

Development of Cell Permeable NanoBRET Probes for the Measurement of PLK1 Target Engagement in Live Cells
 ,
,
Abstract
:
1. Introduction
2. Results
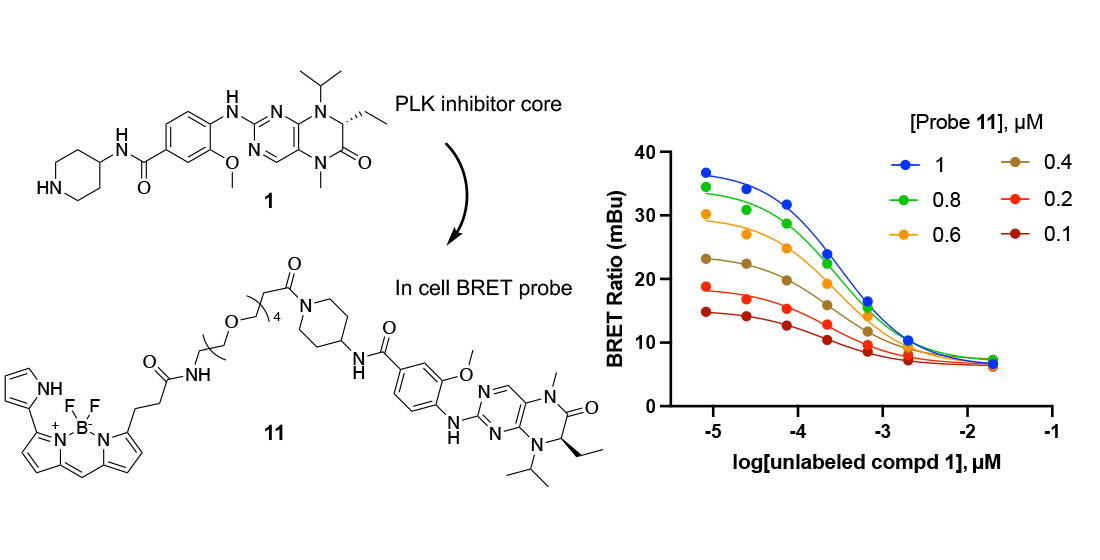
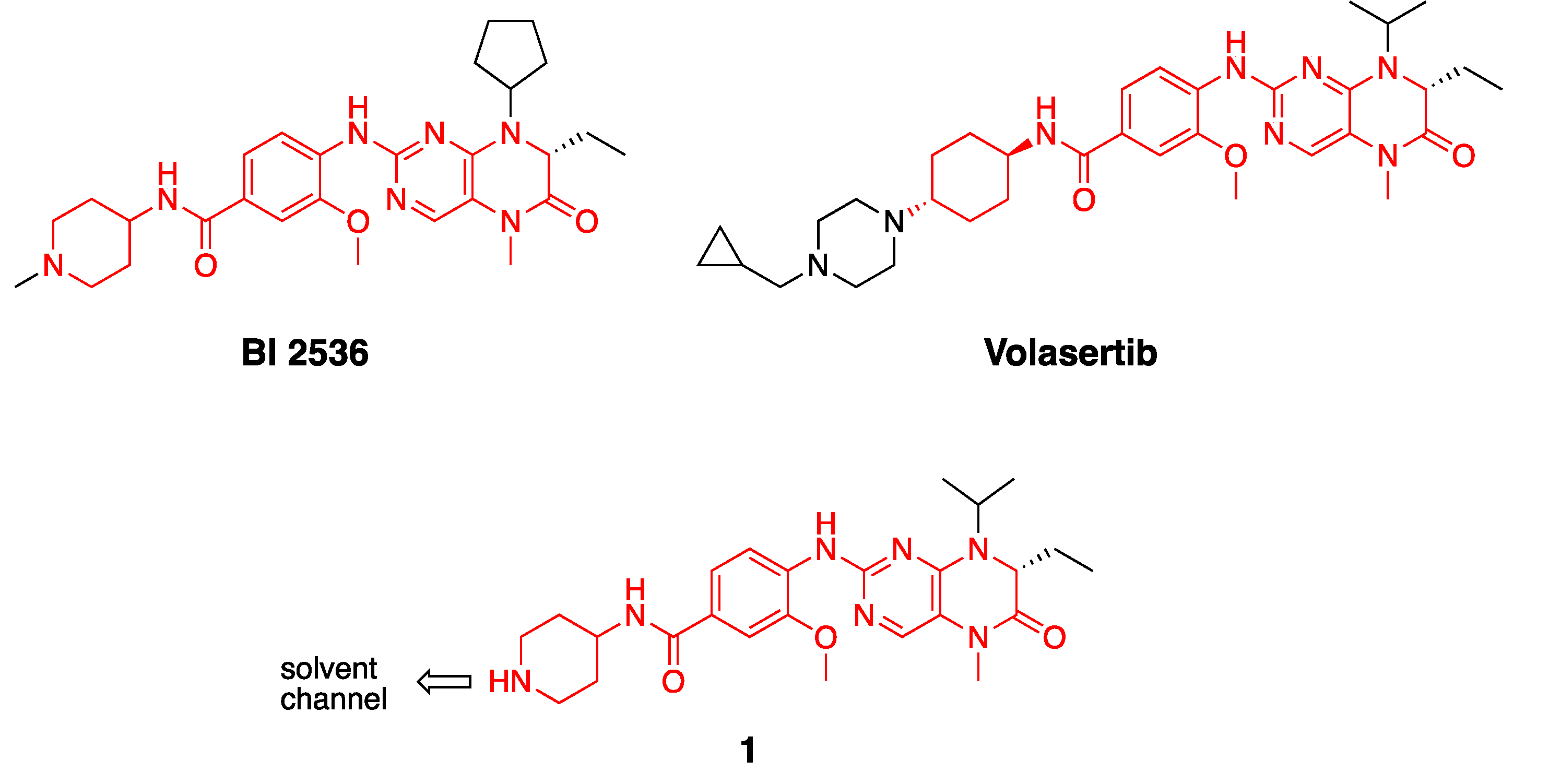
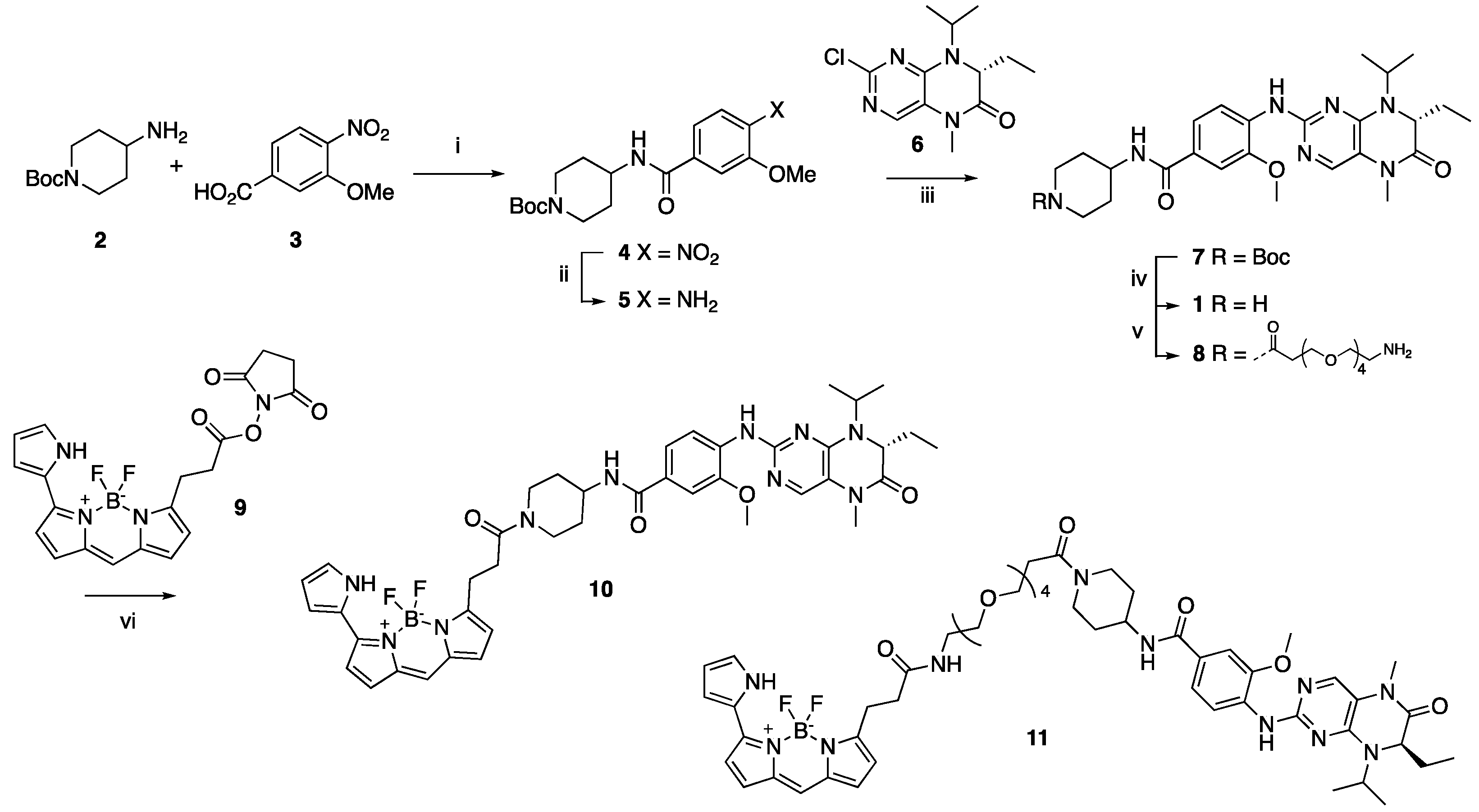
2.1. Synthesis of Fluorescence Energy Transfer Probes 10 and 11
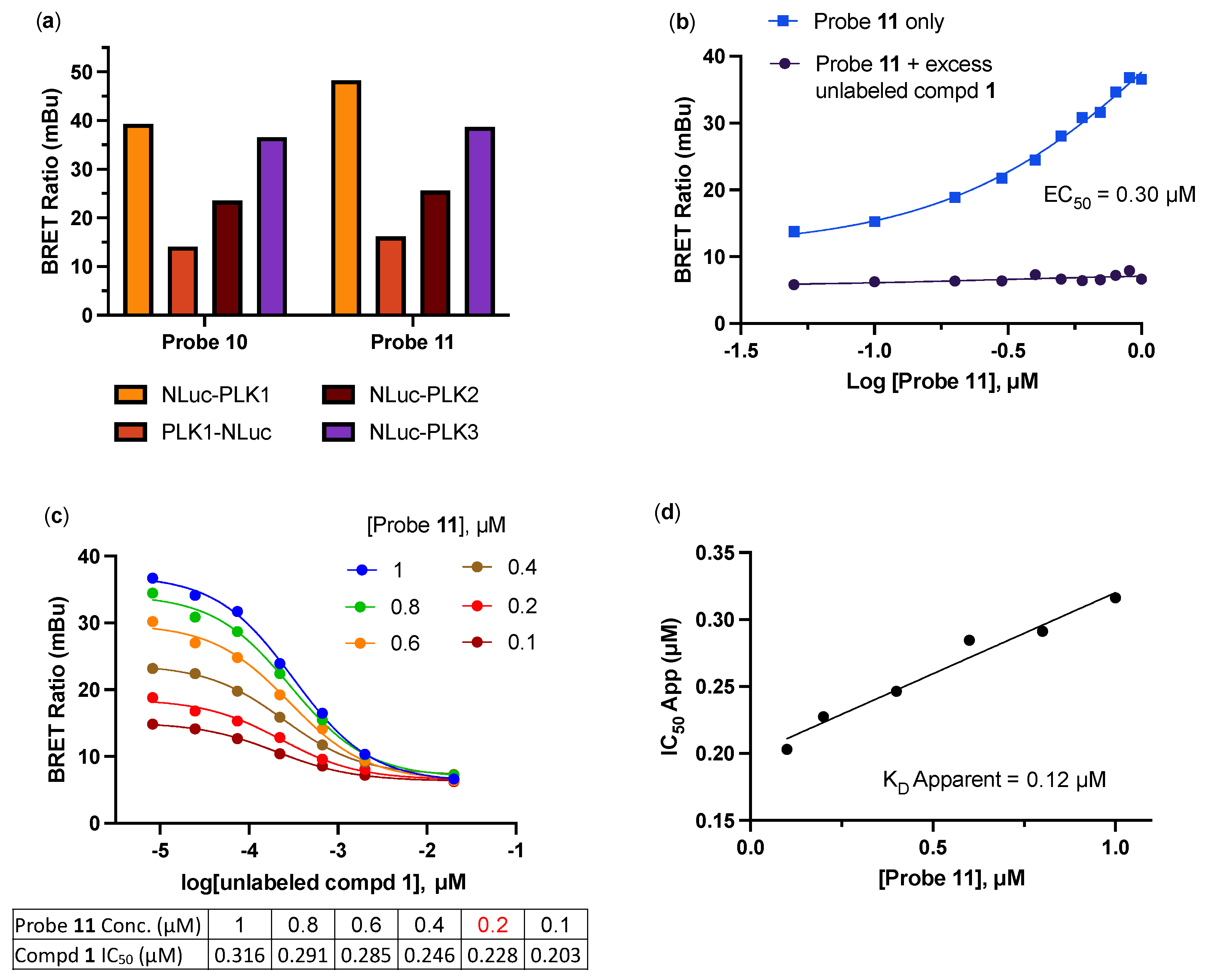
2.2. Development of a PLK1 NanoBRET Assay
2.3. In-Cell Target Engagement of PLK Inhibitors
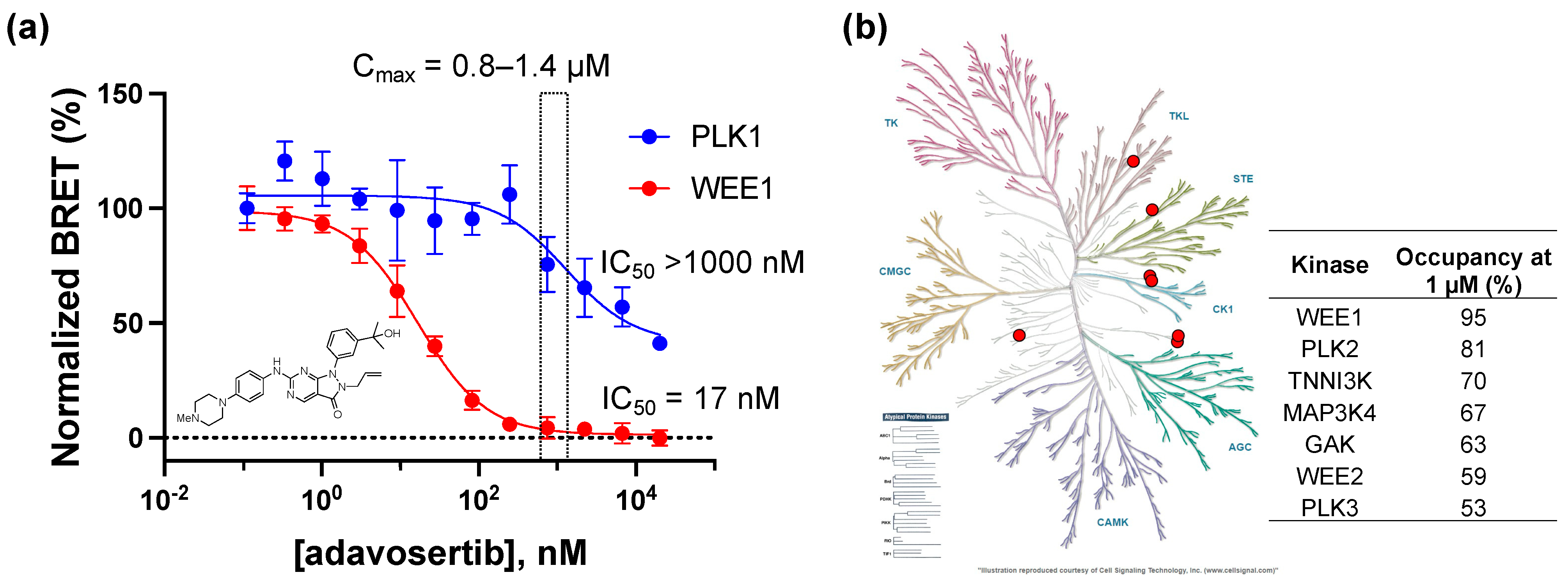
2.4. In-Cell Target Selectivity of Adavosertib
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Methods
4.1.2. Synthesis of Florescence Energy Transfer Probes 10 and 11
Tert-butyl 4-(3-methoxy-4-nitrobenzamido)piperidine-1-carboxylate (4)
Tert-butyl 4-(4-amino-3-methoxybenzamido)piperidine-1-carboxylate (5)
(R)-4-((7-ethyl-8-isopropyl-5-methyl-6-oxo-5,6,7,8-tetrahydropteridin-2-yl)amino)-3-methoxy-N-(piperidin-4-yl)benzamide (1)
(R)-N-(1-(3-(5,5-difluoro-7-(1H-pyrrol-2-yl)-5H-5λ,6λ-dipyrrolo[1,2-c:2’,1’-f][1-3]diazaborinin-3-yl)propanoyl)piperidin-4-yl)-4-((7-ethyl-8-isopropyl-5-methyl-6-oxo-5,6,7,8-tetrahydropteridin-2-yl)amino)-3-methoxybenzamide (10)
(R)-N-(1-(1-amino-3,6,9,12-tetraoxapentadecan-15-oyl)piperidin-4-yl)-4-((7-ethyl-8-isopropyl-5-methyl-6-oxo-5,6,7,8-tetrahydropteridin-2-yl)amino)-3-methoxybenzamide (8)
4.2. Biology
4.2.1. Cell Culture
4.2.2. NanoBRET assays
In-Cell BRET Assays
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Chiappa, M.; Petrella, S.; Damia, G.; Broggini, M.; Guffanti, F.; Ricci, F. Present and Future Perspective on PLK1 Inhibition in Cancer Treatment. Front. Oncol. 2022, 12, 903016. [Google Scholar] [CrossRef] [PubMed]
- Iliaki, S.; Beyaert, R.; Afonina, I.S. Polo-like kinase 1 (PLK1) signaling in cancer and beyond. Biochem. Pharmacol. 2021, 193, 114747. [Google Scholar] [CrossRef] [PubMed]
- Helmke, C.; Becker, S.; Strebhardt, K. The Role of Plk3 in Oncogenesis. Oncogene 2016, 35, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ni, C.; Lu, H. Polo-Like Kinase 2: From Principle to Practice. Front. Oncol 2022, 12, 956225. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, D.; Steegmaier, M.; Hoffmann, M.; Grauert, M.; Baum, A.; Quant, J.; Haslinger, C.; Garin-Chesa, P.; Adolf, G.R. BI 6727, a Polo-like Kinase Inhibitor with Improved Pharmacokinetic Profile and Broad Antitumor Activity. Clin Cancer Res 2009, 15, 3094–3102. [Google Scholar] [CrossRef]
- Steegmaier, M.; Hoffmann, M.; Baum, A.; Lenart, P.; Petronczki, M.; Krssak, M.; Gurtler, U.; Garin-Chesa, P.; Lieb, S.; Quant, J.; et al. BI 2536, a Potent and Selective Inhibitor of Polo-like Kinase 1, Inhibits Tumor Growth in Vivo. Curr Biol 2007, 17, 316–322. [Google Scholar] [CrossRef]
- Gilmartin, A.G.; Bleam, M.R.; Richter, M.C.; Erskine, S.G.; Kruger, R.G.; Madden, L.; Hassler, D.F.; Smith, G.K.; Gontarek, R.R.; Courtney, M.P.; et al. Distinct Concentration-dependent Effects of the Polo-like Kinase 1-specific Inhibitor GSK461364A, Including Differential Effect on Apoptosis. Cancer Res. 2009, 69, 6969–6977. [Google Scholar] [CrossRef]
- Beria, I.; Bossi, R.T.; Brasca, M.G.; Caruso, M.; Ceccarelli, W.; Fachin, G.; Fasolini, M.; Forte, B.; Fiorentini, F.; Pesenti, E.; et al. NMS-P937, a 4,5-Dihydro-1H-pyrazolo[4,3-h]quinazoline Derivative as Potent and Pelective Polo-like Kinase 1 Inhibitor. Bioorg. Med. Chem. Lett. 2011, 21, 2969–2974. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Carceles, J.; Caballero, E.; Gil, C.; Martinez, A. Kinase Inhibitors as Underexplored Antiviral Agents. J. Med. Chem. 2022, 65, 935–954. [Google Scholar] [CrossRef] [PubMed]
- Raghuvanshi, R.; Bharate, S.B. Recent Developments in the Use of Kinase Inhibitors for Management of Viral Infections. J. Med. Chem. 2022, 65, 893–921. [Google Scholar] [CrossRef]
- Meineke, R.; Rimmelzwaan, G.F.; Elbahesh, H. Influenza Virus Infections and Cellular Kinases. Viruses 2019, 11, 171. [Google Scholar] [CrossRef]
- Wei, L.; Adderley, J.; Leroy, D.; Drewry, D.H.; Wilson, D.W.; Kaushansky, A.; Doerig, C. Host-directed Therapy, an Untapped Opportunity for Antimalarial Intervention. Cell Rep. Med. 2021, 2, 100423. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.; Golubeva, V.; Remsing Rix, L.L.; Berndt, N.; Luo, Y.; Ward, G.A.; Gray, J.E.; Schonbrunn, E.; Lawrence, H.R.; Monteiro, A.N.A.; et al. Dual Targeting of WEE1 and PLK1 by AZD1775 Elicits Single Agent Cellular Anticancer Activity. ACS Chem. Biol. 2017, 12, 1883–1892. [Google Scholar] [CrossRef]
- Wells, C.I.; Al-Ali, H.; Andrews, D.M.; Asquith, C.R.M.; Axtman, A.D.; Dikic, I.; Ebner, D.; Ettmayer, P.; Fischer, C.; Frederiksen, M.; et al. The Kinase Chemogenomic Set (KCGS): An Open Science Resource for Kinase Vulnerability Identification. Int. J. Mol. Sci. 2021, 22, 566. [Google Scholar] [CrossRef] [PubMed]
- Vasta, J.D.; Corona, C.R.; Wilkinson, J.; Zimprich, C.A.; Hartnett, J.R.; Ingold, M.R.; Zimmerman, K.; Machleidt, T.; Kirkland, T.A.; Huwiler, K.G.; et al. Quantitative, Wide-Spectrum Kinase Profiling in Live Cells for Assessing the Effect of Cellular ATP on Target Engagement. Cell Chem. Biol. 2018, 25, 206–214 e211. [Google Scholar] [CrossRef]
- Wells, C.I.; Vasta, J.D.; Corona, C.R.; Wilkinson, J.; Zimprich, C.A.; Ingold, M.R.; Pickett, J.E.; Drewry, D.H.; Pugh, K.M.; Schwinn, M.K.; et al. Quantifying CDK Inhibitor Selectivity in Live Cells. Nat. Commun. 2020, 11, 2743. [Google Scholar] [CrossRef]
- Machleidt, T.; Woodroofe, C.C.; Schwinn, M.K.; Mendez, J.; Robers, M.B.; Zimmerman, K.; Otto, P.; Daniels, D.L.; Kirkland, T.A.; Wood, K.V. NanoBRET–A Novel BRET Platform for the Analysis of Protein-Protein Interactions. ACS Chem. Biol. 2015, 10, 1797–1804. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chen, Y.; Zhao, X.; Yu, S.; Yang, B.; Wu, T.; Guo, J.; Hao, C.; Zhao, D.; Cheng, M. Design, Synthesis and Biological Evaluation of Novel 7H-Pyrrolo[2,3-d]pyrimidine Derivatives as Potential FAK Inhibitors and Anticancer Agents. Eur. J. Med. Chem. 2019, 183, 111716. [Google Scholar] [CrossRef]
- Nie, Z.; Feher, V.; Natala, S.; McBride, C.; Kiryanov, A.; Jones, B.; Lam, B.; Liu, Y.; Kaldor, S.; Stafford, J.; et al. Discovery of TAK-960: An Orally Available Small Molecule Inhibitor of Polo-like Kinase 1 (PLK1). Bioorg. Med. Chem. Lett. 2013, 23, 3662–3666. [Google Scholar] [CrossRef]
- Chen, S.; Bartkovitz, D.; Cai, J.; Chen, Y.; Chen, Z.; Chu, X.J.; Le, K.; Le, N.T.; Luk, K.C.; Mischke, S.; et al. Identification of Novel, Potent and Selective Inhibitors of Polo-like Kinase 1. Bioorg. Med. Chem. Lett. 2012, 22, 1247–1250. [Google Scholar] [CrossRef]
- Duffey, M.O.; Vos, T.J.; Adams, R.; Alley, J.; Anthony, J.; Barrett, C.; Bharathan, I.; Bowman, D.; Bump, N.J.; Chau, R.; et al. Discovery of a Potent and Orally Bioavailable Benzolactam-derived Inhibitor of Polo-like Kinase 1 (MLN0905). J. Med. Chem. 2012, 55, 197–208. [Google Scholar] [CrossRef]
- Hirai, H.; Iwasawa, Y.; Okada, M.; Arai, T.; Nishibata, T.; Kobayashi, M.; Kimura, T.; Kaneko, N.; Ohtani, J.; Yamanaka, K.; et al. Small-molecule Inhibition of Wee1 Kinase by MK-1775 Selectively Sensitizes p53-deficient Tumor Cells to DNA-damaging Agents. Mol. Cancer Ther. 2009, 8, 2992–3000. [Google Scholar] [CrossRef]
- Serpico, A.F.; D’Alterio, G.; Vetrei, C.; Della Monica, R.; Nardella, L.; Visconti, R.; Grieco, D. Wee1 Rather Than Plk1 Is Inhibited by AZD1775 at Therapeutically Relevant Concentrations. Cancers (Basel) 2019, 11, 819. [Google Scholar] [CrossRef] [PubMed]
- Robers, M.B.; Wilkinson, J.M.; Vasta, J.D.; Berger, L.M.; Berger, B.T.; Knapp, S. Single Tracer-based Protocol for Broad-spectrum Kinase Profiling in Live Cells with NanoBRET. STAR Protoc. 2021, 2, 100822. [Google Scholar] [CrossRef] [PubMed]
- Do, K.; Wilsker, D.; Ji, J.; Zlott, J.; Freshwater, T.; Kinders, R.J.; Collins, J.; Chen, A.P.; Doroshow, J.H.; Kummar, S. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. J. Clin. Oncol. 2015, 33, 3409–3415. [Google Scholar] [CrossRef] [PubMed]
- Leijen, S.; van Geel, R.M.; Sonke, G.S.; de Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients With TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef]
- Becher, I.; Savitski, M.M.; Savitski, M.F.; Hopf, C.; Bantscheff, M.; Drewes, G. Affinity Profiling of the Cellular Kinome for the Nucleotide Cofactors ATP, ADP, and GTP. ACS Chem. Biol. 2013, 8, 599–607. [Google Scholar] [CrossRef]
- Knight, Z.A.; Shokat, K.M. Features of Selective Kinase Inhibitors. Chem. Biol. 2005, 12, 621–637. [Google Scholar] [CrossRef]
- Gheghiani, L.; Loew, D.; Lombard, B.; Mansfeld, J.; Gavet, O. PLK1 Activation in Late G2 Sets Up Commitment to Mitosis. Cell Rep. 2017, 19, 2060–2073. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.R.; Hall, M.P.; Zimprich, C.A.; Robers, M.B.; Duellman, S.J.; Machleidt, T.; Rodriguez, J.; Zhou, W. Highly Potent Cell-Permeable and Impermeable NanoLuc Luciferase Inhibitors. ACS Chem. Biol. 2017, 12, 1028–1037. [Google Scholar] [CrossRef]
- Leijen, S.; van Geel, R.M.; Pavlick, A.C.; Tibes, R.; Rosen, L.; Razak, A.R.; Lam, R.; Demuth, T.; Rose, S.; Lee, M.A.; et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination With Gemcitabine, Cisplatin, or Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2016, 34, 4371–4380. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Ref. | Reported Enzyme Inhibition IC50 (nM) a | NanoBRET Target Engagement IC50 (nM) c | Reported Cell Proliferation IC50 (nM)a | ||||
|---|---|---|---|---|---|---|---|---|
| PLK1 | PLK2 | PLK3 | PLK1 | PLK2 | PLK3 | |||
| BI2536 | [6] | 0.83 | 3.5 | 9.0 | 3.9 | 9.0 | 42 | 2–25 |
| Volasertib | [5] | 0.87 | 5.0 | 56 | 18 | 23 | 59 | 11–37 |
| TPKI-26 | [14,19] | 2.0 | n.d. | n.d. | 18 | 64 | 1700 | 5 |
| Ro3280 | [20] | 3.0 | n.d. | n.d. | 3.5 | 45 | 180 | 6–45 |
| GSK461364 | [7] | 0.5 b | 860 b | 1000 b | 4.2 | >10,000 | >10,000 | 10–100 |
| MLN0905 | [21] | 2.0 | n.d. | n.d. | 14 | 6600 | 3000 | 9 |
| Onvansertib | [8] | 2.0 | >10,000 | >10,000 | 21 | >10,000 | >10,000 | 42 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Smith, J.L.; Beck, M.T.; Wilkinson, J.M.; Michaud, A.; Vasta, J.D.; Robers, M.B.; Willson, T.M. Development of Cell Permeable NanoBRET Probes for the Measurement of PLK1 Target Engagement in Live Cells. Molecules 2023, 28, 2950. https://doi.org/10.3390/molecules28072950
Yang X, Smith JL, Beck MT, Wilkinson JM, Michaud A, Vasta JD, Robers MB, Willson TM. Development of Cell Permeable NanoBRET Probes for the Measurement of PLK1 Target Engagement in Live Cells. Molecules. 2023; 28(7):2950. https://doi.org/10.3390/molecules28072950
Chicago/Turabian StyleYang, Xuan, Jeffery L. Smith, Michael T. Beck, Jennifer M. Wilkinson, Ani Michaud, James D. Vasta, Matthew B. Robers, and Timothy M. Willson. 2023. "Development of Cell Permeable NanoBRET Probes for the Measurement of PLK1 Target Engagement in Live Cells" Molecules 28, no. 7: 2950. https://doi.org/10.3390/molecules28072950