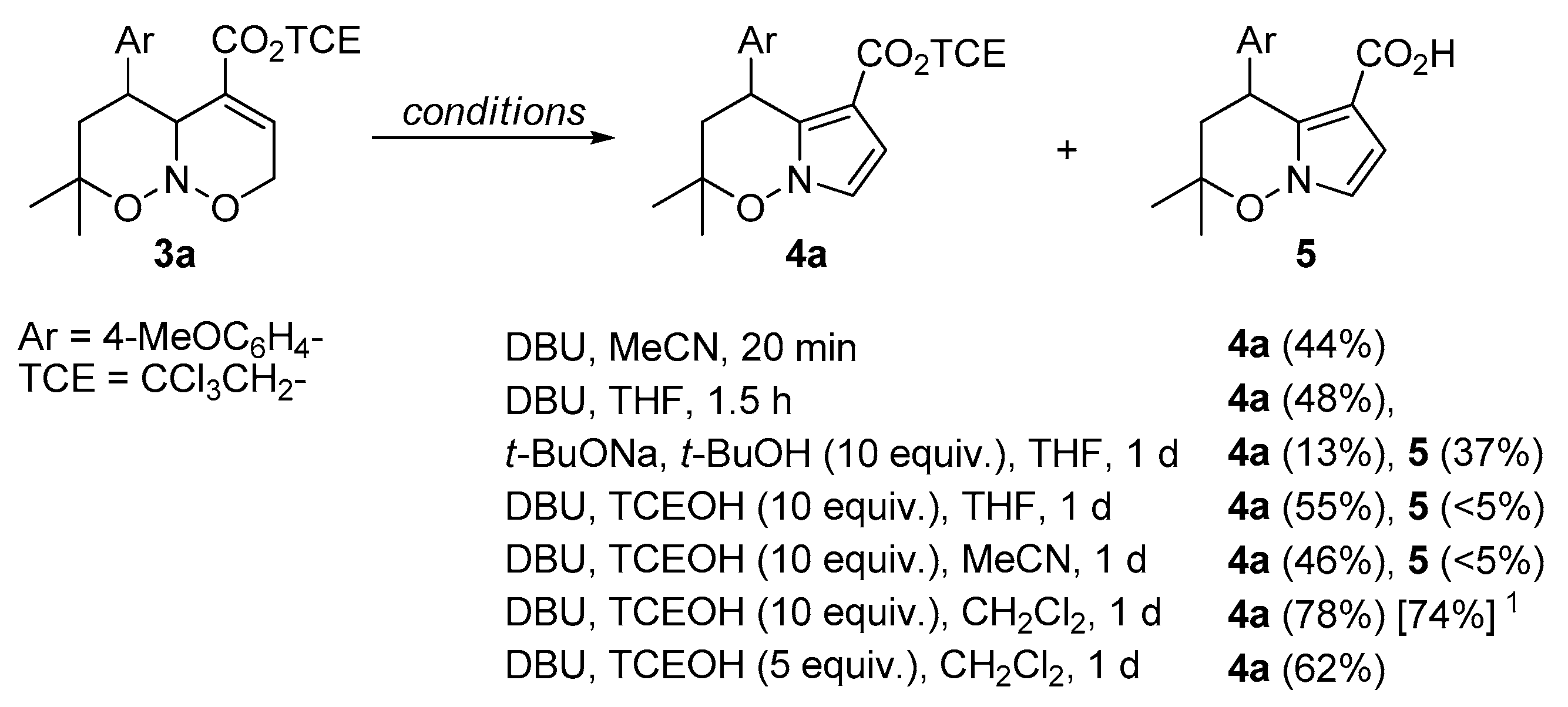
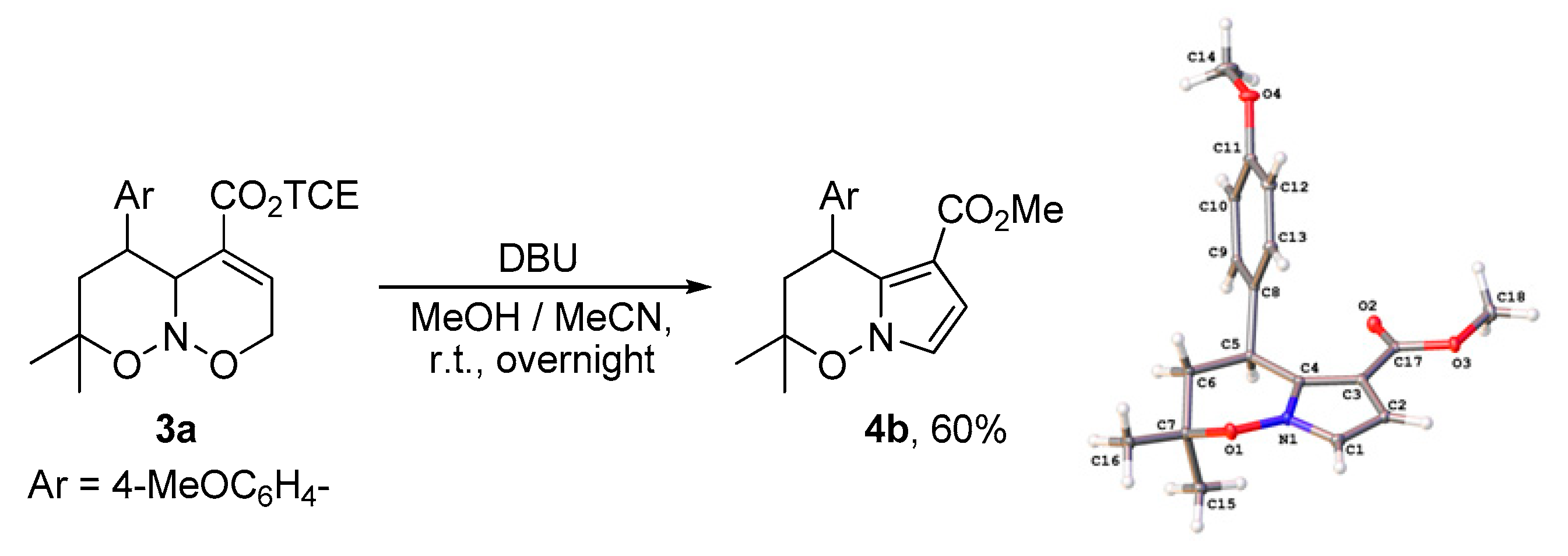
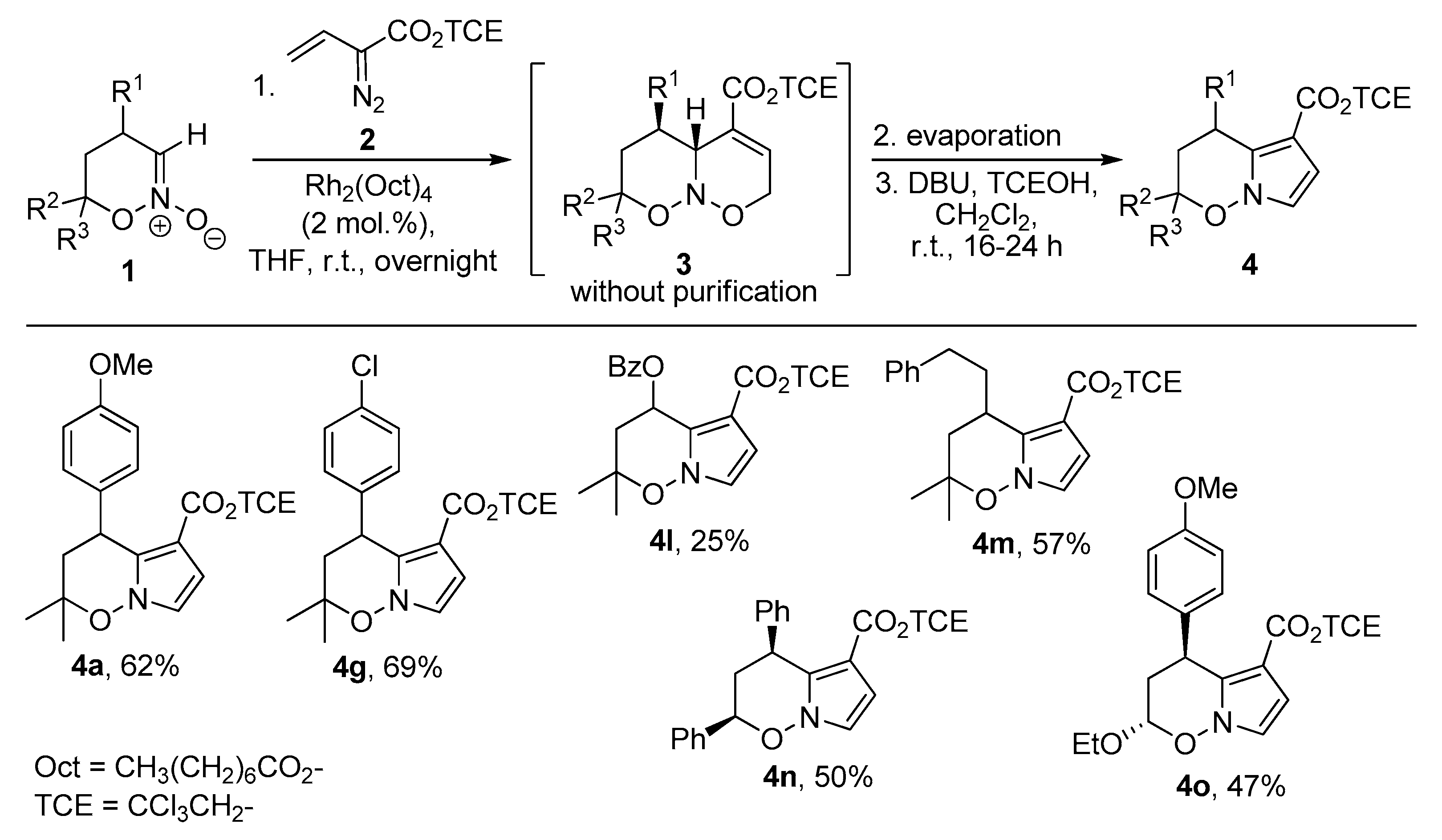
2. Results and Discussion
3. Materials and Methods
3.1. General Experiment
3.2. X-ray Crystallography
3.3. Synthetic Procedures for Products 3–5
3.3.1. General Procedure for the Synthesis of Nitroso Acetals 3 (GP-1)
3.3.2. General Procedure for the Synthesis of Pyrrolooxazines 4 (GP-2)
3.3.3. Preparation of Pyrrolooxazines 4b, 5
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Majireck, M.M.; Bennett, J.M. 1,2-Oxazines and their benzo derivatives. In Comprehensive Heterocyclic Chemistry IV; Weinreb, S.M., Black, D.S.C., Cossy, J., Stevens, C.V., Eds.; Elsevier Ltd.: Amsterdam, The Netherlands, 2022; Volume 8, pp. 283–415. [Google Scholar] [CrossRef]
- Ghosh, P.; Mondal, S.L.; Baidya, M. Ascending of cycloaddition strategy for N–O heterocycles. Synthesis 2022, 54, 1043–1054. [Google Scholar] [CrossRef]
- Sukhorukov, A.Y.; Ioffe, S.L. Chemistry of six-membered cyclic oxime ethers. Application in the synthesis of bioactive compounds. Chem. Rev. 2011, 111, 5004–5041. [Google Scholar] [CrossRef] [PubMed]
- Utecht, G.; Jasinski, M. 3,6-Dihydro-2H-1,2-oxazines (microreview). Chem. Heterocycl. Compd. 2016, 52, 143–145. [Google Scholar] [CrossRef]
- Brulikova, L.; Harrison, A.; Miller, M.J.; Hlavac, J. Stereo- and regioselectivity of the hetero-Diels–Alder reaction of nitroso derivatives with conjugated dienes. Beilstein J. Org. Chem. 2016, 12, 1949–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouche, L.; Reissig, H.-U. Synthesis of novel carbohydrate mimetics via 1,2-oxazines. Pure Appl. Chem. 2012, 84, 23–36. [Google Scholar] [CrossRef]
- Bodnar, B.S.; Miller, M.J. The nitrosocarbonyl hetero-Diels-Alder reaction as a useful tool for organic syntheses. Angew. Chem. Int. Ed. 2011, 50, 5630–5647. [Google Scholar] [CrossRef] [PubMed]
- Pfrengle, F.; Reissig, H.-U. Amino sugars and their mimetics via 1,2-oxazines. Chem. Soc. Rev. 2010, 39, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Saito, T.; Zakarian, A. Approaches to dihydrooxazine ring systems and application in the synthesis of bioactive natural products. Chem. Heterocycl. Compd. 2012, 48, 11–16. [Google Scholar] [CrossRef]
- Wehlauch, R.; Gadermann, K. Securinega alkaloids: Complex structures, potent bioactivities, and efficient total syntheses. Asian J. Org. Chem. 2017, 6, 1146–1159. [Google Scholar] [CrossRef] [Green Version]
- Ohta, S.; Oshimo, S.; Ohta, E.; Nehira, T.; Omura, H.; Uy, M.M.; Ishihara, Y. Asaroidoxazines from the roots of Asarum asaroides induce apoptosis in human neuroblastoma cells. J. Nat. Prod. 2020, 83, 3050–3057. [Google Scholar] [CrossRef]
- Liu, C.; Yang, C.; Zeng, Y.; Shi, J.; Li, L.; Li, W.; Jiao, R.; Tan, R.; Ge, H. Chartrenoline, a novel alkaloid isolated from a marine Streptomyces chartreusis NA02069. Chin. Chem. Lett. 2019, 30, 44–46. [Google Scholar] [CrossRef]
- Li, J.; Hu, Y.; Hao, X.; Tan, J.; Li, F.; Qiao, X.; Chen, S.; Xiao, C.; Chen, M.; Peng, Z.; et al. Raistrickindole A, an anti-HCV oxazinoindole alkaloid from Penicillium raistrickii IMB17-034. J. Nat. Prod. 2019, 82, 1391–1395. [Google Scholar] [CrossRef]
- Wu, Z.-L.; Huang, X.-J.; Xu, M.-T.; Ma, X.; Li, L.; Shi, L.; Wang, W.-J.; Jiang, R.-W.; Ye, W.-C.; Wang, Y. Flueggeacosines A–C, dimeric securinine-type alkaloid analogues with neuronal differentiation activity from Flueggea suffruticosa. Org. Lett. 2018, 20, 7703–7707. [Google Scholar] [CrossRef]
- Kim, Y.; In, Y.; Ishida, T.; Onaka, H.; Igarashi, Y. Biosynthetic origin of alchivemycin A, a new polyketide from Streptomyces and absolute configuration of alchivemycin B. Org. Lett. 2013, 15, 3514–3517. [Google Scholar] [CrossRef]
- Koyama, K.; Hirasawa, Y.; Nugroho, A.E.; Hosoya, T.; Hoe, T.C.; Chan, K.-L.; Morita, H. Alsmaphorazines A and B, novel indole alkaloids from Alstonia pneumatophora. Org. Lett. 2010, 12, 4188–4191. [Google Scholar] [CrossRef]
- Shao, C.-L.; Wang, C.-Y.; Gu, Y.-C.; Cai, J.-W.; Deng, D.-S.; She, Z.-G.; Lin, Y.-C. Revised structure of penicillazine and preparation, bioactivities of penicillazine derivatives. Lett. Org. Chem. 2009, 6, 387–391. [Google Scholar] [CrossRef]
- Garo, E.; Starks, C.M.; Jensen, P.R.; Fenical, W.; Lobkovsky, E.; Clardy, J. Trichodermamides A and B, cytotoxic modified dipeptides from the marine-derived fungus Trichoderma virens. J. Nat. Prod. 2003, 66, 423–426. [Google Scholar] [CrossRef]
- Mfuh, A.M.; Zhang, Y.; Stephens, D.E.; Vo, A.X.T.; Arman, H.D.; Larionov, O.V. Concise total synthesis of trichodermamides A, B and C enabled by an efficient construction of the 1,2-oxazadecaline core. J. Am. Chem. Soc. 2015, 137, 8050–8053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, K.M.; Cox, J.B.; Liu, L.; Jackson, A.C.; Yruegas, S.; Wiberg, K.B.; Wood, J.L. Total synthesis of (±)-phyllantidine: Development and mechanistic evaluation of a novel ring expansion for installation of embedded nitrogen-oxygen bonds. Angew. Chem. Int. Ed. 2020, 59, 9757–9766. [Google Scholar] [CrossRef]
- Doens, D.; Valiente, P.A.; Mfuh, A.M.; Vo, A.X.T.; Tristan, A.; Carreno, L.; Quijada, M.; Nguyen, V.T.; Perry, G.; Larionov, O.V.; et al. Identification of inhibitors of CD36-amyloid beta binding as potential agents for Alzheimer’s disease. ACS Chem. Neurosci. 2017, 8, 1232–1241. [Google Scholar] [CrossRef] [PubMed]
- Jans, P.E.; Mfuh, A.M.; Arman, H.D.; Shaffer, C.V.; Larionov, O.V.; Mooberry, S.L. Cytotoxicity and mechanism of action of the marine-derived fungal metabolite trichodermamide B and synthetic analogues. J. Nat. Prod. 2017, 80, 676–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, T.L.; Sae-Lao, P.; Toh, H.H.M.; Csokas, D.; Bates, R.W. The total synthesis of raistrickindole A. J. Org. Chem. 2022, 87, 16111–16114. [Google Scholar] [CrossRef]
- Li, T.-Z.; Liu, S.-J.; Sun, Y.-W.; Deng, S.; Tan, W.; Jiao, Y.; Zhang, Y.-C.; Shi, F. Regio- and enantioselective (3+3) cycloaddition of nitrones with 2-indolylmethanols enabled by cooperative organocatalysis. Angew. Chem. Int. Ed. 2021, 60, 2355–2363. [Google Scholar] [CrossRef] [PubMed]
- Herrera, L.; Stephens, D.E.; D’Avila, A.; George, K.G.; Arman, H.; Zhang, Y.; Perry, G.; Lleonart, R.; Larionov, O.V.; Fernandez, P.L. Insights into the structural patterns of the antileishmanial activity of bi- and tricyclic N-heterocycles. Org. Biomol. Chem. 2016, 14, 7053–7060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Zollner, T.; Gebhardt, P.; Mollmann, U.; Miller, M.J. Preparation and biological evaluation of novel leucomycin analogs derived from nitroso Diels-Alder reactions. Org. Biomol. Chem. 2010, 8, 691–697. [Google Scholar] [CrossRef] [Green Version]
- Tabolin, A.A.; Sukhorukov, A.Y.; Ioffe, S.L.; Dilman, A.D. Recent advances in the synthesis and chemistry of nitronates. Synthesis 2017, 49, 3255–3268. [Google Scholar] [CrossRef]
- Denmark, S.E.; Thorarensen, A. Tandem [4+2]/[3+2] cycloadditions of nitroalkenes. Chem. Rev. 1996, 96, 137–166. [Google Scholar] [CrossRef]
- Ioffe, S.L. Nitronates. In Nitrile Oxides, Nitrones, and Nitronates in Organic Synthesis, Novel Strategies in Synthesis, 2nd ed.; Feuer, H., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008; pp. 541–604. [Google Scholar]
- Baiazitov, R.Y.; Denmark, S.E. Tandem [4+2]/[3+2] Cycloadditions. In Methods and Applications of Cycloaddition Reactions in Organic Syntheses; Nishiwaki, N., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; pp. 471–550. [Google Scholar]
- Denmark, S.E.; Cottell, J.J. Nitronates. In The Chemistry of Heterocyclic Compounds: Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry toward Heterocycles and Natural Products; Padwa, A., Pearson, W.H., Eds.; Wiley-Interscience: New York, NY, USA, 2002; pp. 83–167. [Google Scholar]
- Denmark, S.E.; Cottell, J.J. Synthesis of (+)-1-epiaustraline. J. Org. Chem. 2001, 66, 4276–4284. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Hurd, A.R. Synthesis of (+)-casuarine. J. Org. Chem. 2000, 65, 2875–2886. [Google Scholar] [CrossRef]
- Denmark, S.E.; Martinborough, E.A. Enantioselective total syntheses of (+)-castanospermine, (+)-6-epicastanospermine, (+)-australine, and (+)-3-epiaustraline. J. Am. Chem. Soc. 1999, 121, 3046–3056. [Google Scholar] [CrossRef]
- Denmark, S.E.; Herbert, B. Synthesis of (1R,2R,3R,7R,7aR)-hexahydro-3-(hydroxymethyl)-1H-pyrrolizine-1,2,7-triol: 7-epiaustraline. J. Am. Chem. Soc. 1998, 120, 7357–7358. [Google Scholar] [CrossRef]
- Denmark, S.E.; Hurd, A.R.; Sacha, H.J. Tandem [4+2]/[3+2] cycloadditions of nitroalkenes. 13. The synthesis of (−)-detoxinine. J. Org. Chem. 1997, 62, 1668–1674. [Google Scholar] [CrossRef]
- Denmark, S.E.; Marcin, L.R. Asymmetric construction of a quaternary carbon center by tandem [4+2]/[3+2] cycloaddition of a nitroalkene. The total synthesis of (−)-mesembrine. J. Org. Chem. 1997, 62, 1675–1686. [Google Scholar] [CrossRef]
- Denmark, S.E.; Thorarensen, A. Tandem [4+2]/[3+2] cycloadditions of nitroalkenes. 11. The synthesis of (+)-crotanecine. J. Am. Chem. Soc. 1997, 119, 125–137. [Google Scholar] [CrossRef]
- Denmark, S.E.; Thorarensen, A.; Middleton, D.S. Tandem [4+2]/[3+2] cycloadditions of nitroalkenes. 9. Synthesis of (−)-rosmarinecine. J. Am. Chem. Soc. 1996, 118, 8266–8277. [Google Scholar] [CrossRef]
- Gorbacheva, E.O.; Tabolin, A.A.; Novikov, R.A.; Khomutova, Y.A.; Nelyubina, Y.V.; Tomilov, Y.V.; Ioffe, S.L. Six-membered cyclic nitronates as 1,3-dipoles in formal [3+3]-cycloaddition with donor-acceptor cyclopropanes. Synthesis of new type of bicyclic nitrosoacetals. Org. Lett. 2013, 15, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Tabolin, A.A.; Gorbacheva, E.O.; Novikov, R.A.; Khoroshutina, Y.A.; Nelyubina, Y.V.; Ioffe, S.L. Synthesis and chemical transformations of six/six-membered bicyclic nitroso acetals. Russ. Chem. Bull. 2016, 65, 2243–2259. [Google Scholar] [CrossRef]
- Cheng, Q.-Q.; Yu, Y.; Yedoyan, J.; Doyle, M.P. Vinyldiazo reagents and metal catalysts: A versatile toolkit for heterocycle and carbocycle construction. ChemCatChem 2018, 10, 488–496. [Google Scholar] [CrossRef]
- Xu, X.-F.; Doyle, M.P. Divergent pathways of β,γ-unsaturated α-diazocarbonyl compounds catalyzed by dirhodium and Lewis acids catalysts separately or in combination. Chin. Chem. Lett. 2015, 26, 227–232. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, Z.; Sun, J. Asymmetric [3+1]-cycloaddition reaction via diazo discrimination. Org. Lett. 2021, 23, 7613–7617. [Google Scholar] [CrossRef] [PubMed]
- Barluenga, J.; Riesgo, L.; Lopez, L.A.; Rubio, E.; Tomas, M. Discrimination of diazo compounds toward carbenoids: Copper(I)-catalyzed synthesis of substituted cyclobutenes. Angew. Chem. Int. Ed. 2009, 48, 7569–7572. [Google Scholar] [CrossRef] [PubMed]
- Dong, K.; Xu, X.; Doyle, M.P. Copper(I)-catalyzed highly enantioselective [3+3]-cycloaddition of γ-alkyl enoldiazoacetates with nitrones. Org. Chem. Front. 2020, 7, 1653–1657. [Google Scholar] [CrossRef]
- Barluenga, J.; Lonzi, G.; Riesgo, L.; Lopez, L.A.; Tomas, M. Pyridine activation via copper(I)-catalyzed annulation toward indolizines. J. Am. Chem. Soc. 2010, 132, 13200–13202. [Google Scholar] [CrossRef]
- Thompson, J.L.; Davies, H.M.L. Enhancement of cyclopropanation chemistry in the silver-catalyzed reactions of aryldiazoacetates. J. Am. Chem. Soc. 2007, 129, 6090–6091. [Google Scholar] [CrossRef]
- Ueda, J.; Harada, S.; Nakayama, H.; Nemoto, T. Silver-catalyzed regioselective hydroamination of alkenyl diazoacetates to synthesize γ-amino acid equivalents. Org. Biomol. Chem. 2018, 16, 4675–4682. [Google Scholar] [CrossRef]
- Xu, G.; Liu, K.; Dai, Z.; Sun, J. Gold/silver-catalyzed controllable regioselective vinylcarbene insertion into O–H bonds. Org. Biomol. Chem. 2017, 15, 2345–2348. [Google Scholar] [CrossRef] [PubMed]
- Pagar, V.V.; Liu, R.-S. Gold-catalyzed α-furanylations of quinoline N-oxides with alkenyldiazo carbonyl species. Org. Biomol. Chem. 2015, 13, 6166–6169. [Google Scholar] [CrossRef]
- Zhou, P.-X.; Zhou, Z.-Z.; Chen, Z.-S.; Ye, Y.-Y.; Zhao, L.-B.; Yang, Y.-F.; Xia, X.-F.; Luo, J.-Y.; Liang, Y.-M. Palladium-catalyzed insertion of α-diazocarbonyl compounds for the synthesis of cyclic amino esters. Chem. Commun. 2013, 49, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Guptill, D.M.; Davies, H.M.L. 2,2,2-Trichloroethyl aryldiazoacetates as robust reagents for the enantioselective C–H functionalization of methyl ethers. J. Am. Chem. Soc. 2014, 136, 17718–17721. [Google Scholar] [CrossRef]
- Negretti, S.; Cohen, C.M.; Chang, J.J.; Guptill, D.M.; Davies, H.M.L. Enantioselective dirhodium(II)-catalyzed cyclopropanations with trimethylsilylethyl and trichloroethyl aryldiazoacetates. Tetrahedron 2015, 71, 7415–7420. [Google Scholar] [CrossRef] [Green Version]
- Shved, A.S.; Tabolin, A.A.; Novikov, R.A.; Nelyubina, Y.V.; Timofeev, V.P.; Ioffe, S.L. Six-membered cyclic nitroso acetals: Synthesis and studies of the nitrogen inversion process of N-silyloxy-3,6-dihydro-2H-1,2-oxazines. Eur. J. Org. Chem. 2016, 2016, 5569–5578. [Google Scholar] [CrossRef]
- Ueda, Y.; Roberge, G.; Vinet, V. A simple method of preparing trimethylsilyl- and tert-butyldimethylsilyl-enol ethers of α-diazoacetoacetates and their use in the synthesis of a chiral precursor to thienamycin analogs. Can. J. Chem. 1984, 62, 2936–2940. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Hu, B.; Saikali, E.; Bruzinski, P.R. Carbenoid versus vinylogous reactivity in rhodium(II)-stabilized vinylcarbenoids. J. Org. Chem. 1994, 59, 4535–4541. [Google Scholar] [CrossRef]
- Crocetti, L.; Floresta, G.; Cilibrizzi, A.; Giovannoni, M.P. An overview of PDE4 inhibitors in clinical trials: 2010 to early 2022. Molecules 2022, 27, 4964. [Google Scholar] [CrossRef] [PubMed]
- Dyke, H.J.; Montana, J.G. Update on the therapeutic potential of PDE4 inhibitors. Expert Opin. Investig. Drugs 2002, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nelyubina, Y.V.; Lyssenko, K.A. Probing stereoelectronic interactions in an O–N–O unit by the atomic energies: Experimental and theoretical electron density study. J. Phys. Chem. A 2013, 117, 3084–3092. [Google Scholar] [CrossRef]
- Tishkov, A.A.; Lesiv, A.V.; Khomutova, Y.A.; Strelenko, Y.A.; Nesterov, I.D.; Antipin, M.Y.; Ioffe, S.L.; Denmark, S.E. 2-Silyloxy-1,2-oxazines, a new type of acetals of conjugated nitroso alkenes. J. Org. Chem. 2003, 68, 9477–9480. [Google Scholar] [CrossRef]
- Zheng, H.; Faghihi, I.; Doyle, M.P. Copper(I)-catalyzed highly enantioselective [3+3]-cycloaddition of β-aryl/alkyl vinyl diazoacetates with nitrones. Helvetica Chim. Acta 2021, 104, e2100081. [Google Scholar] [CrossRef]
- Wang, X.; Abrahams, Q.M.; Zavalij, P.Y.; Doyle, M.P. Highly regio- and stereoselective dirhodium vinylcarbene induced nitrone cycloaddition with subsequent cascade carbenoid aromatic cycloaddition/N–O cleavage and rearrangement. Angew. Chem. Int. Ed. 2012, 51, 5907–5910. [Google Scholar] [CrossRef]
- Qin, C.; Davies, H.M.L. Rh2(R-TPCP)4-catalyzed enantioselective [3+2]-cycloaddition between nitrones and vinyldiazoacetates. J. Am. Chem. Soc. 2013, 135, 14516–14519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagar, V.V.; Liu, R.-S. Gold-catalyzed cycloaddition reactions of ethyl diazoacetate, nitrosoarenes, and vinyldiazo carbonyl compounds: Synthesis of isoxazolidine and benzo[b]azepine derivatives. Angew. Chem. Int. Ed. 2015, 54, 4923–4926. [Google Scholar] [CrossRef]
- Stefkova, K.; Guerzoni, M.G.; van Ingen, Y.; Richards, E.; Melen, R.L. B(C6F5)3-catalyzed diastereoselective and divergent reactions of vinyldiazo esters with nitrones: Synthesis of highly functionalized diazo compounds. Org. Lett. 2023, 25, 500–505. [Google Scholar] [CrossRef]
- Humenny, W.J.; Kyriacou, P.; Sapeta, K.; Karadeolian, A.; Kerr, M.A. Multicomponent synthesis of pyrroles from cyclopropanes: A one-pot palladium(0)-catalyzed dehydrocarbonylation/dehydration. Angew. Chem. Int. Ed. 2012, 51, 11088–11091. [Google Scholar] [CrossRef] [PubMed]
- Firl, J.; Kresze, G. Heterocyclen durch Diensynthese, III. Pyrrole, α-Pyrone und Pyridone aus Diels-Alder-Addukten von Nitrosoverbindungen. Chem. Ber. 1966, 99, 3695–3706. [Google Scholar] [CrossRef]
- Firl, J. Heterocyclen durch Diensynthese, Pyridylpyrrole aus Diels-Alder-Addukten von Nitrosoverbindungen. Chem. Ber. 1968, 101, 218–225. [Google Scholar] [CrossRef]
- Kefalas, P.; Grierson, D.S. Synthesis of pyrrolo-castanospermine analogs: A novel fluoride ion induced rearrangement of a 3-trialkylsilyl substituted dihydro-2H-1,2-oxazine to a pyrrole. Tetrahedron Lett. 1993, 34, 3555–3558. [Google Scholar] [CrossRef]
- Eberlin, L.; Carboni, B.; Whiting, A. Regioisomeric and substituent effects upon the outcome of the reaction of 1-borodienes with nitrosoarene compounds. J. Org. Chem. 2015, 80, 6574–6583. [Google Scholar] [CrossRef] [Green Version]
- Yasukawa, N.; Kuwata, M.; Imai, T.; Monguchi, Y.; Sajiki, H.; Sawama, Y. Copper-catalyzed pyrrole synthesis from 3,6-dihydro-1,2-oxazines. Green Chem. 2018, 20, 4409–4413. [Google Scholar] [CrossRef]
- Jasinski, M.; Watanabe, T.; Reissig, H.-U. Samarium diiodide promoted reduction of 3,6-dihydro-2H-1,2-oxazines: Competition of 1,4-amino alcohol formation and ring contraction to pyrrole derivatives. Eur. J. Org. Chem. 2013, 2013, 605–610. [Google Scholar] [CrossRef]
- Scheiner, P.; Chapman, O.L.; Lassila, J.D. The photolysis of dihydro-l,2-oxazines. J. Org. Chem. 1969, 34, 813–816. [Google Scholar] [CrossRef]
- Givens, R.S.; Choo, D.J.; Merchant, S.N.; Stitt, R.P.; Matuszewski, B. Photochemistry of 3,6-dihydro-1,2-oxazines: A versatile route to substituted pyrroles. Tetrahedron Lett. 1982, 23, 1327–1330. [Google Scholar] [CrossRef]
- Rudchenko, V.F. Synthesis, reactions, and properties of oxygen-nitrogen-oxygen systems. Chem. Rev. 1993, 93, 725–739. [Google Scholar] [CrossRef]
- Lukoyanov, A.A.; Tabolin, A.A.; Nelyubina, Y.V.; Ioffe, S.L.; Sukhorukov, A.Y. Deoxygenative arylation of 5,6-dihydro-4H-1,2-oxazine-N-oxides with arynes. J. Org. Chem. 2022, 87, 6838–6851. [Google Scholar] [CrossRef]
- Kumar, P.; Kumar, R.; Banerjee, P. Accessing dihydro-1,2-oxazine via Cloke−Wilson-type annulation of cyclopropyl carbonyls: Application toward the diastereoselective synthesis of pyrrolo [1,2-b][1,2]oxazine. J. Org. Chem. 2020, 85, 6535–6550. [Google Scholar] [CrossRef]
- Jasinski, M.; Moreno-Clavijo, E.; Reissig, H.-U. Synthesis of a series of enantiopure polyhydroxylated bicyclic N-heterocycles from an L-erythrose-derived nitrone and alkoxyallenes. Eur. J. Org. Chem. 2014, 2014, 442–454. [Google Scholar] [CrossRef]
- Mondal, S.K.; Ali, S.A.; Manna, S.K.; Mandal, A.; Senapati, B.K.; Hossain, M.; Samanta, S. Palladium-catalyzed intramolecular cyclization: Access to rare pentacyclic N-fused heterocycles. ChemistrySelect 2017, 2, 9312–9318. [Google Scholar] [CrossRef]
- Diab, S.; Noel-Duchesneau, L.; Sanselme, M.; Kondo, Y.; De Paolis, M.; Chataigner, I. High pressure elicits unexpected transformations of plain nitroaromatics with 4-(cyclohex-1-en-1-yl)morpholine. Eur. J. Org. Chem. 2018, 2018, 2048–2052. [Google Scholar] [CrossRef]
- Xu, C.; Du, J.; Ma, L.; Li, G.; Tao, M.; Zhang, W. Tertiary amine functionalized polyacrylonitrile fiber catalyst for the synthesis of tetrahydrothiophenes. Tetrahedron 2013, 69, 4749–4757. [Google Scholar] [CrossRef]
- Ramirez-Osuna, M.; Chavez, D.; Hernandez, L.; Mollins, E.; Somanathan, R.; Aguirre, G. Synthesis of analogs of amathamide A and their preliminary antimicrobial activity. Molecules 2005, 10, 295–301. [Google Scholar] [CrossRef] [Green Version]
- Jakubec, P.; Cockfield, D.M.; Hynes, P.S.; Cleator, E.; Dixon, D.J. Enantio- and diastereoselective Michael additions of C-succinimidyl esters to nitro olefins using cinchonine-derived bifunctional organocatalysts. Tetrahedron Asymm. 2011, 22, 1147–1155. [Google Scholar] [CrossRef]
- Wen, L.; Tang, F.; Ge, C.; Wang, X.; Han, Z.; Wu, J. Practical large-scale preparation of (R)-rolipram using chiral nickel catalyst. Synth. Commun. 2012, 42, 3288–3295. [Google Scholar] [CrossRef]
- Fadeeva, A.A.; Ioffe, S.L.; Tabolin, A.A. Chlorination of conjugated nitroalkenes with PhICl2 and SO2Cl2 for the synthesis of α-chloronitroalkenes. Synthesis 2020, 52, 2679–2688. [Google Scholar] [CrossRef]
- Smirnov, V.O.; Ioffe, S.L.; Tishkov, A.A.; Khomutova, Y.A.; Nesterov, I.D.; Antipin, M.Y.; Smit, W.A.; Tartakovsky, V.A. New C-C coupling reaction of cyclic nitronates with carbon nucleophiles. Umpolung of the conventional reactivity of nitronates. J. Org. Chem. 2004, 69, 8485–8488. [Google Scholar] [CrossRef] [PubMed]
- Ushakov, P.Y.; Tabolin, A.A.; Ioffe, S.L.; Sukhorukov, A.Y. In situ generated magnesium cyanide as an efficient reagent for nucleophilic cyanation of nitrosoalkenes and parent nitronates. Eur. J. Org. Chem. 2019, 2019, 1888–1892. [Google Scholar] [CrossRef]
- Mikhaylov, A.A.; Dilman, A.D.; Khomutova, Y.A.; Arkhipov, D.E.; Korlyukov, A.A.; Ioffe, S.L. Stereoselective amine addition to six-membered cyclic nitronates promoted by silyl triflate. Eur. J. Org. Chem. 2013, 2013, 5670–5677. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Hougland, P.W.; Cantrell, W.R., Jr. Convenient Synthesis of Vinyldiazomethanes from α-Diazo-β-Keto Esters and Related Systems. Synth. Commun. 1992, 22, 971–978. [Google Scholar] [CrossRef]
- Wu, J.-Q.; Yang, Z.; Zhang, S.-S.; Jiang, C.-Y.; Li, Q.; Huang, Z.-S.; Wang, H. From indoles to carbazoles: Tandem Cp*Rh(III)-catalyzed C–H activation/Brønsted acid-catalyzed cyclization reactions. ACS Catal. 2015, 5, 6453–6457. [Google Scholar] [CrossRef]
- Korte, F.; Wusten, F. Über den Enolgehalt von Acetessigsäureestern mit verschiedenen Esteralkyl-Komponenten. Justus Liebigs Ann. Chem. 1961, 647, 18–22. [Google Scholar] [CrossRef]
- Dolle, F.; Hinnen, F.; Valette, H.; Fuseau, C.; Duval, R.; Peglion, J.-L.; Crouzel, C. Synthesis of two optically active calcium channel antagonists labelled with carbon-11 for in vivo cardiac PET imaging. Bioorg. Med. Chem. Lett. 1997, 5, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.; Li, B.; Dikarev, E.; Autschbach, J.; Davies, H.M.L. Combined experimental and computational studies of heterobimetallic Bi−Rh paddlewheel carboxylates as catalysts for metal carbenoid transformations. J. Org. Chem. 2009, 74, 6564–6571. [Google Scholar] [CrossRef]
- Chinthapally, K.; Massaro, N.P.; Ton, S.; Gardner, E.D.; Sharma, I. Trapping rhodium vinylcarbenoids with aminochalcones for the synthesis of medium-sized azacycles. Tetrahedron Lett. 2019, 60, 151253. [Google Scholar] [CrossRef]
- Rossbach, J.; Baumeister, J.; Harms, K.; Koert, U. Regio- and diastereoselective crotylboration of vic-tricarbonyl compounds. Eur. J. Org. Chem. 2013, 2013, 662–665. [Google Scholar] [CrossRef]
- Sarabia, F.J.; Li, Q.; Ferreira, E.M. Cyclopentene annulations of alkene radical cations with vinyl diazo species using photocatalysis. Angew. Chem. Int. Ed. 2018, 57, 11015–11019. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Solvent | Diazo 2a, Equiv. | Catalyst (Equiv.) | Recovery of 1a, % 2 | Yield of 3a, % 2 |
|---|---|---|---|---|---|
| 1 | THF | 2 | Rh2Oct4 (0.02) | 0 | 85 |
| 2 | Glyme | 2 | Rh2Oct4 (0.02) | 9 | 54 |
| 3 | Et2O | 2 | Rh2Oct4 (0.02) | 0 | 65 |
| 4 | 1,4-dioxane | 2 | Rh2Oct4 (0.02) | 0 | 62 |
| 5 | EtOAc | 2 | Rh2Oct4 (0.02) | 0 | 66 |
| 6 | MeCN | 2 | Rh2Oct4 (0.02) | 23 | 30 |
| 7 | CH2Cl2 | 2 | Rh2Oct4 (0.02) | 28 | 31 |
| 8 | Hexane | 2 | Rh2Oct4 (0.02) | 49 | 22 |
| 9 | Tol | 2 | Rh2Oct4 (0.02) | 0 | 54 |
| 10 | THF | 2 | Rh2(OAc)4 (0.02) | 0 | 74 |
| 11 | THF | 2 | Rh2(OAcF)4 (0.02) | 19 | 7 |
| 12 | THF | 2 | Rh2(esp)2 3 | 7 | 64 |
| 13 | THF | 2 | Cu(MeCN)4PF6 (0.05) | 0 | 0 |
| 14 | THF | 2 | CuOTf·½C6H6 (2) | 0 | 0 |
| 15 | THF | 2 | AgSbF6 (5) | 0 | 0 |
| 16 | THF | 2 | Pd(OAc)2 (5) | 4 | 0 |
| 17 | THF | 2 | Pd(PPh3)4 (2) | 49 | 0 |
| 18 | THF | 3 | Rh2Oct4 (0.02) | 0 | 81 |
| 19 | THF | 1.5 | Rh2Oct4 (0.02) | 4 | 68 |
| 20 | THF | 2 | Rh2Oct4 (0.01) | 10 | 66 |
| 21 4 | THF | 2 | Rh2Oct4 (0.02) | 0 | 73 |
| 22 5 | THF | 2 | Rh2Oct4 (0.02) | 4 | 77 |
| 23 6 | THF | 2 | Rh2Oct4 (0.02) | 0 | 75 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antonova, Y.A.; Nelyubina, Y.V.; Ioffe, S.L.; Tabolin, A.A. [3+3]-Annulation of Cyclic Nitronates with Vinyl Diazoacetates: Diastereoselective Synthesis of Partially Saturated [1,2]Oxazino[2,3-b][1,2]oxazines and Their Base-Promoted Ring Contraction to Pyrrolo[1,2-b][1,2]oxazine Derivatives. Molecules 2023, 28, 3025. https://doi.org/10.3390/molecules28073025
Antonova YA, Nelyubina YV, Ioffe SL, Tabolin AA. [3+3]-Annulation of Cyclic Nitronates with Vinyl Diazoacetates: Diastereoselective Synthesis of Partially Saturated [1,2]Oxazino[2,3-b][1,2]oxazines and Their Base-Promoted Ring Contraction to Pyrrolo[1,2-b][1,2]oxazine Derivatives. Molecules. 2023; 28(7):3025. https://doi.org/10.3390/molecules28073025
Chicago/Turabian StyleAntonova, Yulia A., Yulia V. Nelyubina, Sema L. Ioffe, and Andrey A. Tabolin. 2023. "[3+3]-Annulation of Cyclic Nitronates with Vinyl Diazoacetates: Diastereoselective Synthesis of Partially Saturated [1,2]Oxazino[2,3-b][1,2]oxazines and Their Base-Promoted Ring Contraction to Pyrrolo[1,2-b][1,2]oxazine Derivatives" Molecules 28, no. 7: 3025. https://doi.org/10.3390/molecules28073025