
Synthesis of Mono- and Polyazole Hybrids Based on Polyfluoroflavones


Abstract
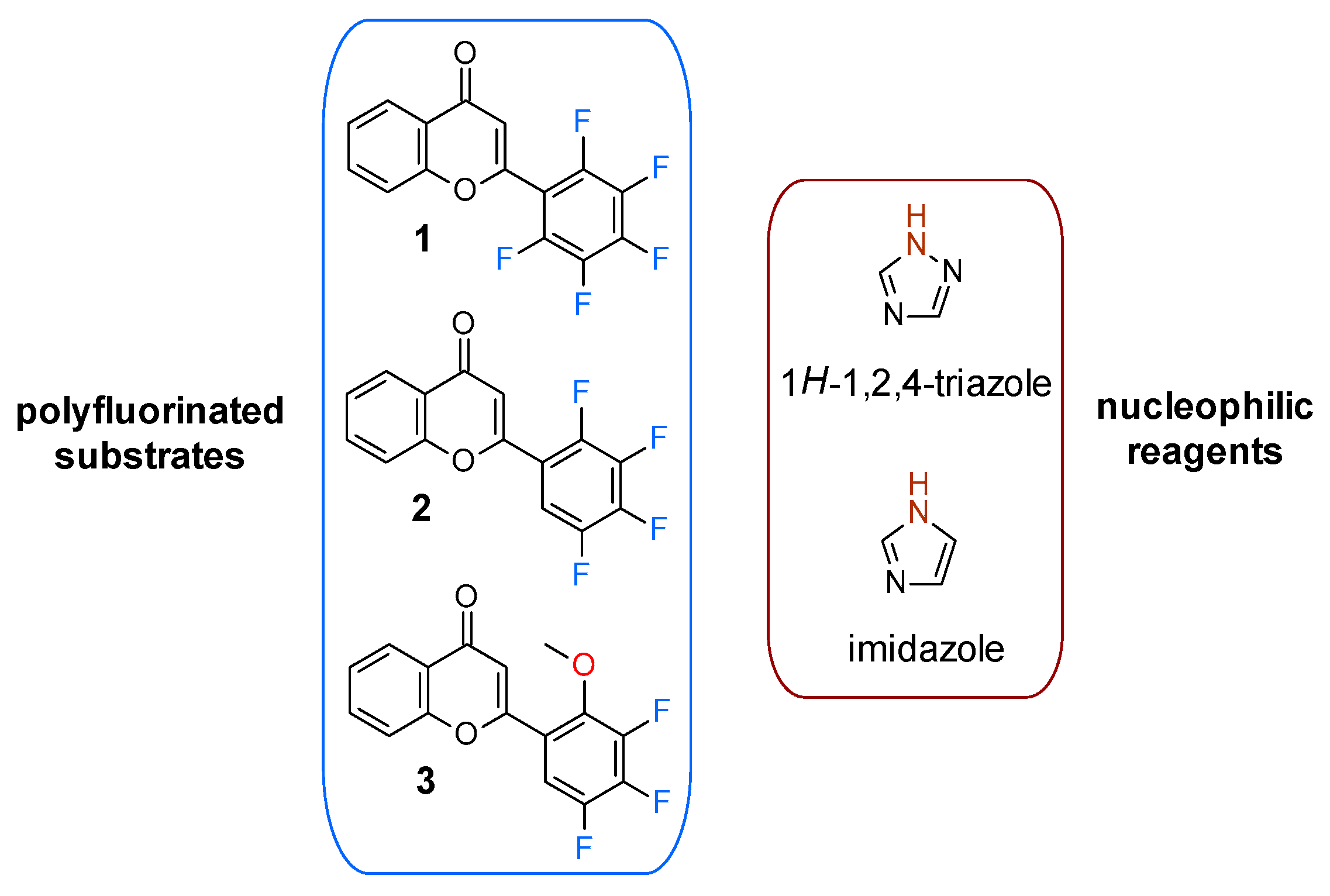
:1. Introduction
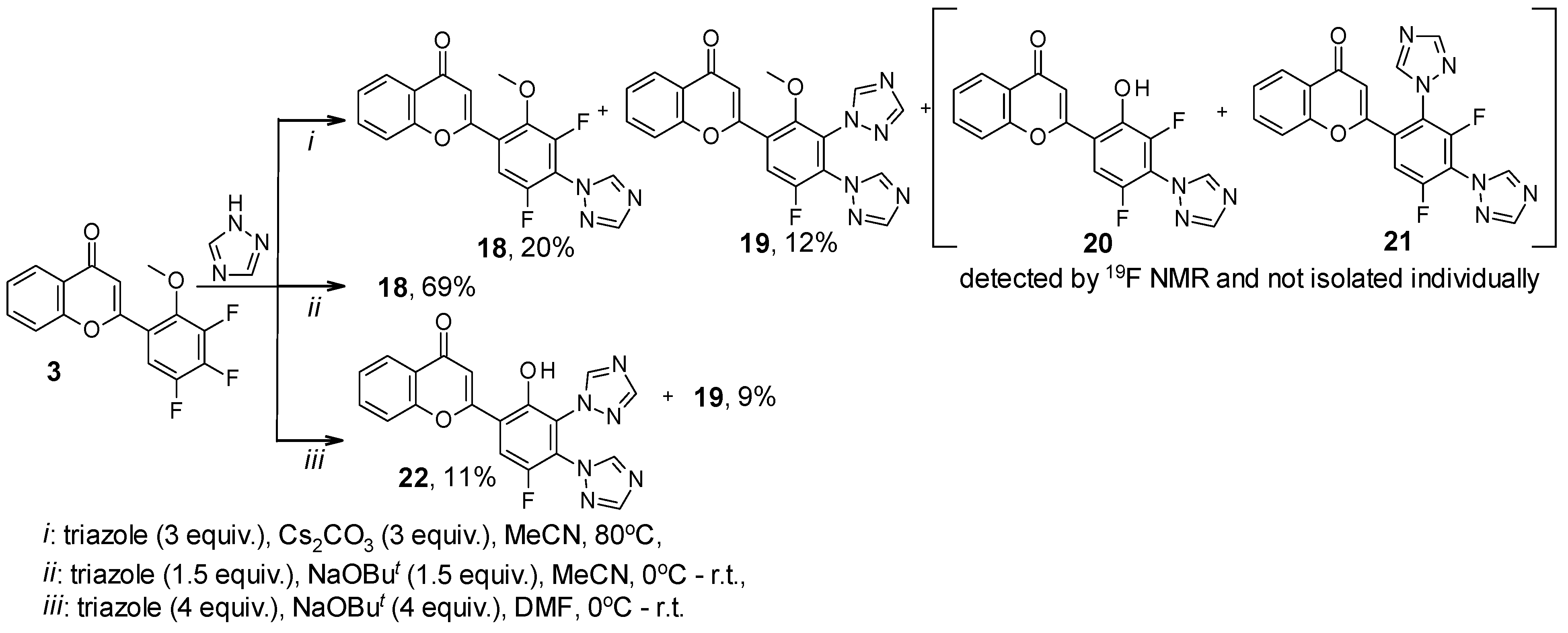
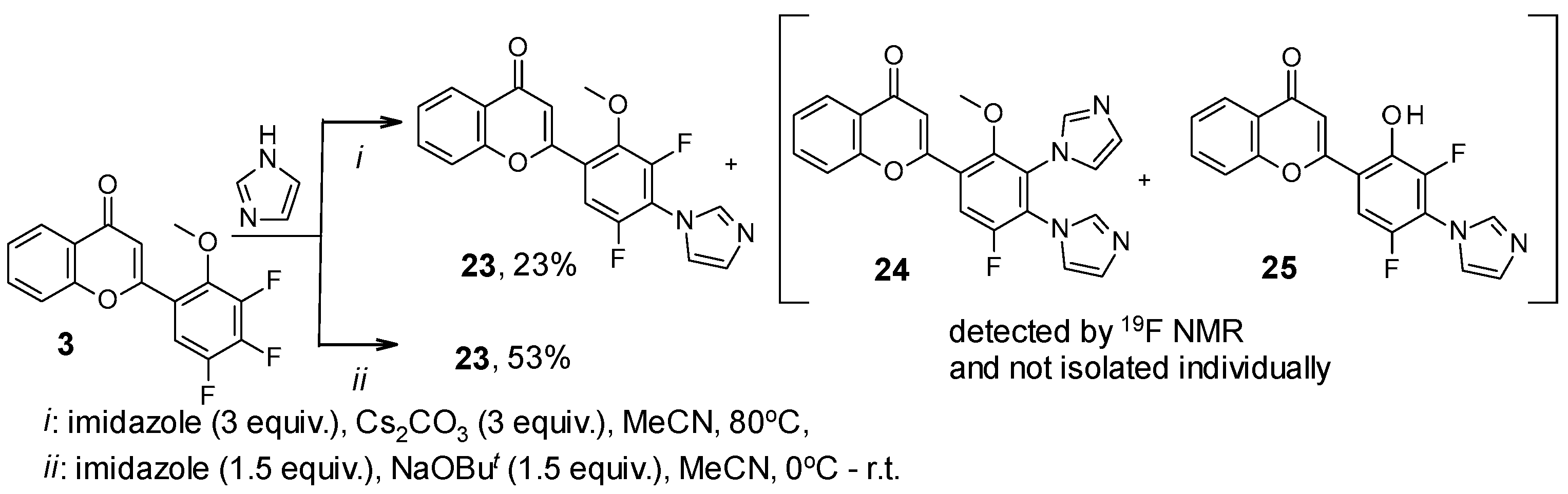
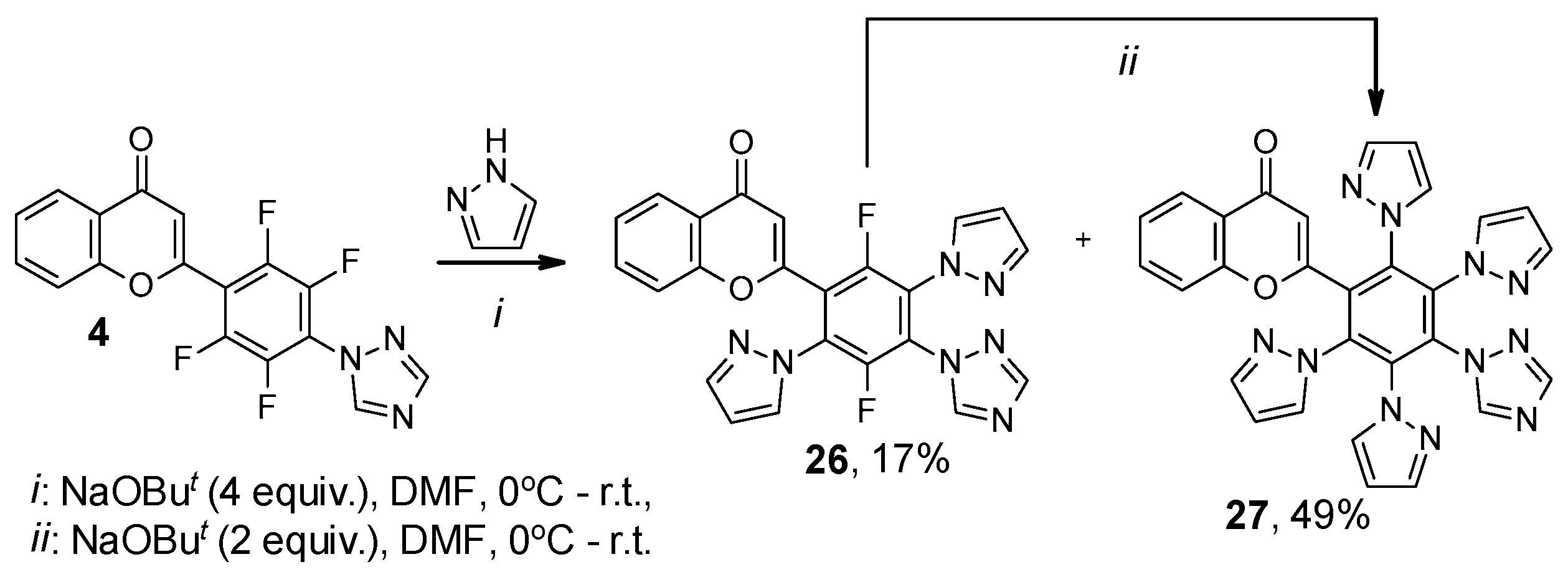
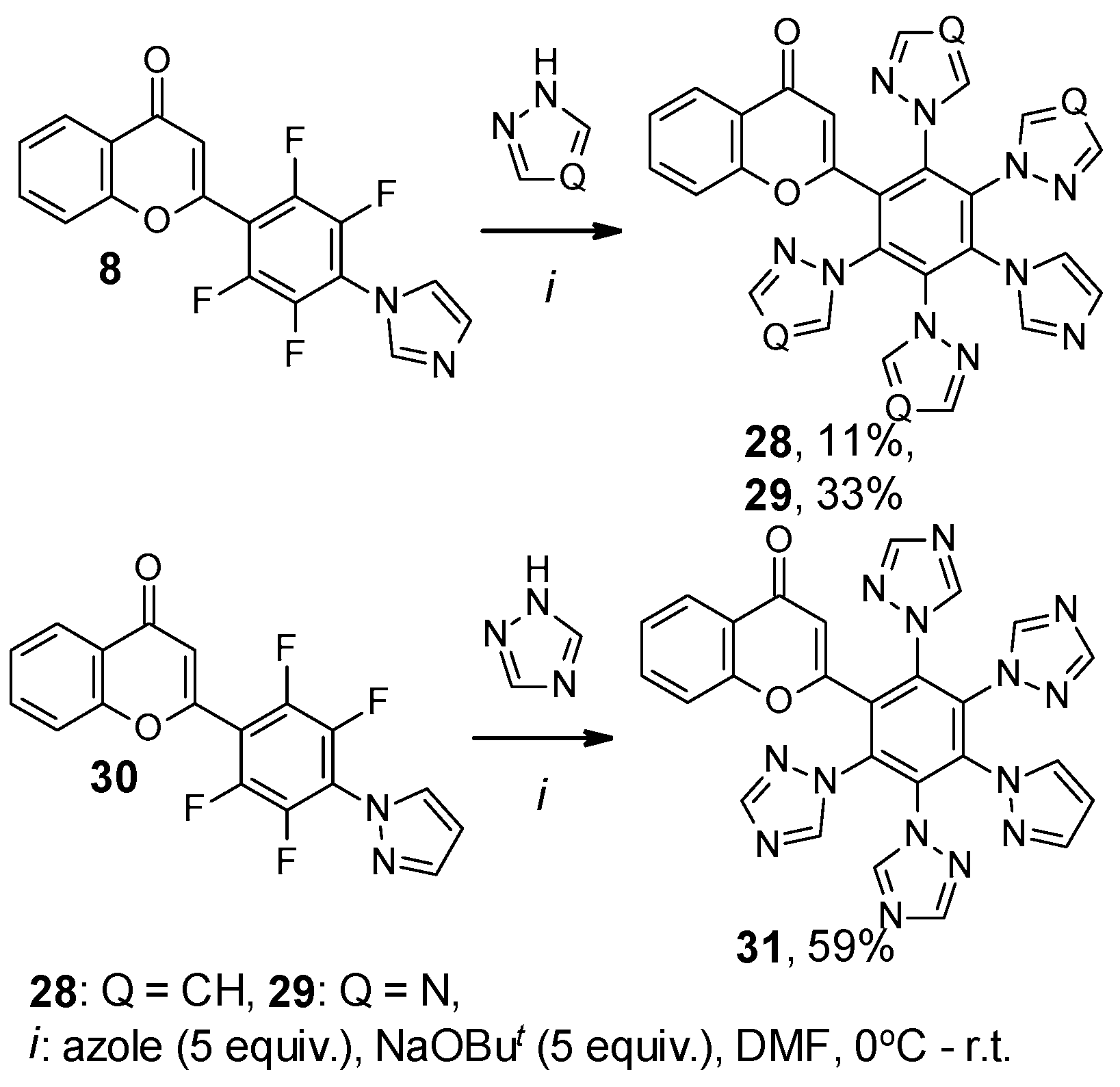
2. Results
3. Materials and Methods
3.1. Chemistry: General Information and Synthetic Techniques
3.2. Spectral and Elemental Analysis Data of Synthesized Compounds
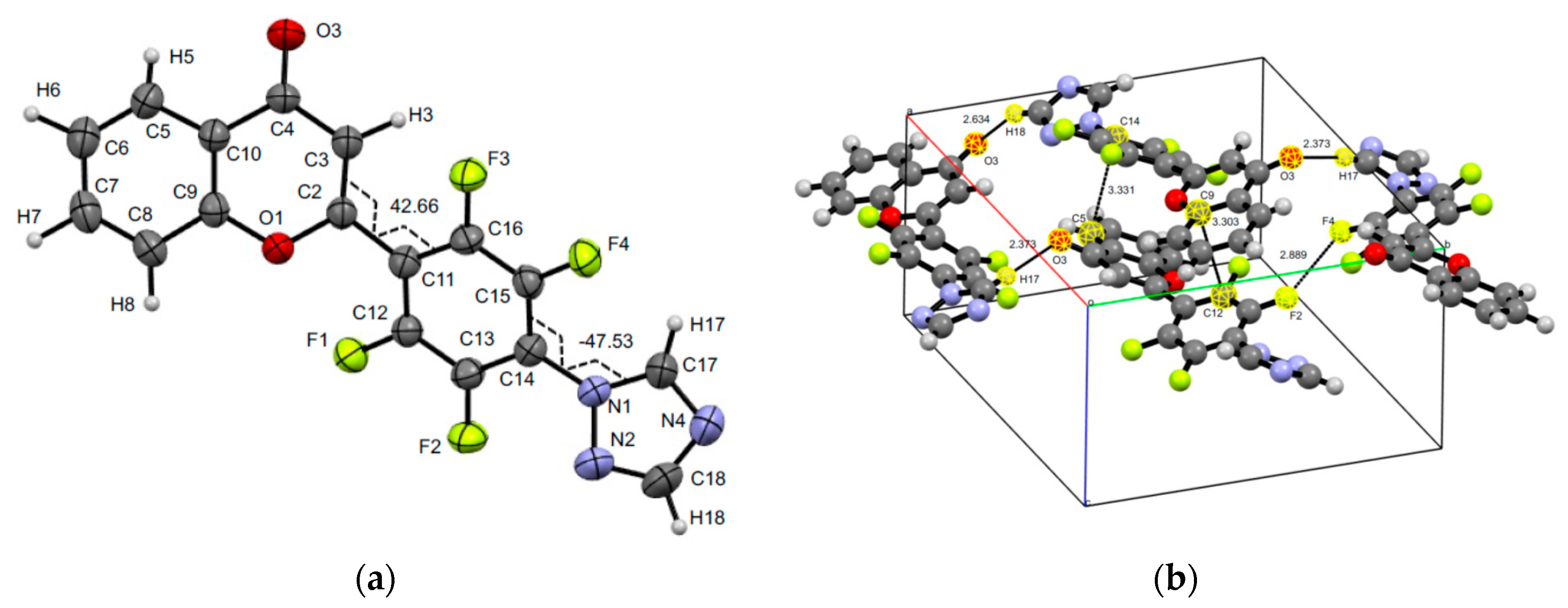
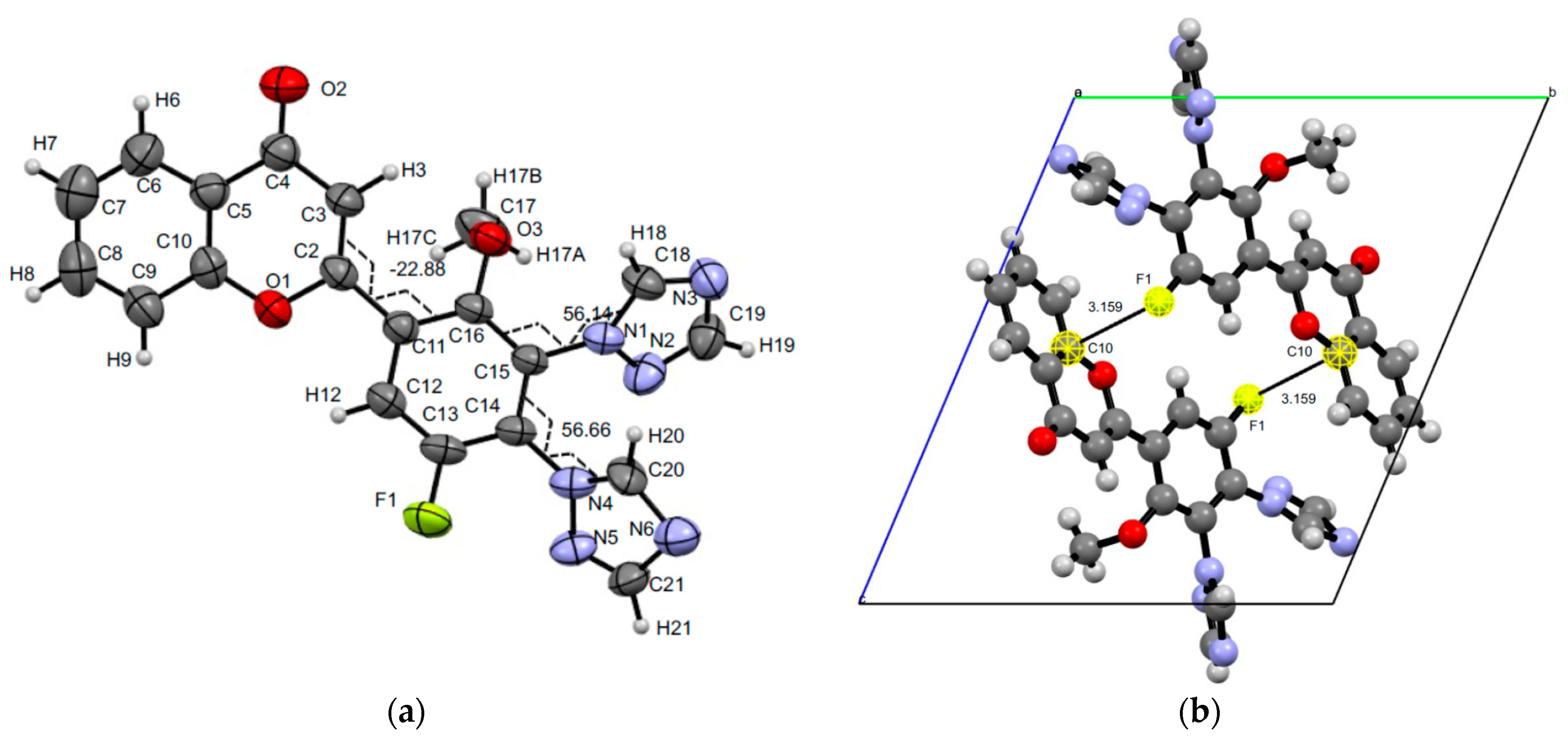
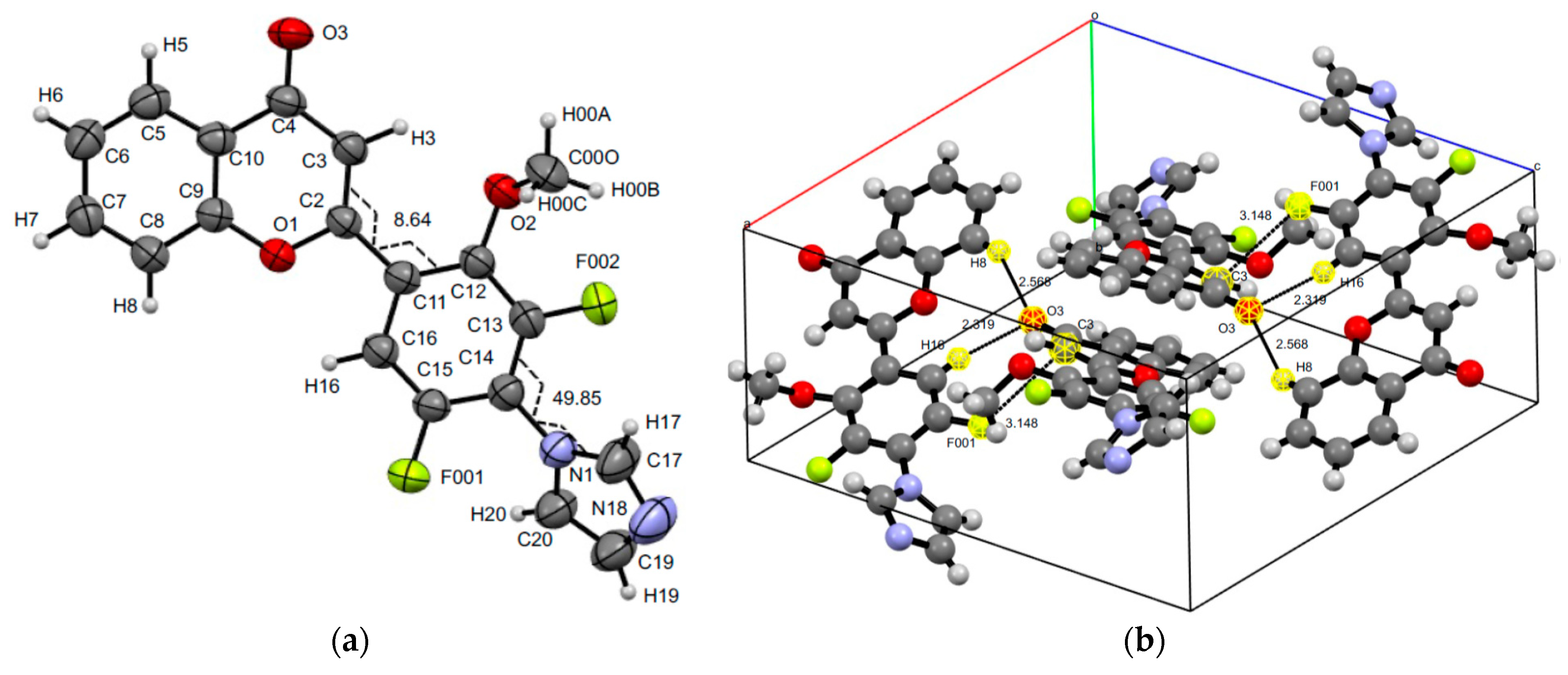
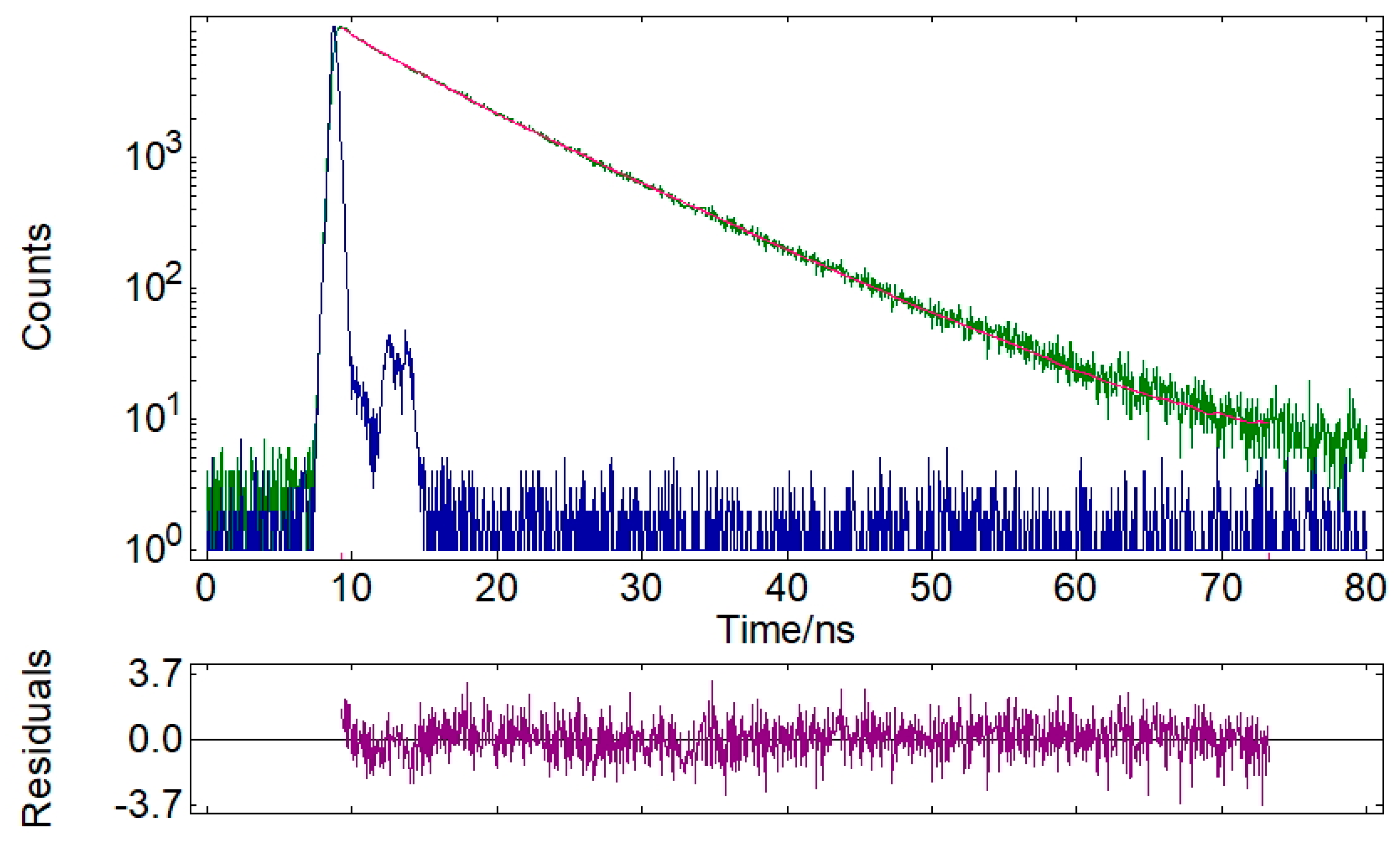
3.3. XRD Experiments
3.4. Fungistatic Activity Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | H3 | F2 | F3 | F5 | ||||
| δ | J | δ | J | δ | J | δ | J | |
| 9 | 6.52, d | 5JHF 1.4 | 26.33 ddd | 3JFF 22.6 5JFF 13.0 5JFH 1.4 | 23.60 d | 3JFF 22.4 | 30.57 d | 5JFF 13.1 |
| 10 | 6.51, d | 5JHF 1.3 | 41.18 dd | 5JFF 14.7 5JFH 1.3 | – | – | 31.47 d | 5JFF 14.7 |
| A | 6.44, d | 5JHF 1.1 | 29.51 dd | 5JFF 14.4 5JFH 1.1 | – | – | 39.83 d | 5JFF 14.3 |
Appendix B
| Compound | Solid | |||||
|---|---|---|---|---|---|---|
| τ1, ns | f1, % | τ2, ns | f2, % | τ avg, ns | χ2 | |
| 28 | 3.57 | 16.5 | 8.53 | 83.5 | 7.72 | 1.109 |
| 32 | 2.84 | 21.3 | 7.88 | 78.7 | 6.81 | 1.206 |


Appendix C
| No | Compounds | MIC (μg/mL)/MIC50 (μg/mL) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | R5 | T. rubrum | E. floccosum | M. canis | C. parapsilosis | |
| 4 | F | F | trz | F | F | >200 | >200 | >200 | >200 |
| 8 | F | F | imz | F | F | 100/25 | 200/100 | 100/50 | >200 |
| 11 | H | F | trz | F | F | >200 | >200 | >200 | >200 |
| 14 | H | trz | trz | trz | trz | 100/25 | 200/100 | 100/50 | >200 |
| 18 | OMe | F | trz | F | H | >200 | >200 | >200 | >200 |
| 19 | OMe | trz | trz | F | H | - | >200/50 | >200/200 | >200/200 |
| 23 | OMe | F | imz | F | H | >200/12.5 | >200/12.5 | >200/6.25 | >200 |
| 26 | F | pz | trz | F | pz | >200 | >200 | >200 | >200 |
| 33 | OMe | pz | pz | F | H | >100/1.56 | >100/1.56 | >100 | >100 |
| 34 | F | pz | pz | F | pz | >100 | >100 | >100 | >100 |
| 35 | pz | pz | pz | pz | pz | >100 | >100 | >100 | >100 |
| Fluconazole | 3.12 | 1.56 | 3.12 | 0.5–2 | |||||
References
- Tian, S.; Luo, T.; Zhu, Y.; Wan, J.-P. Recent advances in diversification of chromones and flavones by direct C–H bond activation or functionalization. Chin. Chem. Lett. 2020, 31, 3073–3082. [Google Scholar] [CrossRef]
- Kshatriya, R.; Jejurkar, V.P.; Saha, S. Recent advances in the synthetic methodologies of flavones. Tetrahedron 2018, 74, 811–833. [Google Scholar] [CrossRef]
- Spagnuolo, C.; Moccia, S.; Russo, G.L. Anti-inflammatory effects of flavonoids in neurodegenerative disorders. Eur. J. Med. Chem. 2018, 153, 105–115. [Google Scholar] [CrossRef]
- Singh, M.; Kaur, M.; Silakari, O. Flavones: An important scaffold for medicinal chemistry. Eur. J. Med. Chem. 2014, 84, 206–239. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.K.; Singh, H.; Satyanarayana, M.; Srivastava, S.P.; Tiwari, P.; Singh, A.B.; Dwivedi, A.K.; Singh, S.K.; Srivastava, M.; Nath, C.; et al. Flavone-based novel antidiabetic and antidyslipidemic agents. J. Med. Chem. 2012, 55, 4551–4567. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.-M.; Benfodda, Z.; Dunyach-Rémy, C.; Bénimélis, D.; Roulard, R.; Fontaine, J.-X.; Mathiron, D.; Quéro, A.; Molinié, R.; Meffre, P. Isolation and identification of flavones responsible for the antibacterial activities of Tillandsia bergeri extracts. ACS Omega 2022, 7, 35851–35862. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Wu, M.; Xiang, S.; Song, T.; Li, Y.; Long, B.; Feng, C.; Shi, Z. Total syntheses and antibacterial evaluations of neocyclomorusin and related flavones. J. Nat. Prod. 2022, 85, 2217–2225. [Google Scholar] [CrossRef] [PubMed]
- Rubin, D.; Sansom, C.E.; Lucas, N.T.; McAdam, J.C.; Simpson, J.; Lord, J.M.; Perry, N.B. O-Acylated flavones in the alpine daisy Celmisia viscosa: Intraspecific variation. J. Nat. Prod. 2022, 85, 1904–1911. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cao, Y.; Chen, S.; Lin, J.; Yang, X.; Huang, D. Structure–activity relationship (SAR) of flavones on their anti-inflammatory activity in murine macrophages in culture through the NF-κB pathway and c-Src kinase receptor. J. Agric. Food Chem. 2022, 70, 8788–8798. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-Y.; Chen, M.-Y.; Hsu, C.; Kuan, K.-Y.; Chang, C.-F.; Wang, C.-W.; Hsu, C.-P.; Su, N.-W. Luteolin phosphate derivatives generated by cultivating Bacillus subtilis var. Natto BCRC 80517 with luteolin. J. Agric. Food Chem. 2022, 70, 8738–8745. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Wu, M.; Li, Y.; Lu, L.; Qin, J.; He, Y.; Shi, Z. Total syntheses and anti-inflammatory evaluations of pongamosides A–C, natural furanoflavonoid glucosides from fruit of Pongamia pinnata (L.) Pierre. J. Nat. Prod. 2022, 85, 1118–1127. [Google Scholar] [CrossRef]
- Li, J.; Tan, L.-H.; Zou, H.; Zou, Z.-X.; Long, H.-P.; Wang, W.-X.; Xu, P.-S.; Liu, L.-F.; Xu, K.-P.; Tan, G.-S. Palhinosides A–H: Flavone glucosidic truxinate esters with neuroprotective activities from Palhinhaea cernua. J. Nat. Prod. 2020, 83, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Koh, J.-J.; Aung, T.T.; Sin, W.L.W.; Lim, F.; Wang, L.; Lakshminarayanan, R.; Zhou, L.; Tan, D.T.H.; Cao, D.; et al. Semisynthetic flavone-derived antimicrobials with therapeutic potential against methicillin-resistant Staphylococcus aureus (MRSA). J. Med. Chem. 2017, 60, 6152–6165. [Google Scholar] [CrossRef]
- Pajtás, D.; Kónya, K.; Kiss-Szikszai, A.; Džubák, P.; Pethő, Z.; Varga, Z.; Panyi, G.; Patonay, T. Optimization of the synthesis of flavone–amino acid and flavone–dipeptide hybrids via Buchwald–Hartwig reaction. J. Org. Chem. 2017, 82, 4578–4587. [Google Scholar] [CrossRef] [PubMed]
- Byun, Y.; Moon, K.; Park, J.; Ghosh, P.; Mishra, N.K.; Kim, I.S. Methylene thiazolidinediones as alkylation reagents in catalytic C–H functionalization: Rapid access to glitazones. Org. Lett. 2022, 24, 8578–8583. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Schaus, S.E.; Porco, J.A., Jr. Metal-catalyzed cascade rearrangements of 3-alkynyl flavone ethers. Org. Lett. 2013, 15, 1962–1965. [Google Scholar] [CrossRef] [Green Version]
- Hosseini, S.; Thapa, B.; Medeiros, M.J.; Pasciak, E.M.; Pence, M.A.; Twum, E.B.; Karty, J.A.; Gao, X.; Raghavachari, K.; Peters, D.G.; et al. Electrosynthesis of a baurone by controlled dimerization of flavone: Mechanistic insight and large-scale application. J. Org. Chem. 2020, 85, 10658–10669. [Google Scholar] [CrossRef]
- Tang, Q.; Bian, Z.; Wu, W.; Wang, J.; Xie, P.; Pittman, C.U., Jr.; Zhou, A. Making flavone thi-oethers using halides and powdered sulfur or Na2S2O3. J. Org. Chem. 2017, 82, 10617–10622. [Google Scholar] [CrossRef]
- Tokárová, Z.; Balogh, R.; Tisovský, P.; Hrnčariková, K.; Végh, D. Direct nucleophilic substitution of polyfluorobenzenes with pyrrole and 2,5-dimethylpyrrole. J. Fluorine Chem. 2017, 204, 59–64. [Google Scholar] [CrossRef]
- Gerencsér, J.; Balázs, A.; Dormán, G. Synthesis and modification of heterocycles by metal-catalyzed cross-coupling reactions. In Topics in Heterocyclic Chemistry; Patonay, T., Kónya, K., Eds.; Springer International Publishing AG: Cham, Switzerland, 2016; Volume 45. [Google Scholar]
- Kong, X.; Zhang, H.; Cao, C.; Shi, Y.; Pang, G. Effective transition metal free and selective C–F activation under mild conditions. RSC Adv. 2015, 5, 7035–7048. [Google Scholar] [CrossRef]
- Boelke, A.; Sadat, S.; Lork, E.; Nachtsheim, B.J. Pseudocyclic bis-N-heterocycle-stabilized iodanes—Synthesis, characterization and applications. Chem. Commun. 2021, 57, 7434–7437. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Wang, L.; Qin, D.; Zhou, J.; Duan, H. Two novel fluorescent probes as systematic sensors for multiple metal ions: Focus on detection of Hg2+. ACS Omega 2020, 5, 24285–24295. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Hong, X.; Cui, H.-Z.; Huang, S.; Yi, Y.; Hou, X.-F. The construction of C–N, C–O, and C(sp2)–C(sp3) bonds from fluorine-substituted 2-aryl benzazoles for direct synthesis of N-, O-, C-functionalized 2-aryl benzazole derivatives. J. Org. Chem. 2018, 83, 6363–6372. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.M.M.; Silva, V.L.M.; Silva, A.M.S. Synthesis of chromone-related pyrazole compounds. Molecules 2017, 22, 1665–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shcherbakov, K.V.; Panova, M.A.; Burgart, Y.V.; Saloutin, V.I. Selective nucleophilic aromatic substitution of 2-(polyfluorophenyl)-4H-chromen-4-ones with pyrazole. J. Fluorine Chem. 2022, 263, 110034. [Google Scholar] [CrossRef]
- Shcherbakov, K.V.; Artemyeva, M.A.; Burgart, Y.V.; Saloutin, V.I.; Volobueva, A.S.; Misiurina, M.A.; Esaulkova, Y.L.; Sinegubova, E.O.; Zarubaev, V.V. 7-Imidazolylsubstituted 4’-methoxy and 3’,4’-dimethoxy-containing polyfluoroflavones as promising antiviral agents. J. Fluorine Chem. 2020, 240, 109657. [Google Scholar] [CrossRef]
- Podlech, J. Elimination of fluorine to form C–N bonds. In Organo-Fluorine Compounds, 4th ed.; Baasner, B., Hagemann, H., Tatlow, J.C., Eds.; Georg Thieme Verlag: Stuttgart, Germany, 1999; pp. 449–464. [Google Scholar]
- Shcherbakov, K.V.; Panova, M.A.; Burgart, Y.V.; Zarubaev, V.V.; Gerasimova, N.A.; Evstigneeva, N.P.; Saloutin, V.I. The synthesis and biological evaluation of A- and B-ring fluorinated flavones and their key intermediates. J. Fluorine Chem. 2021, 249, 109857. [Google Scholar] [CrossRef]
- Shcherbakov, K.V.; Artemyeva, M.A.; Burgart, Y.V.; Evstigneeva, N.P.; Gerasimova, N.A.; Zilberberg, N.V.; Kungurov, N.V.; Saloutin, V.I.; Chupakhin, O.N. Transformations of 3-acyl-4H-polyfluorochromen-4-ones under the action of amino acids and biogenic amines. J. Fluorine Chem. 2019, 226, 109354. [Google Scholar] [CrossRef]
- Shcherbakov, K.V.; Burgart, Y.V.; Saloutin, V.I.; Chupakhin, O.N. Modification of polyfluoro-containing 3-(ethoxycarbonyl)flavones by biogenic amines and amino acids. Curr. Org. Synth. 2018, 15, 707–714. [Google Scholar] [CrossRef]
- Shcherbakov, K.V.; Burgart, Y.V.; Saloutin, V.I.; Chupakhin, O.N. Polyfluorinecontaining chromen-4-ones: Synthesis and transformations. Russ. Chem. Bull. 2016, 65, 2151–2162. [Google Scholar] [CrossRef]
- Ahmadi, A.; Mohammadnejadi, E.; Karami, P.; Razzaghi-Asi, N. Current status and structure activity relationship of privileged azoles as antifungal agents. Int. J. Antimicrob. Agents 2022, 59, 106518. [Google Scholar] [CrossRef]
- Seck, I.; Nguemo, F. Triazole, imidazole, and thiazole-based compounds as potential agents against coronavirus. Results Chem. 2021, 3, 100132. [Google Scholar] [CrossRef]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A review on recent advances in nitrogen-containing molecules and their biological applications. Molecules 2020, 25, 1909–1951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, R.; Sumran, G. An insight on medicinal attributes of 1,2,4-triazoles. Eur. J. Med. Chem. 2020, 205, 112652. [Google Scholar] [CrossRef] [PubMed]
- Prasher, P.; Sharma, M. “Azole” as privileged heterocycle for targeting the inducible cyclooxygenase enzyme. Drug Dev. Res. 2020, 82, 167–197. [Google Scholar] [CrossRef]
- Hou, Y.; Shang, C.; Wang, H.; Yun, J. Isatin-azole hybrids and their anticancer activities. Arch. Pharm. Chem. Life Sci. 2019, 353, e1900272. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Peng, Y.; Wang, S.; Ji, J.; Rakesh, K.P. Triazole derivatives as inhibitors of Alzheimer’s disease: Current developments and structure-activity relationships. Eur. J. Med. Chem. 2019, 180, 656–672. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.-L.; Jin, X.-H.; Huang, Z.-P.; Yu, H.-F.; Zeng, Z.-G.; Gao, T.; Feng, L.-S. Resent advances of imidazole-containing derivatives as anti-tubercular agents. Eur. J. Med. Chem. 2018, 150, 347–365. [Google Scholar] [CrossRef]
- Yan, M.; Xu, L.; Wang, Y.; Wan, J.; Liu, T.; Liu, W.; Wan, Y.; Zhang, B.; Wang, R. Opportunities and challenges of using five-membered ring compounds as promising antitubercular agents. Drug Dev. Res. 2020, 81, 402–418. [Google Scholar] [CrossRef]
- Gao, F.; Wang, T.; Xiao, J.; Huang, G. Antibacterial activity study of 1,2,4-triazole derivatives. Eur. J. Med. Chem. 2019, 173, 274–281. [Google Scholar] [CrossRef]
- Teli, G.; Chawla, P.A. Hybridization of imidazole with various heterocycles in targeting cancer. ChemistrySelect 2021, 6, 4803–4836. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, S.; Ba, Y.; Xu, Z. 1,2,4-Triazole-quinoline/ quinolone hybrids as potential anti-bacterial agents. Eur. J. Med. Chem. 2019, 174, 1–8. [Google Scholar] [CrossRef]
- Fan, Y.-L.; Liu. M. Coumarin-triazole hybrids and their biological activities. J. Heterocycl. Chem. 2018, 55, 791–802. [Google Scholar] [CrossRef]
- Zhang, T.; Zhu, M.; Li, J.; Zhang, Y.; Wang, X. Bipolar host materials comprising carbazole, pyridine and triazole moieties for efficient and stable phosphorescent OLEDs. Dyes Pigm. 2021, 192, 109426. [Google Scholar] [CrossRef]
- Ye, S.; Zhuang, S.; Pan, B.; Guo, R.; Wang, L. Imidazole derivatives for efficient organic light-emitting diodes. J. Inf. Disp. 2020, 21, 173–196. [Google Scholar] [CrossRef]
- Xu, H.; Zhao, Y.; Zhang, J.; Zhang, D.; Miao, Y.; Shinar, J.; Shinar, R.; Wang, H.; Xu, B. Low efficiency rol-off phosphorescent organic light-emitting devices using thermally activated delayed fluorescence hosts materials based 1,2,4-triazole acceptor. Org. Electron. 2019, 74, 13–22. [Google Scholar] [CrossRef]
- Tao, Y.; Yang, C.; Qin, J. Organic host materials for phosphorescent organic light-emitting diodes. Chem. Soc. Rev. 2011, 40, 2943–2970. [Google Scholar] [CrossRef]
- Emami, L.; Faghih, Z.; Ataollahi, E. Azole derivatives: Recent advances as potent antibacterial and antifungal agents. Curr. Med. Chem. 2022, 129, 220–249. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Palatinus, L.; Chapuis, G. SUPERFLIP–A computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Crystallogr. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2007, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubanova, A.A.; Stepanova, Z.V.; Gus’kova, T.A.; Pushkina, T.V.; Krylova, L.Y.; Shilova, I.B.; Trenin, A.S. Metodicheskie rekomendacii po izucheniyu protivogribkovoi aktivnosti lekarstvennykh sredstv (Guidelines for the study of the antifungal activity of medicines). In Rukovodstvo po Provedeniyu Doklinicheskikh Issledovanii Lekarstvennykh Sredstv (A Guide to Preclinical Trials of Medicines); Mironov, A.N., Ed.; Grif i K: Moscow, Russia, 2012; pp. 578–586. [Google Scholar]
- Koldobskii, G.I.; Ostrovskii, V.A. Acid-base properties of five-membered nitrogen-containing heterocycles. Chem. Heterocycl. Compds. 1988, 24, 469–480. [Google Scholar] [CrossRef]
- Krishnan, R.; Parthiban, A. Regioselective preparation of functional aryl ethers and esters by stepwise nucleophilic aromatic substitution reaction. J. Fluorine Chem. 2014, 162, 17–25. [Google Scholar] [CrossRef]
- Chambers, R.D.; Martin, P.A.; Sandford, G.; Williams, L.H. Mechanisms of reactions of halogenated compounds: Part 7. Effects of fluorine and other groups as substituents on nucleophilic aromatic substitution. J. Fluorine Chem. 2008, 129, 998–1002. [Google Scholar] [CrossRef]















| Compound | 19F NMR Data | |
|---|---|---|
| δ, ppm | Products Ratio, % | |
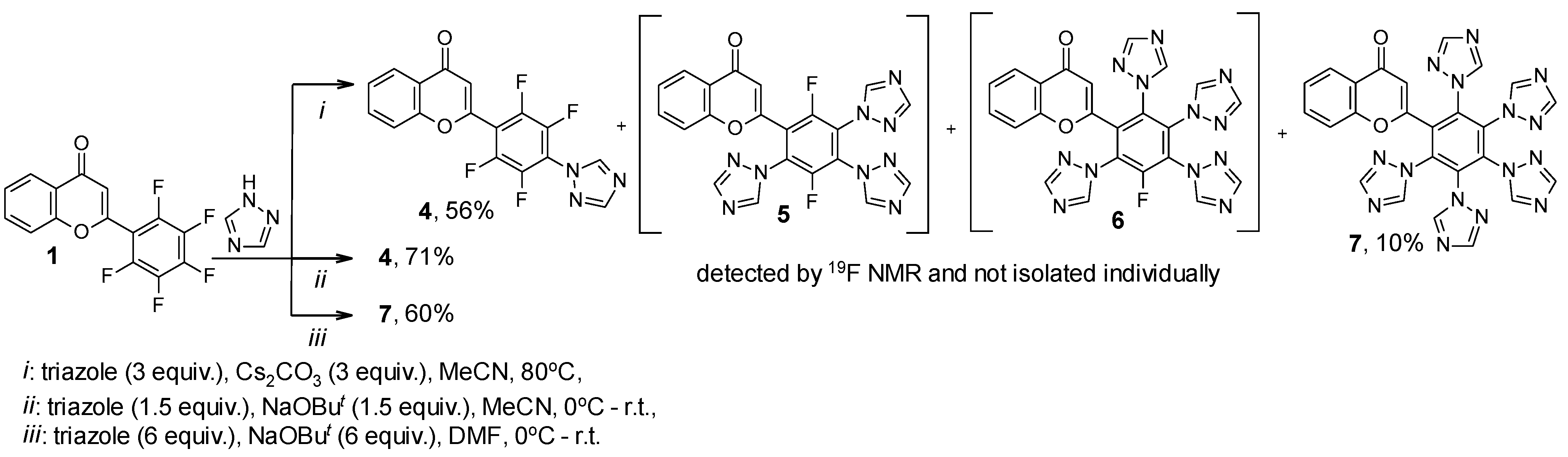
| 4 | 16.66, m; 24.82, m | 97 |
| 5 | 15.95, m; 26.03, m; 26.86, m | 1 |
| 6 | 31.53, m | 2 |
| Compound | 19F NMR Data | |
|---|---|---|
| δ, ppm, J, Hz | Products Ratio, % | |
| 8 | 14.88, m; 24.52, m | 63 |
| 9 | 23.57, m; 26.31, m; 30.56, m | 6 |
| 10 | 31.46, dm, J = 14.2 Hz; 41.17, dd, J = 14.9, 1.1 Hz | 31 |
| Compound | 19F NMR Data | |
|---|---|---|
| δ, ppm | Products Ratio, % | |
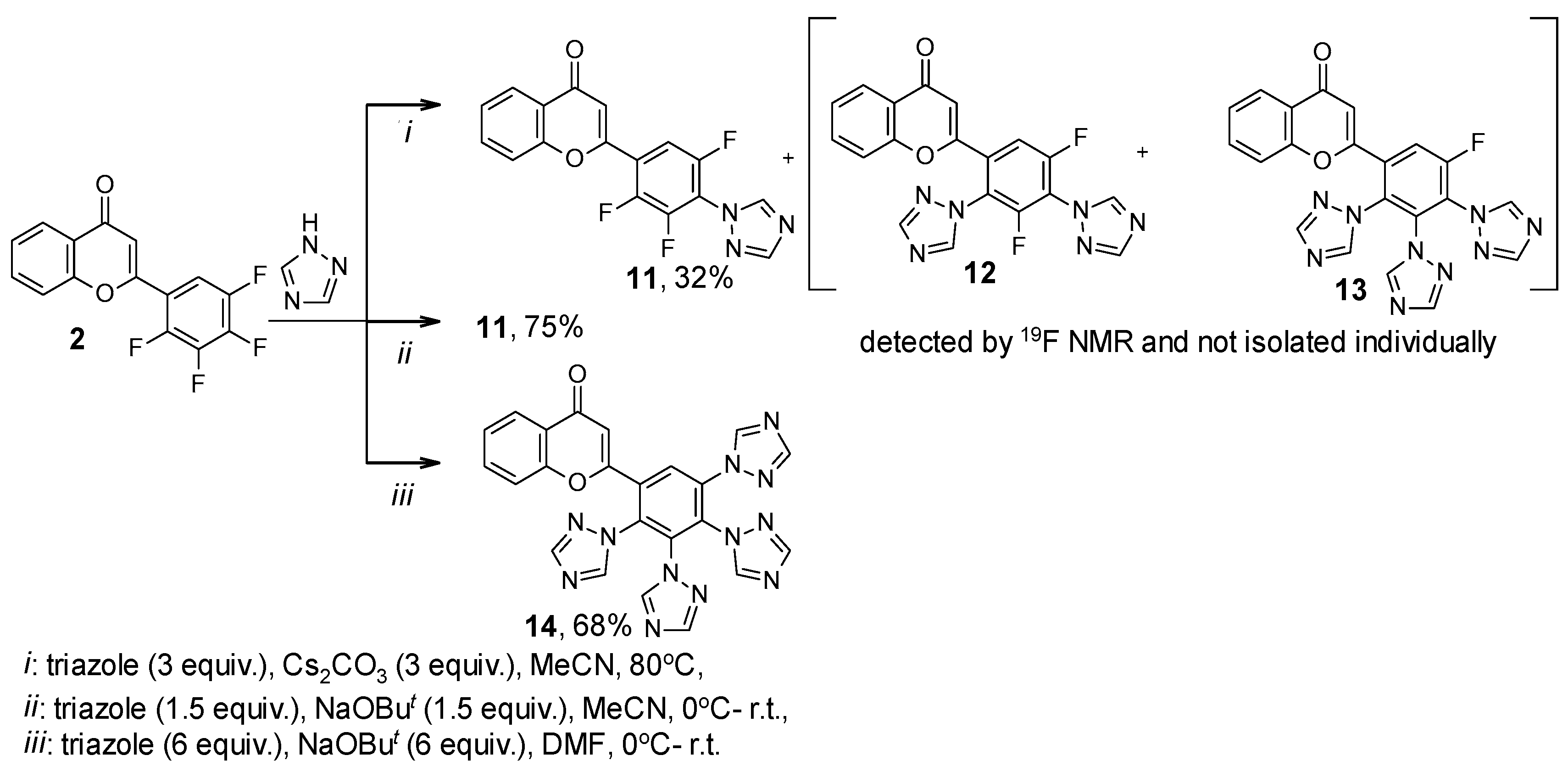
| 11 | 23.72, d, J = 19.9 Hz; 24.92, ddd, J = 20.1, 14.7, 5.6 Hz; 39.24, dd, J = 14.1, 10.7 Hz | 56 |
| 12 | 39.54, dd, J = 15.2, 9.8 Hz; 39.86, dd, J = 16.0, 5.7 Hz | 34 |
| 13 | 49.03, d, J = 9.2 Hz | 10 |
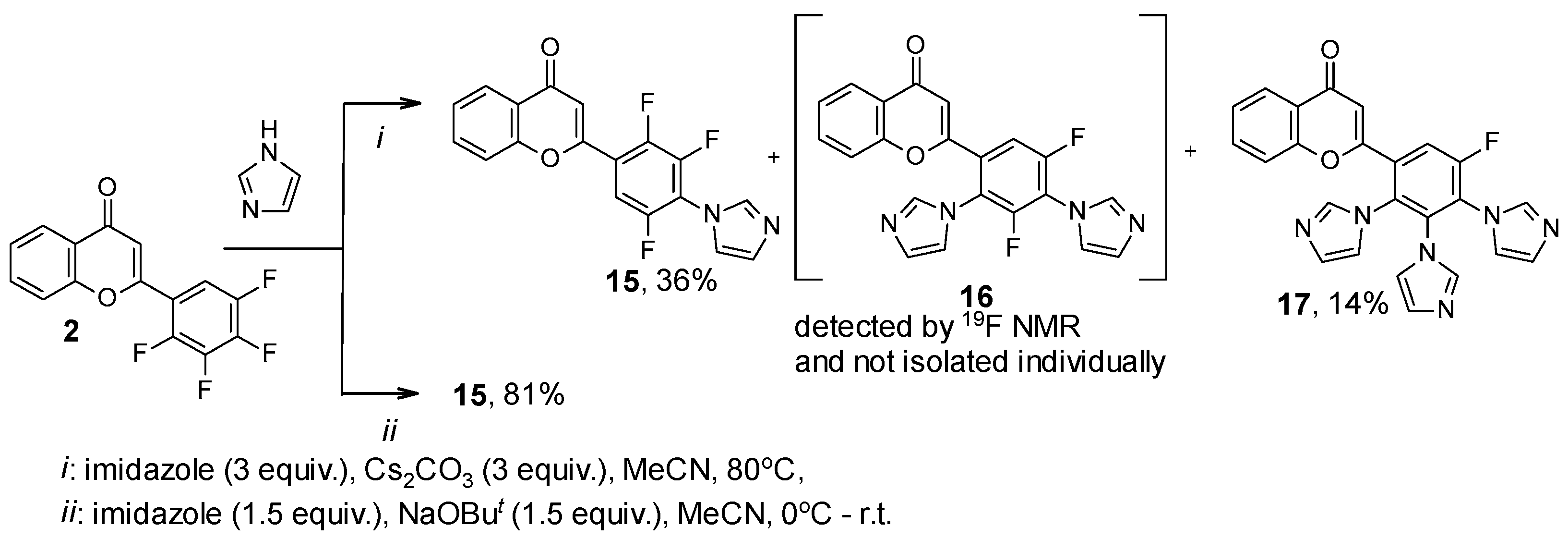
| 15 | 21.76, dd, J = 20.0, 1.9 Hz; 24.80, ddd, J = 20.1, 14.4, 6.1 Hz, 38.16, m | 35 |
| 16 | 39.41, m; 39.71, m | 20 |
| 17 | 47.34, d, J = 9.1 Hz | 45 |
| Compound | 19F NMR Data | |
|---|---|---|
| δ, ppm | Products Ratio, % | |
| 18 | 29.29, m; 37.18, m | 31 |
| 19 | 38.83, dd, J = 9.9, 1.9 Hz | 64 |
| 20 | 38.77, m; 39.82, m | 2 |
| 21 | 37.13, m; 39.63, m | 3 |
| Compound | 19F NMR Data | |
|---|---|---|
| δ, ppm | Products Ratio, % | |
| 23 | 27.60, m; 36.46, m | 31 |
| 24 | 38.70, m | 67 |
| 25 | 28.22, m; 38.63, m | 2 |
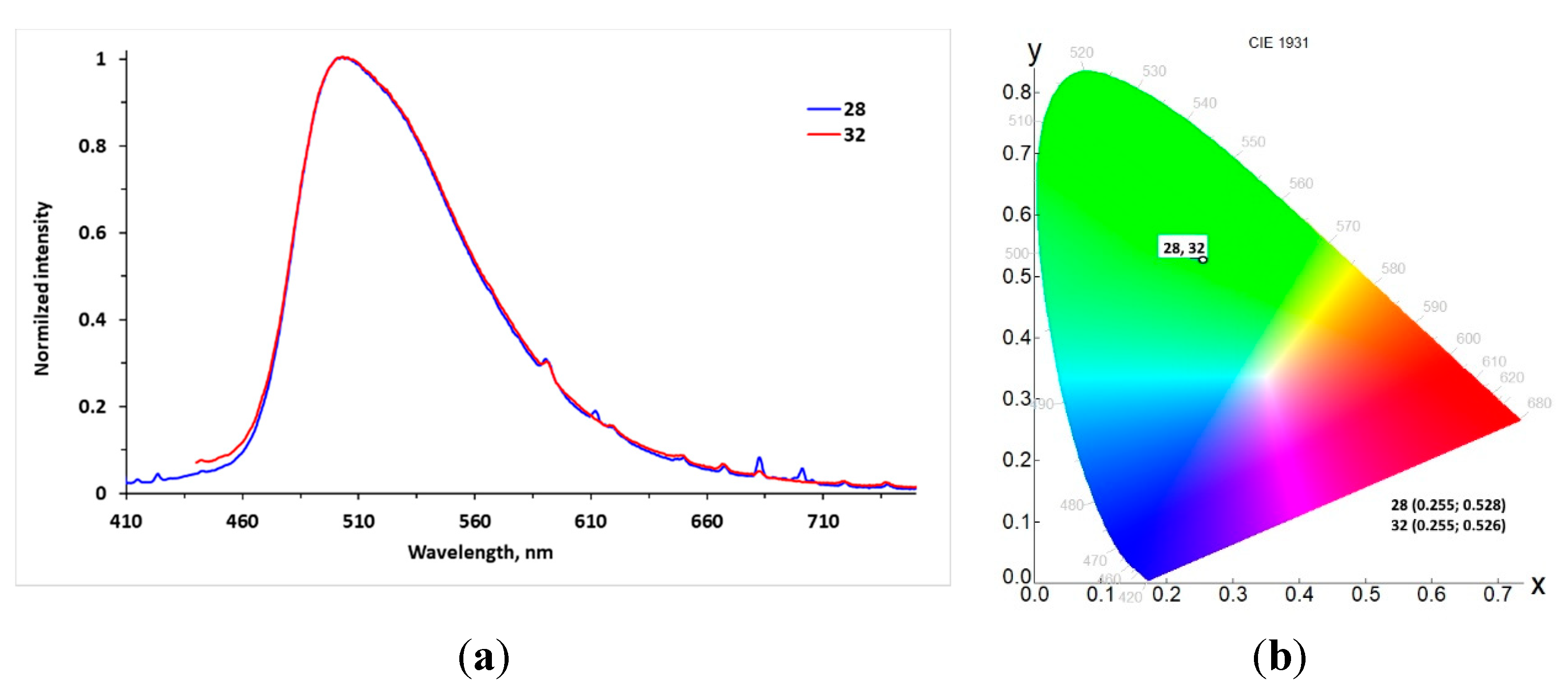
| Compound | Emission, λem, nm | τavg, [ns]/χ2 | ΦF |
|---|---|---|---|
| 28 | 504 | 7.72/ 1.109 | 0.29 |
| 32 | 504 | 6.81/ 1.206 | 0.18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panova, M.A.; Shcherbakov, K.V.; Zhilina, E.F.; Burgart, Y.V.; Saloutin, V.I. Synthesis of Mono- and Polyazole Hybrids Based on Polyfluoroflavones. Molecules 2023, 28, 869. https://doi.org/10.3390/molecules28020869
Panova MA, Shcherbakov KV, Zhilina EF, Burgart YV, Saloutin VI. Synthesis of Mono- and Polyazole Hybrids Based on Polyfluoroflavones. Molecules. 2023; 28(2):869. https://doi.org/10.3390/molecules28020869
Chicago/Turabian StylePanova, Mariya A., Konstantin V. Shcherbakov, Ekaterina F. Zhilina, Yanina V. Burgart, and Victor I. Saloutin. 2023. "Synthesis of Mono- and Polyazole Hybrids Based on Polyfluoroflavones" Molecules 28, no. 2: 869. https://doi.org/10.3390/molecules28020869