
Molecular Modelling Study and Antibacterial Evaluation of Diphenylmethane Derivatives as Potential FabI Inhibitors
 , , ,
, , ,  , , and
, , and
Abstract
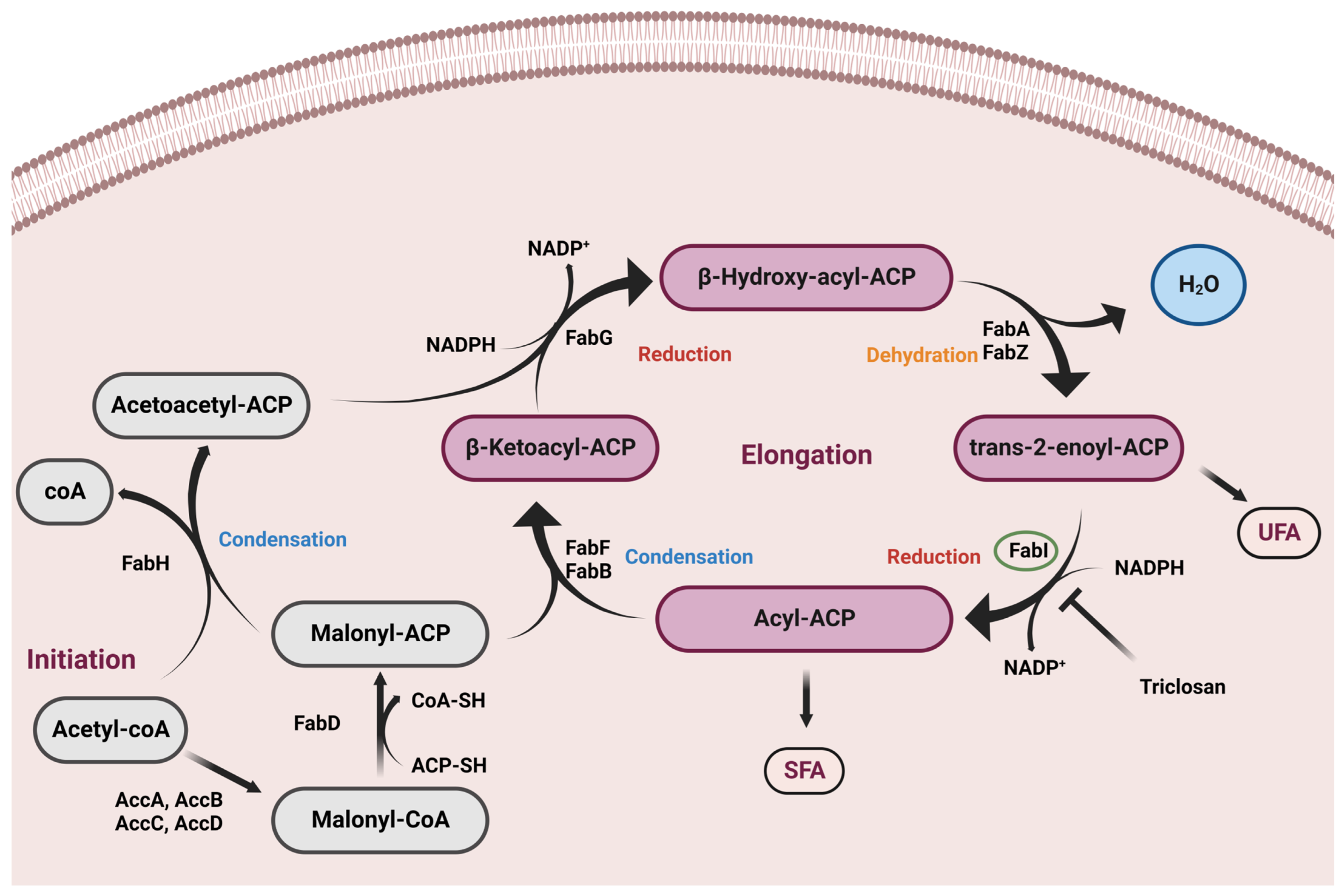
:1. Introduction
2. Results and Discussion
2.1. MN02 Derivatives
2.2. Antimicrobial Assay
2.3. Assessment of Pharmacokinetic and Druglike Characteristics
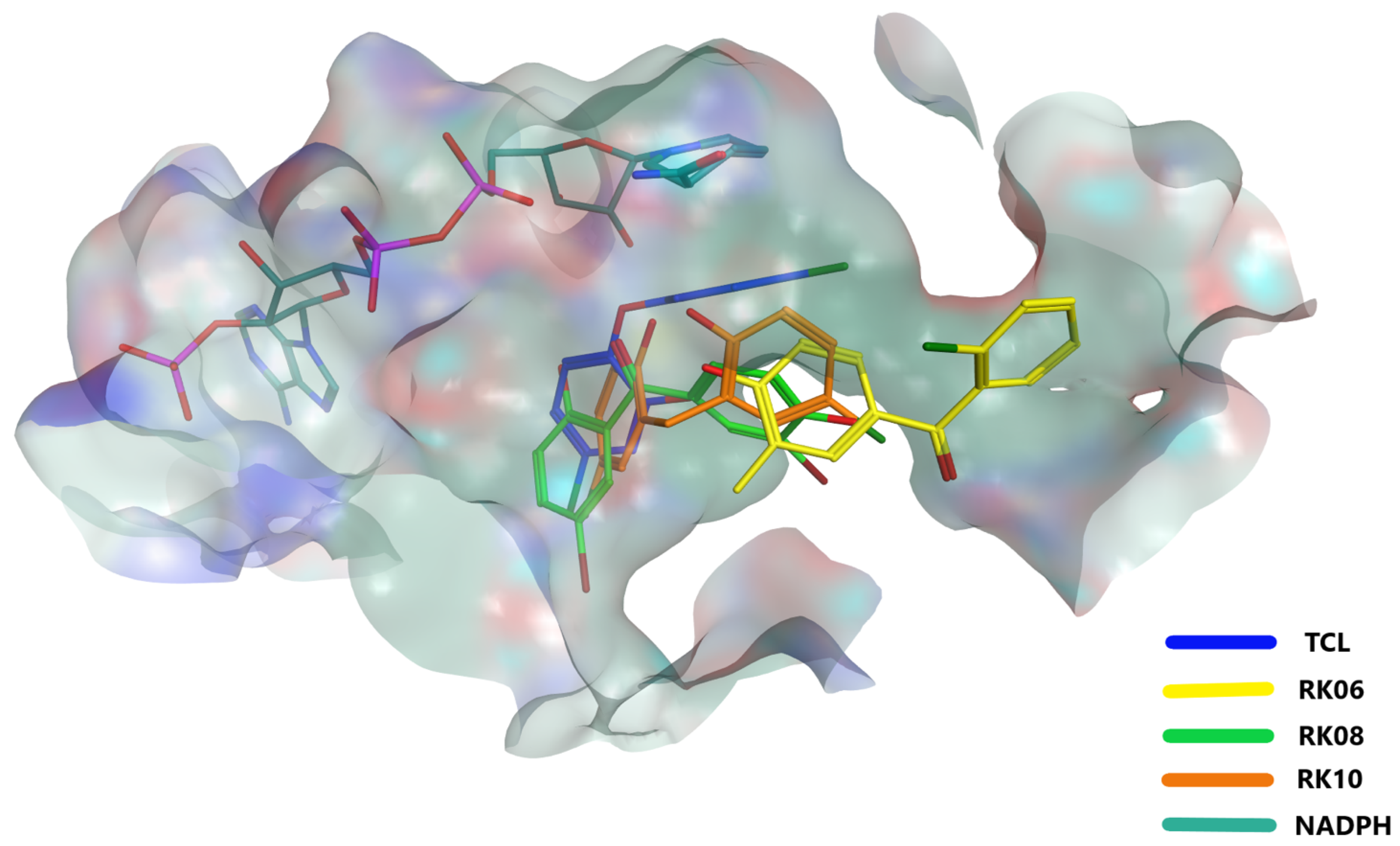
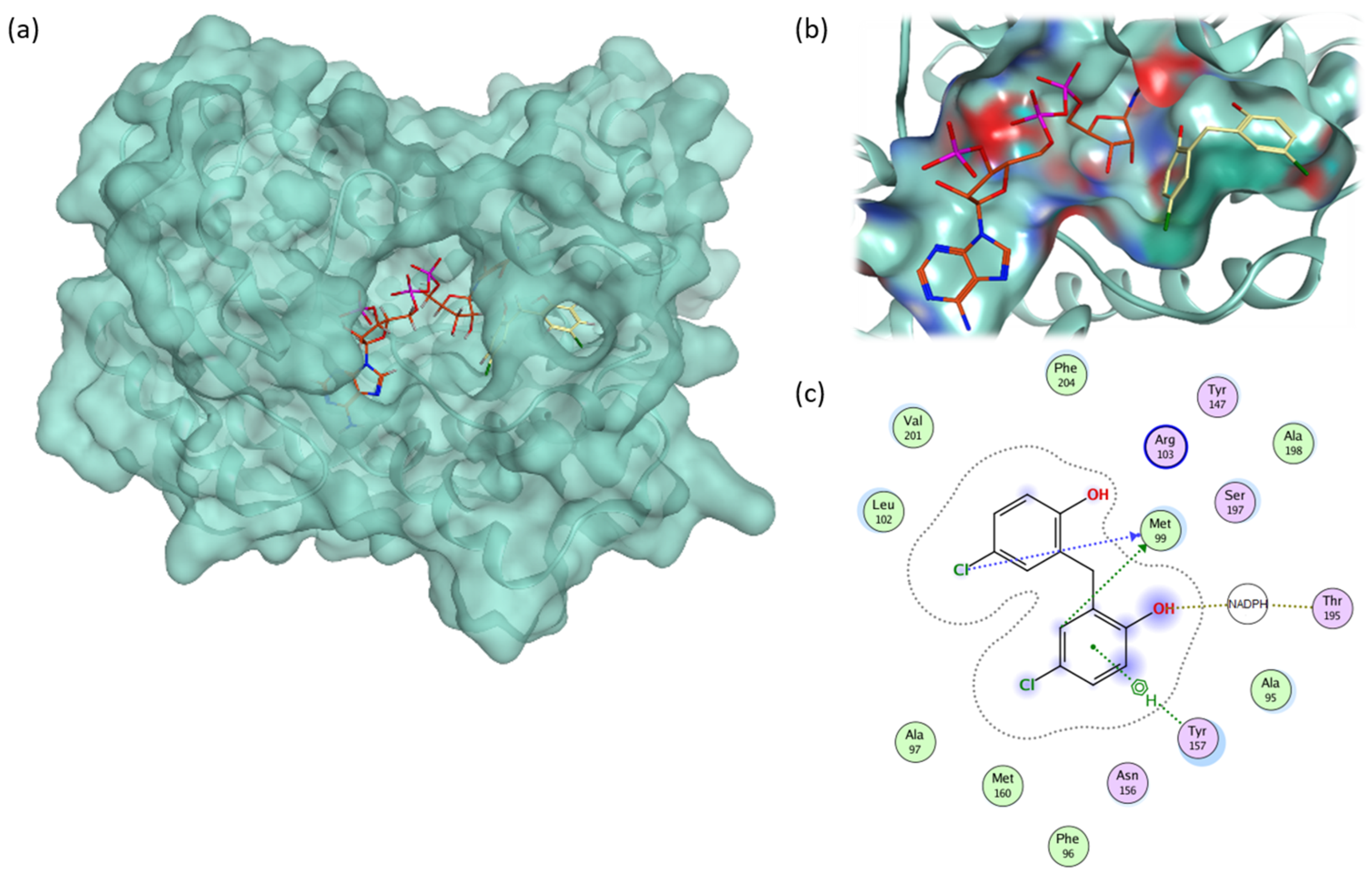
2.4. Glide Docking and MD Simulation
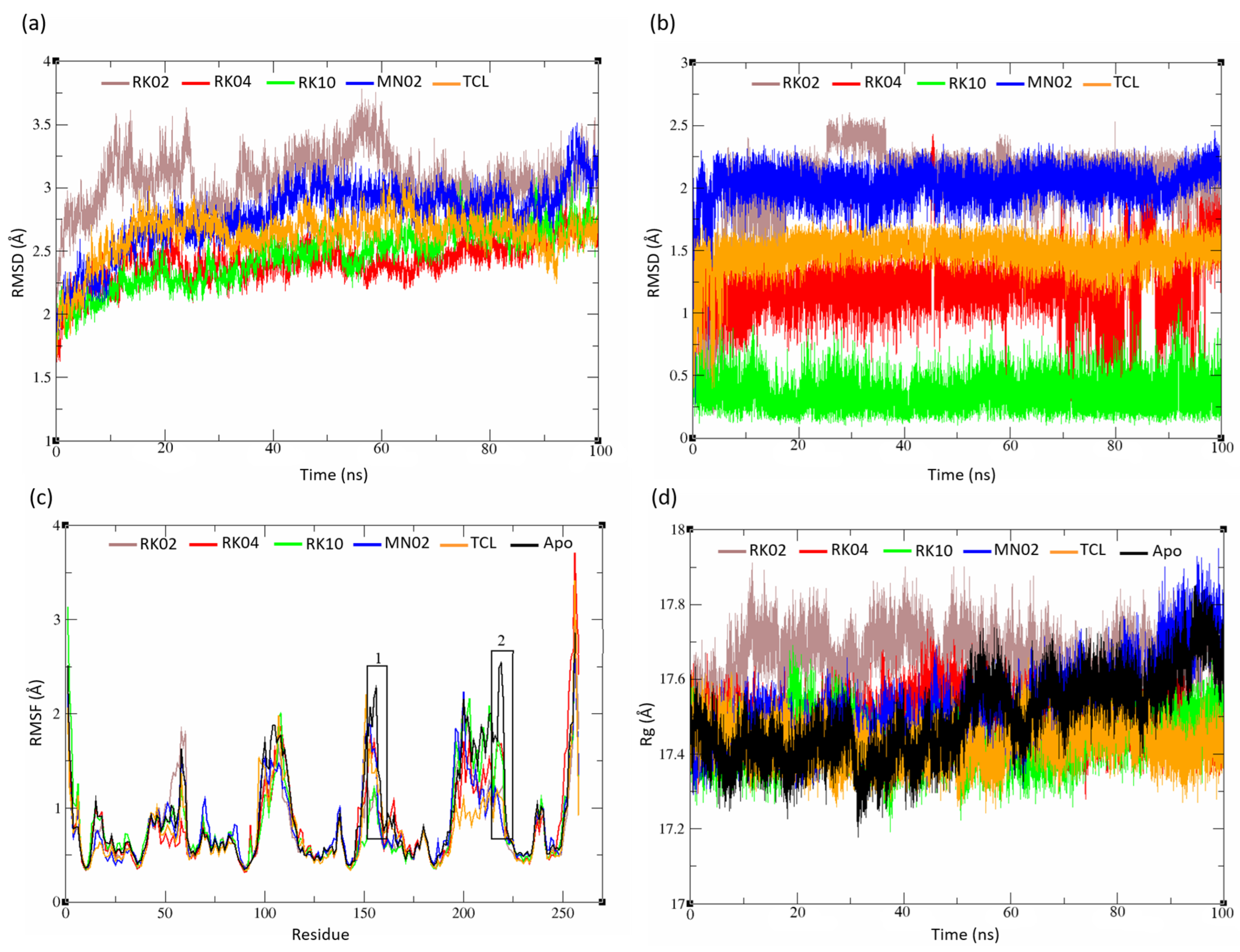
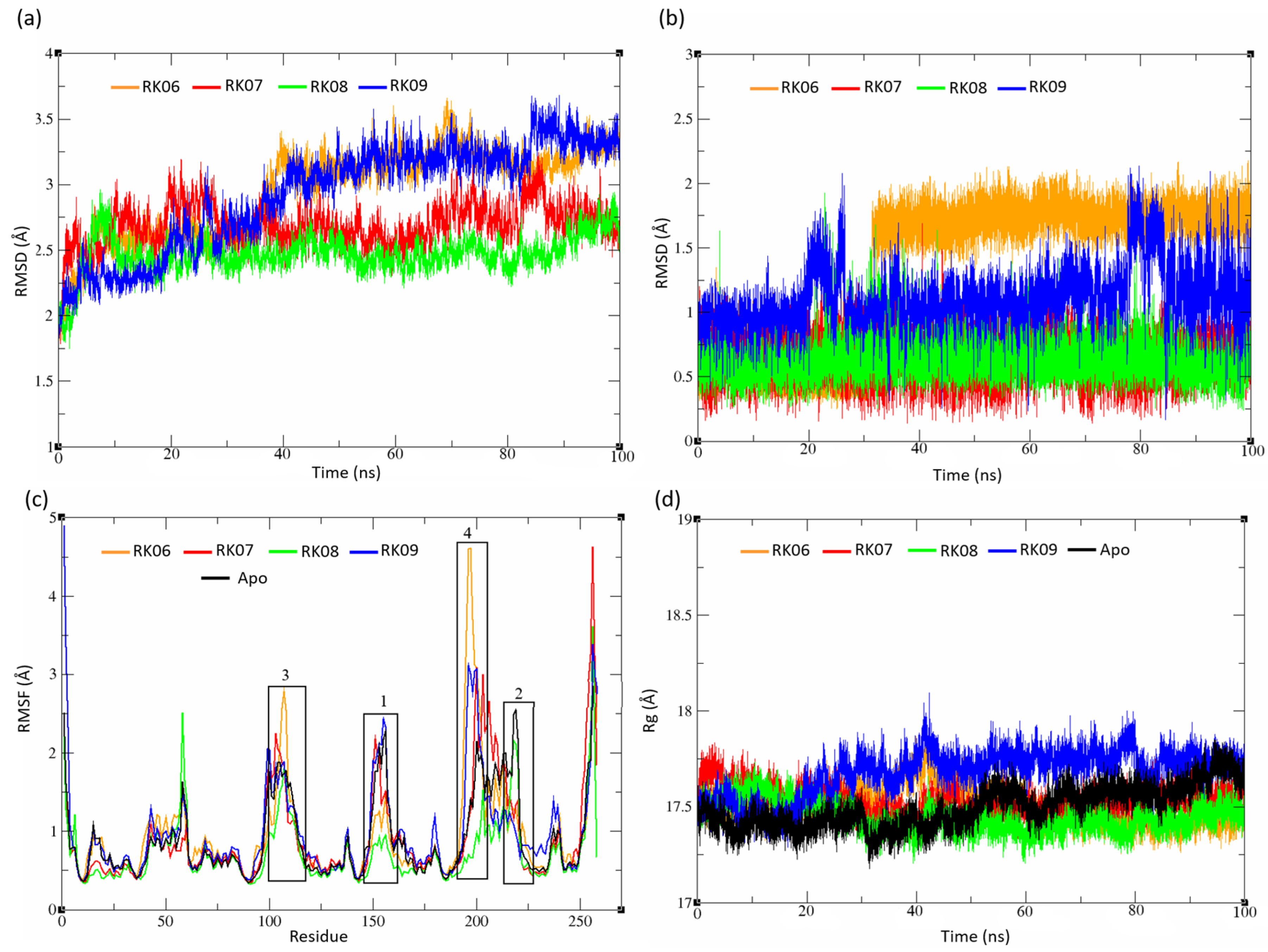
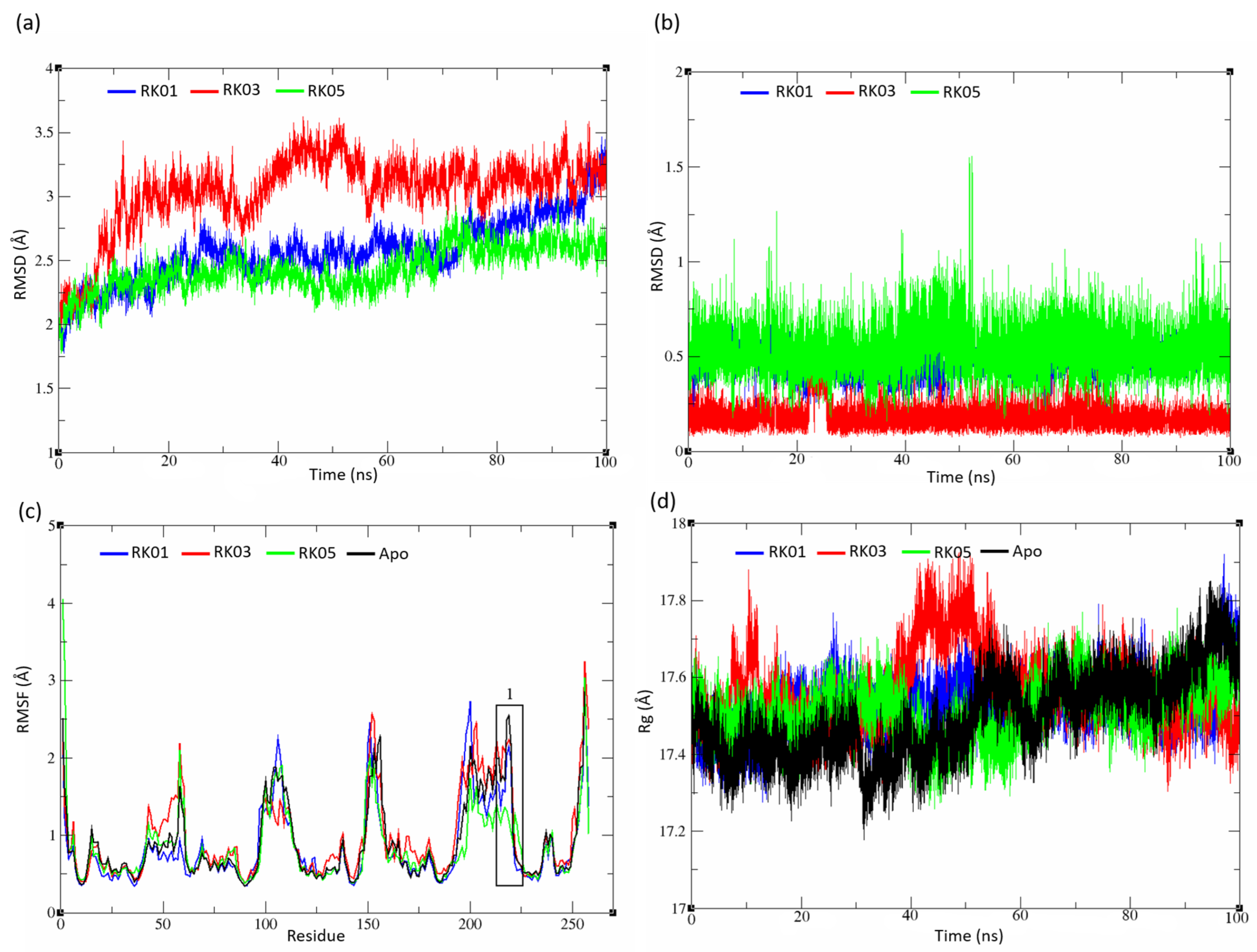
2.5. MD Analysis
2.5.1. Unsubstituted Diphenylmethane
2.5.2. Substituted Diphenylmethane Set
2.5.3. Diverse Set
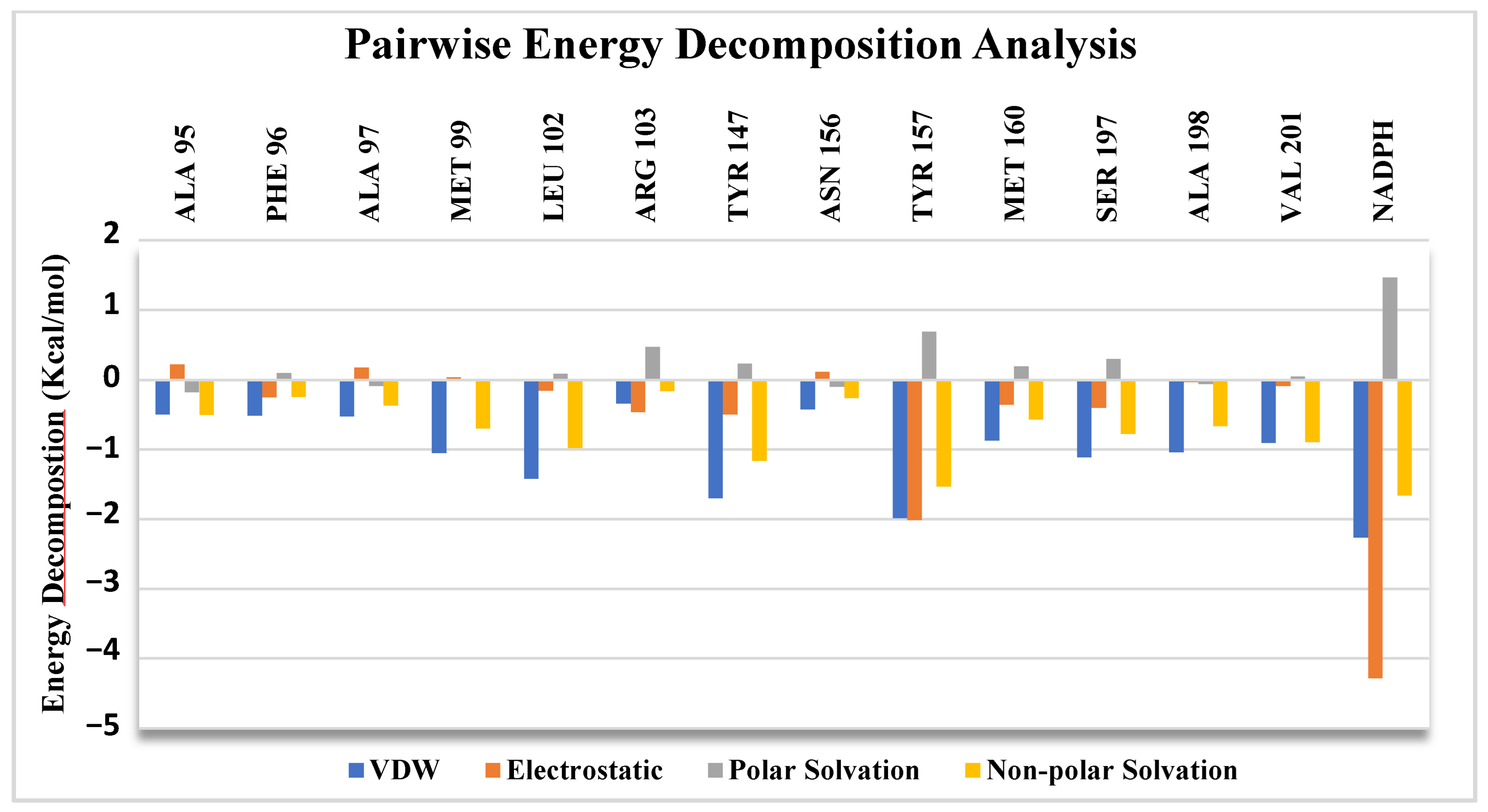
2.6. Pairwise Energy Decomposition Analysis
3. Method
3.1. Similarity Search
3.2. Antimicrobial Testing
3.2.1. Disk Diffusion
3.2.2. Minimum Inhibitory Concentrations
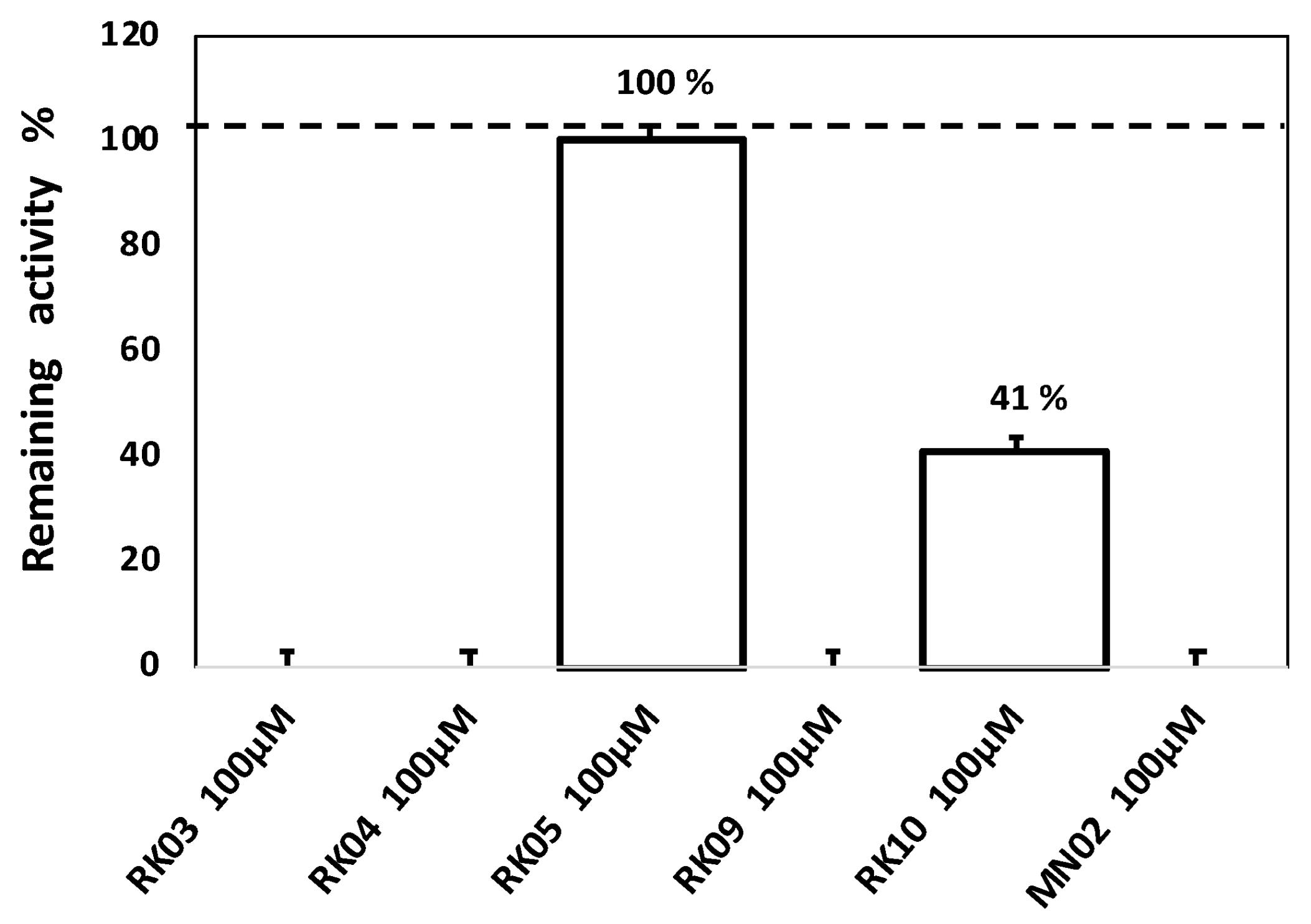
3.3. Enzyme Inhibition Assay
3.4. Protein Preparation and Grid Generation
3.5. Ligand Preparation and Molecular Docking of MN02 Derivatives
3.6. Molecular Dynamic Simulation and MM-GBSA Scoring
3.7. ADME Screening
3.8. MD Simulation Analysis
4. Conclusions
5. Limitations
- In order to prove that the observed antibacterial activity of the RKs compounds is due to their binding to FabI enzyme, assessing the inhibitory effect of all compounds on FabI was important. However, only RKs 3, 4, 5, 9 and 10 were tested.
- Only four bacterial species were tested (MRSA, E. coli, B. subtilis and S. aureus), three Gram positive and one Gram negative. Even though testing only four strains with only one Gram-negative bacteria was published in previous studies assessing antibacterial effects of drugs/compounds [68], a more extensive study of the antibacterial activities of the selected compounds would be needed to draw reliable conclusions on their potential use as antibiotics.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Herruzo, R.; Vizcaíno, M.J.; Herruzo, I.; De La Cruz, J.J. Can the Antibiotic Resistance of a Microorganism Predict Decreased Bactericidal Efficacy of Disinfectants? Application to OPA and Other Products. Eur. J. Clin. Microbiol. Infect. Dis. 2009, 28, 539–541. [Google Scholar] [CrossRef] [PubMed]
- Stratton, C.W. Mechanisms of Bacterial Resistance to Antimicrobial Agents. J. Med. Liban. 2000, 48, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.C. Reducing Inappropriate Prescribing of Antibiotics in English Primary Care: Evidence and Outlook. J. Antimicrob. Chemother. 2018, 73, 833–834. [Google Scholar] [CrossRef] [Green Version]
- Palmer, A.C.; Kishony, R. Understanding, Predicting and Manipulating the Genotypic Evolution of Antibiotic Resistance. Nat. Rev. Genet. 2013, 14, 243–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iredell, J. Antimicrobial Resistance. Microbiol. Aust. 2019, 40, 55. [Google Scholar] [CrossRef]
- Talebi Bezmin Abadi, A.; Rizvanov, A.A.; Haertlé, T.; Blatt, N.L. World Health Organization Report: Current Crisis of Antibiotic Resistance. Bionanoscience 2019, 9, 778–788. [Google Scholar] [CrossRef]
- Hofer, U. The Cost of Antimicrobial Resistance. Nat. Rev. Microbiol. 2019, 17, 3. [Google Scholar] [CrossRef] [PubMed]
- Massengo-Tiassé, R.P.; Cronan, J.E. Diversity in Enoyl-Acyl Carrier Protein Reductases. Cell. Mol. Life Sci. 2009, 66, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.Y.; Cronan, J.E. β-Ketoacyl-Acyl Carrier Protein Synthase III (FabH) Is Essential for Bacterial Fatty Acid Synthesis. J. Biol. Chem. 2003, 278, 51494–51503. [Google Scholar] [CrossRef] [Green Version]
- Revill, W.P.; Bibb, M.J.; Scheu, A.K.; Kieser, H.J.; Hopwood, D.A. β-Ketoacyl Acyl Carrier Protein Synthase III (FabH) Is Essential for Fatty Acid Biosynthesis in Streptomyces Coelicolor A3(2). J. Bacteriol. 2001, 183, 3526–3530. [Google Scholar] [CrossRef] [Green Version]
- Schujman, G.E.; Choi, K.H.; Altabe, S.; Rock, C.O.; De Mendoza, D. Response of Bacillus Subtilis to Cerulenin and Acquisition of Resistance. J. Bacteriol. 2001, 183, 3032–3040. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Leeds, J.A.; Meredith, T.C. Pseudomonas Aeruginosa Directly Shunts β-Oxidation Degradation Intermediates into de Novo Fatty Acid Biosynthesis. J. Bacteriol. 2012, 194, 5185–5196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, D.M.; Ngoc, T.M.; Dat, N.T.; Ha, D.T.; Kim, Y.H.; Van Luong, H.; Ahn, J.S.; Bae, K.H. Protein Tyrosine Phosphatase 1B Inhibitors Isolated from Morus Bombycis. Bioorganic Med. Chem. Lett. 2009, 19, 6759–6761. [Google Scholar] [CrossRef]
- Tasdemir, D.; Lack, G.; Brun, R.; Rüedi, P.; Scapozza, L.; Perozzo, R. Inhibition of Plasmodium Falciparum Fatty Acid Biosynthesis: Evaluation of FabG, FabZ, and FabI as Drug Targets for Flavonoids. J. Med. Chem. 2006, 49, 3345–3353. [Google Scholar] [CrossRef] [PubMed]
- Kimber, M.S.; Martin, F.; Lu, Y.; Houston, S.; Vedadi, M.; Dharamsi, A.; Fiebig, K.M.; Schmid, M.; Rock, C.O. The Structure of (3R)-Hydroxyacyl-Acyl Carrier Protein Dehydratase (FabZ) from Pseudomonas Aeruginosa. J. Biol. Chem. 2004, 279, 52593–52602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waag, H.; Cronan, J.E. Functional Replacement of the Faba and Fabb Proteins of Escherichia Coli Fatty Acid Synthesis by Enterococcus Faecalis FabZ and FabF Homologues. J. Biol. Chem. 2004, 279, 34489–34495. [Google Scholar] [CrossRef] [Green Version]
- Bergler, H.; Fuchsbichler, S.; Högenauer, G.; Turnowsky, F. The Enoyl-[Acyl-Carrier-Protein] Reductase (FabI) of Escherichia Coli, Which Catalyzes a Key Regulatory Step in Fatty Acid Biosynthesis, Accepts NADH and NADPH as Cofactors and Is Inhibited by Palmitoyl-CoA. Eur. J. Biochem. 1996, 242, 689–694. [Google Scholar] [CrossRef]
- Heath, R.J.; Rock, C.O. Microbiology: A Triclosan-Resistant Bacterial Enzyme. Nature 2000, 406, 145–146. [Google Scholar] [CrossRef]
- Heath, R.J.; Rock, C.O. Regulation of Fatty Acid Elongation and Initiation by Acyl-Acyl Carrier Protein in Escherichia Coli. J. Biol. Chem. 1996, 271, 1833–1836. [Google Scholar] [CrossRef] [Green Version]
- Heath, R.J.; Rock, C.O. Enoyl-Acyl Carrier Protein Reductase (FabI) Plays a Determinant Role in Completing Cycles of Fatty Acid Elongation in Escherichia Coli. J. Biol. Chem. 1995, 270, 26538–26542. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Tonge, P.J. Inhibitors of FabI, an Enzyme Drug Target in the Bacterial Fatty Acid Biosynthesis Pathway. Acc. Chem. Res. 2008, 41, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Asturias, F.J.; Chadick, J.Z.; Cheung, I.K.; Stark, H.; Witkowski, A.; Joshi, A.K.; Smith, S. Structure and Molecular Organization of Mammalian Fatty Acid Synthase. Nat. Struct. Mol. Biol. 2005, 12, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Fage, C.D.; Lathouwers, T.; Vanmeert, M.; Gao, L.J.; Vrancken, K.; Lammens, E.M.; Weir, A.N.M.; Degroote, R.; Cuppens, H.; Kosol, S.; et al. The Kalimantacin Polyketide Antibiotics Inhibit Fatty Acid Biosynthesis in Staphylococcus Aureus by Targeting the Enoyl-Acyl Carrier Protein Binding Site of FabI. Angew. Chemie Int. Ed. 2020, 59, 10549–10556. [Google Scholar] [CrossRef]
- Ghattas, M.A.; Eissa, N.A.; Tessaro, F.; Perozzo, R.; Scapozza, L.; Obaid, D.; Atatreh, N. Structure-Based Drug Design and in Vitro Testing Reveal New Inhibitors of Enoyl-Acyl Carrier Protein Reductases. Chem. Biol. Drug Des. 2019, 94, 1545–1555. [Google Scholar] [CrossRef] [PubMed]
- Heath, R.J.; Rubin, J.R.; Holland, D.R.; Zhang, E.; Snow, M.E.; Rock, C.O. Mechanism of Triclosan Inhibition of Bacterial Fatty Acid Synthesis. J. Biol. Chem. 1999, 274, 11110–11114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghattas, M.A.; Mansour, R.A.; Atatreh, N.; Bryce, R.A. Analysis of Enoyl-Acyl Carrier Protein Reductase Structure and Interactions Yields an Efficient Virtual Screening Approach and Suggests a Potential Allosteric Site. Chem. Biol. Drug Des. 2016, 87, 131–142. [Google Scholar] [CrossRef]
- National Cancer Institute NCI Open Database Compounds. Available online: https://cactus.nci.nih.gov/download/nci/ (accessed on 11 February 2021).
- MDL Information Systems [MACCS] MACCS Keys. Available online: https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&cad=rja&uact=8&ved=2ahUKEwjO7vvXrf39AhUNVWwGHZfhARAQFnoECAkQAQ&url=https%3A%2F%2Fwww.bloomberg.com%2Fprofile%2Fcompany%2F2675Q%3AUS&usg=AOvVaw0JmVJJe9bCOQGdr6VotvUH (accessed on 11 February 2021).
- Breijyeh, Z.; Jubeh, B.; Karaman, R. Resistance of Gram-Negative Bacteria to Current Antibacterial Agents and Approaches to Resolve It. Molecules 2020, 25, 1340. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Montanari, F.; Ecker, G.F. Prediction of Drug–ABC-Transporter Interaction—Recent Advances and Future Challenges. Adv. Drug Deliv. Rev. 2015, 86, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple Selection Criteria for Drug-like Chemical Matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenk, R.; Schipani, A.; James, D.; Krasowski, A.; Gilbert, I.H.; Frearson, J.; Wyatt, P.G. Lessons Learnt from Assembling Screening Libraries for Drug Discovery for Neglected Diseases. ChemMedChem 2008, 3, 435–444. [Google Scholar] [CrossRef]
- Guimarães, C.R.W.; Cardozo, M. MM-GB/SA Rescoring of Docking Poses in Structure-Based Lead Optimization. J. Chem. Inf. Model. 2008, 48, 958–970. [Google Scholar] [CrossRef]
- Gerusz, V.; Denis, A.; Faivre, F.; Bonvin, Y.; Oxoby, M.; Briet, S.; LeFralliec, G.; Oliveira, C.; Desroy, N.; Raymond, C.; et al. From Triclosan toward the Clinic: Discovery of Nonbiocidal, Potent FabI Inhibitors for the Treatment of Resistant Bacteria. J. Med. Chem. 2012, 55, 9914–9928. [Google Scholar] [CrossRef]
- Kronenberger, T.; de Oliveira Fernades, P.; Drumond Franco, I.; Poso, A.; Gonçalves Maltarollo, V. Ligand- and Structure-Based Approaches of Escherichia Coli FabI Inhibition by Triclosan Derivatives: From Chemical Similarity to Protein Dynamics Influence. ChemMedChem 2019, 14, 1995–2004. [Google Scholar] [CrossRef] [Green Version]
- Priyadarshi, A.; Kim, E.E.; Hwang, K.Y. Structural Insights into Staphylococcus Aureus Enoyl-ACP Reductase (Fabl), in Complex with NADP End Triclosan. Proteins Struct. Funct. Bioinforma. 2010, 78, 480–486. [Google Scholar] [CrossRef]
- Sharma, A.; Vora, J.; Patel, D.; Sinha, S.; Jha, P.C.; Shrivastava, N. Identification of Natural Inhibitors against Prime Targets of SARS-CoV-2 Using Molecular Docking, Molecular Dynamics Simulation and MM-PBSA Approaches. J. Biomol. Struct. Dyn. 2022, 40, 3296–3311. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Panwar, A.; Sharma, A.K. Molecular Dynamic Simulation Study on Chromones and Flavonoids for the In Silico Designing of a Potential Ligand Inhibiting MTOR Pathway in Breast Cancer. Curr. Pharmacol. Reports 2020, 6, 373–379. [Google Scholar] [CrossRef]
- Al Qaraghuli, M.; Kubiak-Ossowska, K.; Mulheran, P. Thinking Outside the Laboratory: Analyses of Antibody Structure and Dynamics within Different Solvent Environments in Molecular Dynamics (MD) Simulations. Antibodies 2018, 7, 21. [Google Scholar] [CrossRef] [Green Version]
- Gorai, S.; Junghare, V.; Kundu, K.; Gharui, S.; Kumar, M.; Patro, B.S.; Nayak, S.K.; Hazra, S.; Mula, S. Synthesis of Dihydrobenzofuro[3,2-b]Chromenes as Potential 3CLpro Inhibitors of SARS-CoV-2: A Molecular Docking and Molecular Dynamics Study. ChemMedChem 2022, 17, e202100782. [Google Scholar] [CrossRef]
- Parikh, S.; Moynihan, D.P.; Xiao, G.; Tonge, P.J. Roles of Tyrosine 158 and Lysine 165 in the Catalytic Mechanism of InhA, the Enoyl-ACP Reductase from Mycobacterium Tuberculosis. Biochemistry 1999, 38, 13623–13634. [Google Scholar] [CrossRef] [PubMed]
- Schiebel, J.; Chang, A.; Merget, B.; Bommineni, G.R.; Yu, W.; Spagnuolo, L.A.; Baxter, M.V.; Tareilus, M.; Tonge, P.J.; Kisker, C.; et al. An Ordered Water Channel in Staphylococcus Aureus FabI: Unraveling the Mechanism of Substrate Recognition and Reduction. Biochemistry 2015, 54, 1943–1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, H.T.; Reynolds, K.A. Antibacterial Targets in Fatty Acid Biosynthesis. Curr. Opin. Microbiol. 2007, 10, 447–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Lu, J.; Ying, M.; Mu, J.; Li, P.; Liu, Y. Docking and Molecular Dynamics Studies on Triclosan Derivatives Binding to FabI. J. Mol. Model. 2017, 23, 25. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE). 2022.02 Chemical Computing Group ULC: 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7. 2022. Available online: https://www.chemcomp.com/Research-Citing_MOE.htm (accessed on 1 March 2022).
- Jorgensen, J.H.; Ferraro, M.J. Antimicrobial Susceptibility Testing: A Review of General Principles and Contemporary Practices. Clin. Infect. Dis. 2009, 49, 1749–1755. [Google Scholar] [CrossRef]
- National Committee for Clinical Laboratory Standards (NCCLS). Method for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, 11th ed.; National Committee for Clinical Laboratory Standards: Wayne, PA, USA, 2018. [Google Scholar]
- Cockerill, F.R.; Wikler, M.A.; Alder, J.; Dudley, M.N.; Eliopoulos, G.M.; Ferraro, M.J.; Hardy, D.J.; Hecht, D.W.; Hindler, J.A.; Patel, J. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically: Approved Standard, 9th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2012; Volume 32. [Google Scholar]
- Kaplan, N.; Albert, M.; Awrey, D.; Bardouniotis, E.; Berman, J.; Clarke, T.; Dorsey, M.; Hafkin, B.; Ramnauth, J.; Romanov, V.; et al. Mode of Action, in Vitro Activity, and in Vivo Efficacy of AFN-1252, a Selective Antistaphylococcal Fabi Inhibitor. Antimicrob. Agents Chemother. 2012, 56, 5865–5874. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Sharma, A.; Nandi, S.P. Identification of Potent Inhibitors of COVID-19 Main Protease Enzyme By Molecular Docking Study. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- LigPreP. Schrödinger; LLC: New York, NY, USA, 2021. [Google Scholar]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Śledź, P.; Caflisch, A. Protein Structure-Based Drug Design: From Docking to Molecular Dynamics. Curr. Opin. Struct. Biol. 2018, 48, 93–102. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An Overview of the Amber Biomolecular Simulation Package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Le Grand, S.; Götz, A.W.; Walker, R.C. SPFP: Speed without Compromise—A Mixed Precision Model for GPU Accelerated Molecular Dynamics Simulations. Comput. Phys. Commun. 2013, 184, 374–380. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Swiss institute of bioinformatics, SwissADME. Available online: http://www.swissadme.ch/index.php (accessed on 1 March 2022).
- Zhu, Y.; Zhang, S. Antibacterial Activity and Mechanism of Lacidophilin From Lactobacillus Pentosus Against Staphylococcus Aureus and Escherichia Coli. Front. Microbiol. 2020, 11, 2639. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Zone of Inhibition (mm) | |||
|---|---|---|---|---|
| MRSA | E. coli | B. subtilis | S. aureus | |
| RK01 | ++ | - | ++ | + |
| RK02 | ++ | ++ | +++ | ++ |
| RK03 | +++ | ++ | +++ | +++ |
| RK04 | +++ | - | ++++ | +++ |
| RK05 | ++ | - | - | + |
| RK06 | - | - | - | - |
| RK07 | - | - | - | - |
| RK08 | ++++ | - | +++ | +++ |
| RK09 | ++++ | - | ++++ | ++++ |
| RK10 | ++++ | ++ | ++++ | ++++ |
| Chloramphenicol | + | ++++ | ++++ | +++ |
| TCL † | ++++ | +++ | ++++ | ++++ |
| Dimethyl Sulfoxide | - | - | - | - |
| ID | MIC (μg/mL) | |||
|---|---|---|---|---|
| MRSA | E. coli | B. subtillis | S. aureus | |
| MN02 | 8.00 | 32.00 | 4.00 | 4.00 |
| TCL | 64.00 † | 0.50 † | 0.40 † | 0.13 † |
| Chloramphenicol | 74.00 † | 4.00 † | 4.00 † | 4.00 † |
| RK01 | 13.39 | - | 13.39 | 27.45 |
| RK02 | 41.54 | - | 41.54 | 41.54 |
| RK03 | 2.29 | - | 9.64 | 9.64 |
| RK04 | 20.10 | - | 9.81 | 9.81 |
| RK05 | 22.34 | - | - | 10.90 |
| RK06 | - | - | - | - |
| RK07 | - | - | - | - |
| RK08 | 15.67 | - | 7.64 | 15.67 |
| RK09 | 2.84 | - | 1.39 | 2.84 |
| RK10 | 2.70 | 47.64 | 1.32 | 1.32 |
| Molecule ID | Docking Score (kcal/mol) | MM-GBSA (kcal/mol) | MIC (μg/mL) S. aureus |
|---|---|---|---|
| MN02 | −7.94 | −34.60 | 4.00 |
| TCL | −8.70 | −32.32 | 0.13 † |
| RK01 | −6.03 | −27.22 | 27.45 |
| RK02 | −9.18 | −25.97 | 41.54 |
| RK03 | −8.71 | −23.47 | 9.64 |
| RK04 | −8.87 | −29.71 | 9.81 |
| RK05 | −7.48 | −28.08 | 10.90 |
| RK06 | −8.33 | −24.95 | - |
| RK07 | −9.21 | −22.62 | - |
| RK08 | −8.53 | −29.63 | 15.67 |
| RK09 | −8.88 | −30.26 | 2.84 |
| RK10 | −8.00 | −32.92 | 1.32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasan, S.; Kayed, K.; Ghemrawi, R.; Bataineh, N.A.; Mahgoub, R.E.; Audeh, R.; Aldulaymi, R.; Atatreh, N.; Ghattas, M.A. Molecular Modelling Study and Antibacterial Evaluation of Diphenylmethane Derivatives as Potential FabI Inhibitors. Molecules 2023, 28, 3000. https://doi.org/10.3390/molecules28073000
Hasan S, Kayed K, Ghemrawi R, Bataineh NA, Mahgoub RE, Audeh R, Aldulaymi R, Atatreh N, Ghattas MA. Molecular Modelling Study and Antibacterial Evaluation of Diphenylmethane Derivatives as Potential FabI Inhibitors. Molecules. 2023; 28(7):3000. https://doi.org/10.3390/molecules28073000
Chicago/Turabian StyleHasan, Shaima, Kawthar Kayed, Rose Ghemrawi, Nezar Al Bataineh, Radwa E. Mahgoub, Rola Audeh, Raghad Aldulaymi, Noor Atatreh, and Mohammad A. Ghattas. 2023. "Molecular Modelling Study and Antibacterial Evaluation of Diphenylmethane Derivatives as Potential FabI Inhibitors" Molecules 28, no. 7: 3000. https://doi.org/10.3390/molecules28073000