
Synthesis and Molecular Docking of Some Novel 3-Thiazolyl-Coumarins as Inhibitors of VEGFR-2 Kinase
by
 , , and
, , and
Tariq Z. Abolibda
1 ,
,
Maher Fathalla
1,2,
Basant Farag
2,
Magdi E. A. Zaki
3 and
Sobhi M. Gomha
1,4,*
1
Department of Chemistry, Faculty of Science, Islamic University of Madinah, Madinah 42351, Saudi Arabia
2
Department of Chemistry, Faculty of Science, Zagazig University, Zagazig 44519, Egypt
3
Department of Chemistry, Faculty of Science, Imam Mohammad Ibn Saud Islamic University (IMSIU), Riyadh 11623, Saudi Arabia
4
Department of Chemistry, Faculty of Science, Cairo University, Cairo 12613, Egypt
*
Author to whom correspondence should be addressed.
Molecules 2023, 28(2), 689; https://doi.org/10.3390/molecules28020689
Submission received: 10 December 2022
/
Revised: 4 January 2023
/
Accepted: 6 January 2023
/
Published: 10 January 2023
(This article belongs to the Special Issue Synthesis of Heteroaromatic Compounds)
Abstract
:One crucial strategy for the treatment of breast cancer involves focusing on the Vascular Endothelial Growth Factor Receptor (VEGFR-2) signaling system. Consequently, the development of new (VEGFR-2) inhibitors is of the utmost importance. In this study, novel 3-thiazolhydrazinylcoumarins were designed and synthesized via the reaction of phenylazoacetylcoumarin with various hydrazonoyl halides and α-bromoketones. By using elemental and spectral analysis data (IR, 1H-NMR, 13C-NMR, and Mass), the ascribed structures for all newly synthesized compounds were clarified, and the mechanisms underlying their formation were delineated. The molecular docking studies of the resulting 6-(phenyldiazenyl)-2H-chromen-2-one (3, 6a–e, 10a–c and 12a–c) derivatives were assessed against VEGFR-2 and demonstrated comparable activities to that of Sorafenib (approved medicine) with compounds 6d and 6b showing the highest binding scores (−9.900 and −9.819 kcal/mol, respectively). The cytotoxicity of the most active thiazole derivatives 6d, 6b, 6c, 10c and 10a were investigated for their human breast cancer (MCF-7) cell line and normal cell line LLC-Mk2 using MTT assay and Sorafenib as the reference drug. The results revealed that compounds 6d and 6b exhibited greater anticancer activities (IC50 = 10.5 ± 0.71 and 11.2 ± 0.80 μM, respectively) than the Sorafenib reference drug (IC50 = 5.10 ± 0.49 μM). Therefore, the present study demonstrated that thiazolyl coumarins are potential (VEGFR-2) inhibitors and pave the way for the synthesis of additional libraries based on the reported scaffold, which could eventually lead to the development of efficient treatment for breast cancer.
1. Introduction
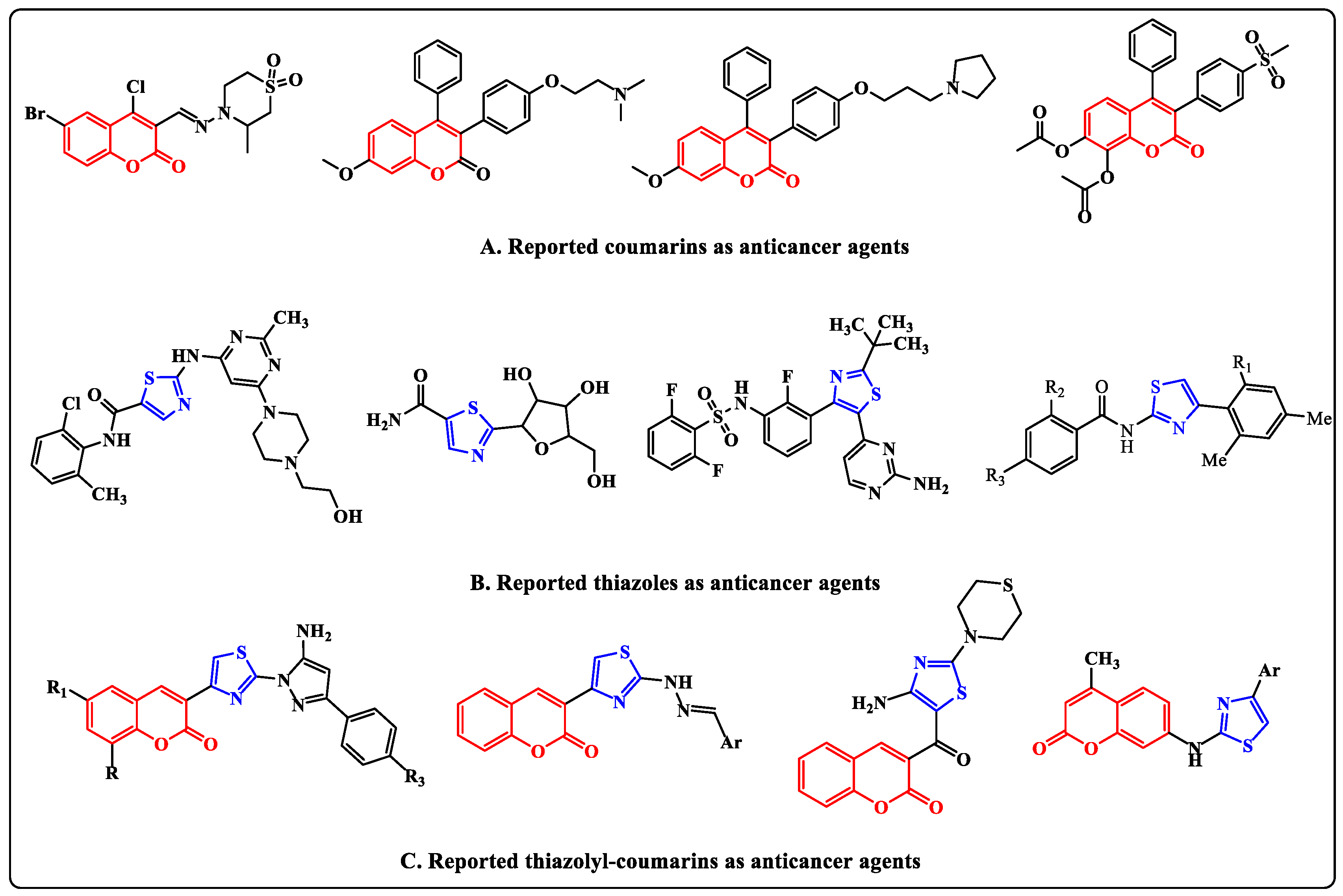
According to statistics, among all cancers affecting women, breast cancer accounts for 33.1 percent, making it the most prevalent disease of either gender [1]. Breast cancer early detection programs were a valuable resource for the tens of thousands of women who were diagnosed with cervical cancer or malignant or premalignant breast cancer [2]. The percentages of incidence and fatality, however, remain at historically high levels [3,4]. Invasive breast cancer has been used to express a number of angiogenic factors at all tumor stages [5]. Additionally, vascular endothelial growth factor receptor-2 (VEGFR-2) has been discovered to be significantly expressed in both primary and metastatic invasive breast carcinomas, suggesting a role for the VEGF signaling pathway in the regulation of breast tumor angiogenesis [6]. The C-terminal and N-terminal lobes each contributed residues to the region of the VEGFR-2 of protein kinases, which actively bind adenosine triphosphate (ATP), which is placed in the gap between the two lobes [7]. At the lobe, the C-terminal is an activation loop that has a conserved aspartate-phenylalanine-glycine (DFG) motif at the start of it [8]. Type I through III inhibitors are the three classes of VEGFR-2 inhibitors. Type II inhibitors maintain the DFG motif-containing DFG-out conformation of inactive VEGFR2 kinase; Type II inhibitors create a hydrophobic allosteric pocket next to the ATP-binding site. Improved kinase selectivity and high cellular potency are just two benefits of type II inhibitors [9]. Through the suppression of the Ras/MAPK pathway, VEGFR-2 inhibitors also delayed the development of selective estrogen receptor modulator (SERM) resistance in breast cancer [10]. An important field of study in the fight against cancer is the discovery and creation of novel anticancer drugs with high efficacy and low toxicity. According to reports in the literature, compounds comprising coumarins, thiazoles, or thiazolylcoumarins have drawn a lot of attention from drug research due to their potential anticancer action with good IC50 [11,12] (Figure 1).
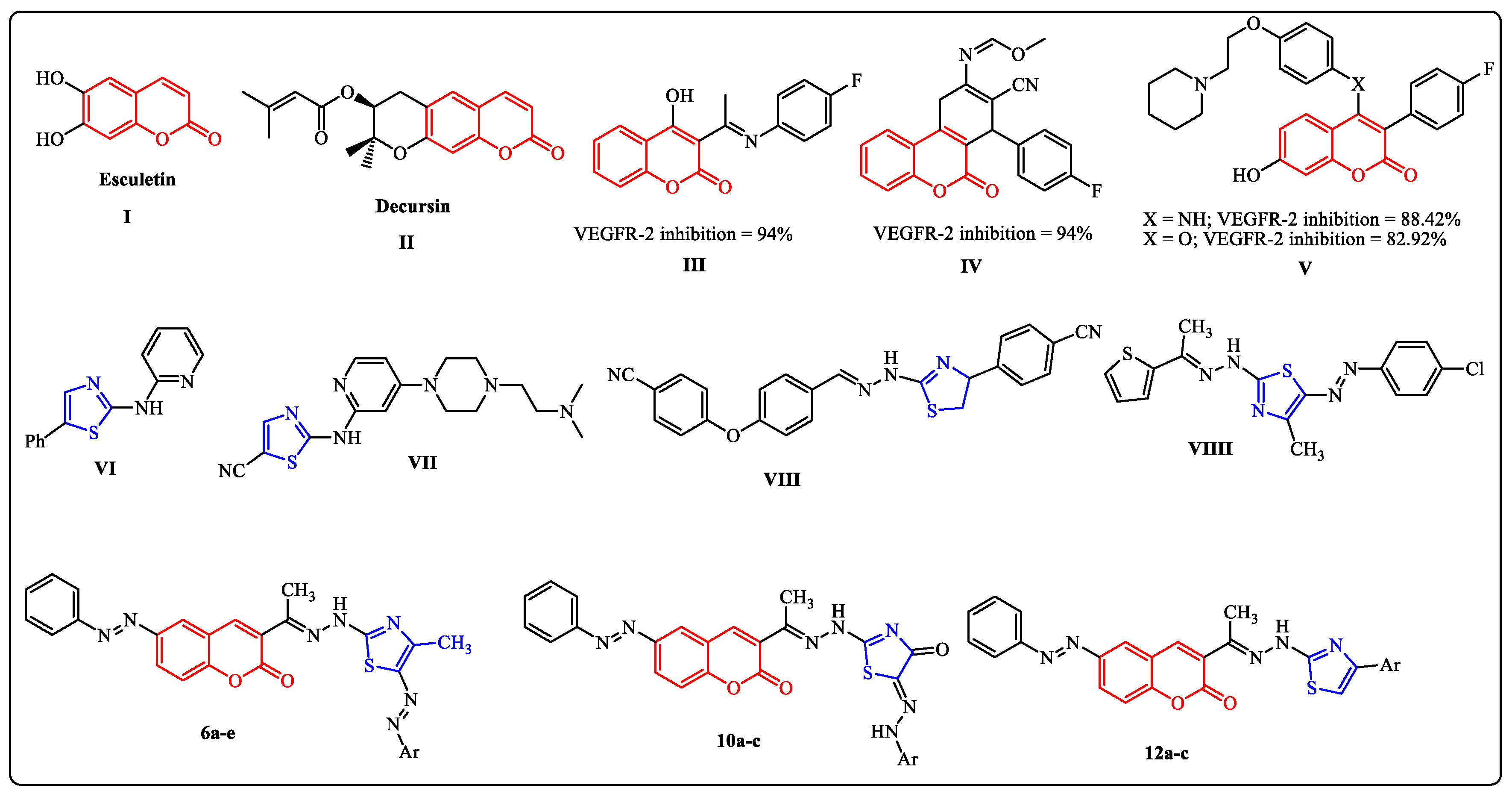
Coumarin is a versatile molecule that serves as the pharmacological and biological building block for a wide range of naturally occurring chemicals [13,14,15]. It has been regarded as an intriguing framework for the development of anticancer drugs [16,17,18,19]. Furthermore, recent research revealed that a variety of coumarin compounds, both natural and synthetic, have antiproliferative properties via inhibiting VEGFR-2-mediated signaling pathways (Figure 2) [20,21,22,23]. On the other hand, thiazoles are considered to be important chemical synthons found in a variety of pharmacologically active compounds [24]. They possess a wide range of biological activities as anticancer, antimicrobial and anti-inflammatory agents [25,26,27]. Some thiazole derivatives were reported as type II VEGFR-2 inhibitors with similar activity compared with the Sorafenib reference drug (Figure 2) [28,29,30,31,32,33,34,35,36,37,38].
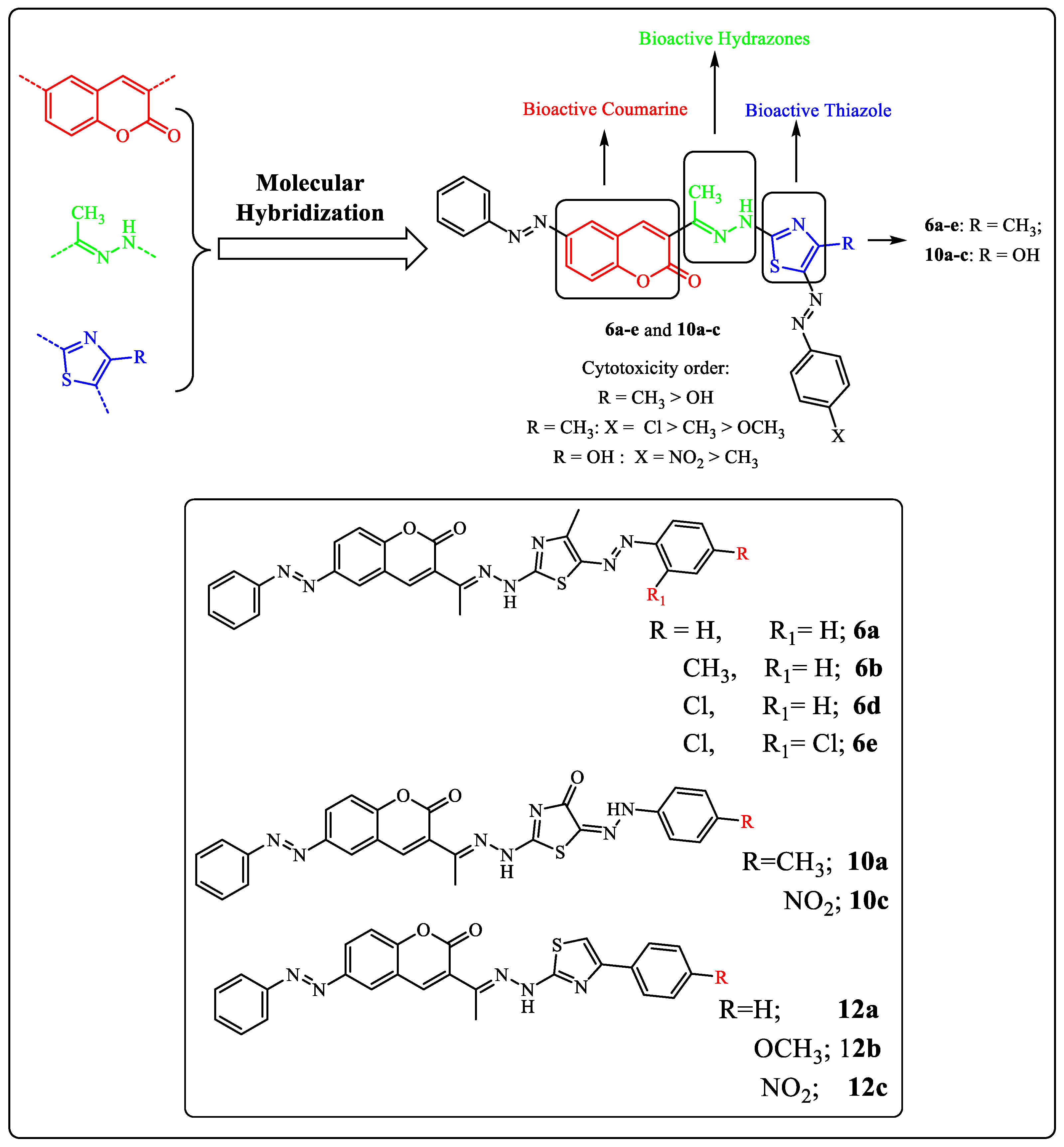
In light of our previous work on the synthesis of novel antitumor heterocycles [39,40,41,42,43,44,45,46] and with consideration of the aforementioned results, a new sort of VEGFR-2 inhibitors has been developed as prospective anti-breast cancer agents by hybridizing the coumarin and 1,3-thiazole moieties, which have been found to inhibit kinases and have antiproliferative properties. In this study, we developed and synthesized new 3-thiazolhydrazinylcoumarins in an effort to enhance the target compounds’ synergistic pharmacological significance and assess their anti-breast cancer activity targeting VEGFR-2. Finally, in order to occupy the hydrophobic back pocket of VEGFR-2, a side phenyl ring was maintained, either mono or di, substituted with a wide variety consisting of hydrophobic groups, such as chloro, methyl, and phenyl. The molecular docking studies of these compounds were performed to confirm their ability to satisfy the pharmacophoric features. Moreover, it also determines the binding mode interaction that occurred with the desired VEGFR-2 inhibition.
2. Results and Discussion
2.1. Chemistry
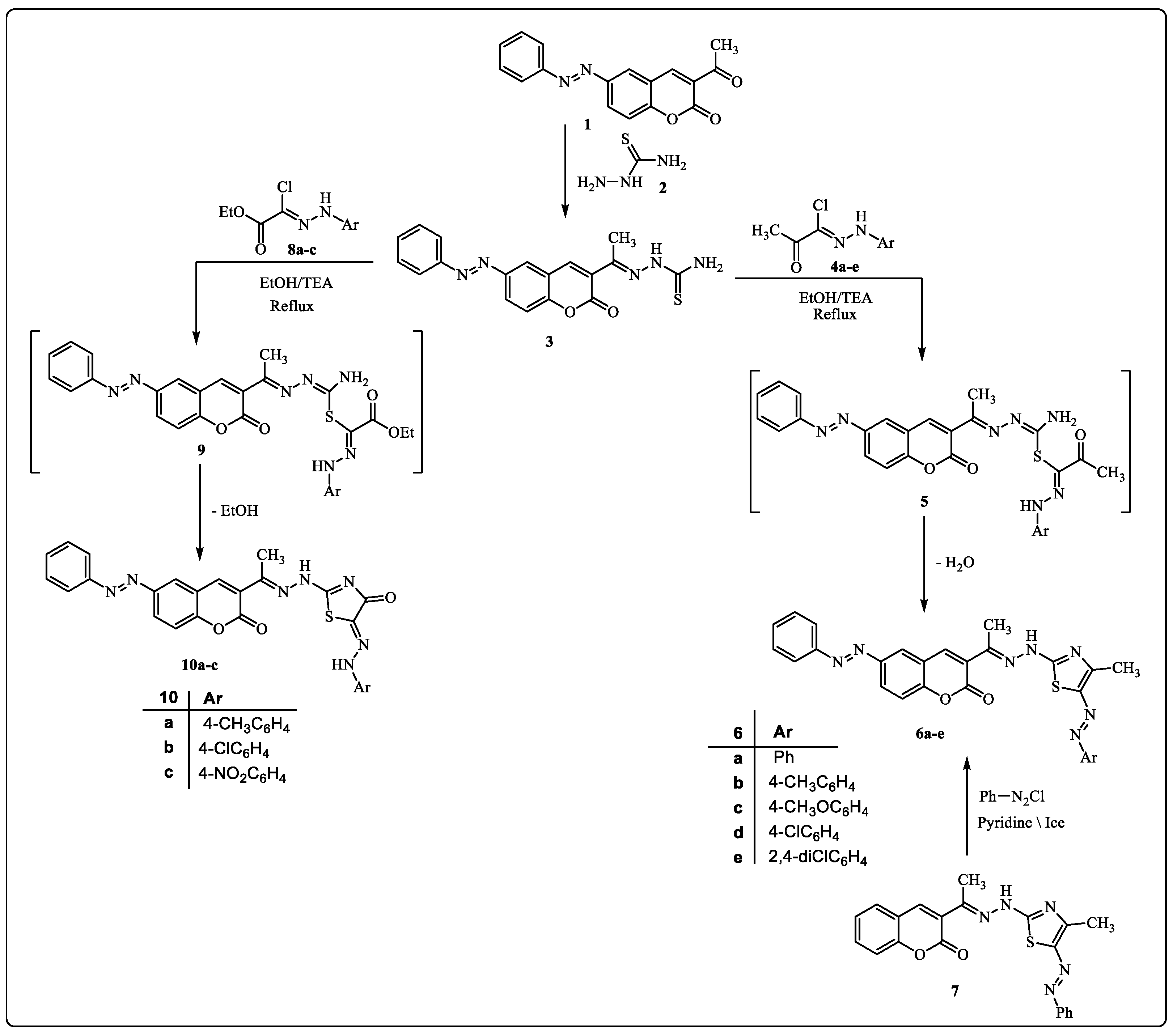
Our research aims to synthesize a new series of bioactive thiazole derivatives, and this may be performed by synthesizing the starting derivative 2-(1-(2-oxo-6-(phenyldiazenyl)-2H-chromen-3-yl)ethylidene)hydrazine-1-carbothioamide (3) via the reaction of 3-acetyl-6-(phenyldiazenyl)-2H-chromen-2-one (1) [47] and hydrazinecarbothioamide 2 in EtOH in the presence of a catalytic amount of HCl under reflux as depicted in Scheme 1. Element and spectral data techniques (IR, 1H-NMR, mass) were used to determine structure 3 (see Section 3 experimental section).
The reaction of compound 3 with hydrazonoyl chlorides 4a–e [48] in EtOH containing Et3N yielded the thiazole derivatives 6a–e via cyclization with the removal of the H2O molecule from intermediate 5 (Scheme 1). The structure of product 6 was proved by spectral (IR, mass, 1H-NMR, 13C-NMR) and elemental data. The 1H-NMR spectrum of product 6a showed a singlet signal at δ = 10.36 ppm assigned to the -NH proton, in addition to the usual signals of the fourteen aromatic protons and the two CH3 group protons. Furthermore, the IR spectrum showed two stretching bands at υ = 1725 and 3347 cm−1 due to the C=O and the NH groups. Its 13C-NMR spectrum showed δ = 8.4, 14.7 (2CH3), 116.3–162.1 (20Ar-C and C=N) and 163.5 (C=O) ppm. Moreover, the mass spectra of all derivatives of compound 6 showed molecular ion peaks at the right molecular weight for the corresponding molecules (see the Section 3 experimental section, supporting information).
It is envisioned that the nucleophilic attack of the thiol group of compound 3 at the electron-deficient carbon of the hydrazone group of compound 4 creates an intermediate 5, which would then undergo a dehydrative cyclization to produce the final products of compound 6.
Alternative synthetic techniques might be used to create authentic samples of 6a. Product 6a was obtained as a result of the reaction of phenyl diazonium chloride and 3-(1-(2-(4-methyl-5-(phenyldiazenyl)thiazol-2-yl)hydrazineylidene)ethyl)-2H-chromen-2-one (7) [49] in pyridine (Scheme 1).
On the other hand, the reaction of compound 3 with ethyl 2-chloro-2-(2-arylhydrazineylidene)acetate 8 [50] in refluxing EtOH containing TEA as a basic catalyst afforded products 10 (Scheme 1). The structures of product 10 were confirmed based on spectral (IR, mass, 1H-NMR, 13C-NMR) and elemental data. The 1H-NMR spectrum of 10a revealed the expected signals at δ = 2.26, 2.27 (2s, 2CH3), 7.02–7.92 (m, 13 Ar-H), 10.54, 10.79 and (2 br s, 2 NH) ppm. Also, the 13C-NMR spectrum showed the expected signals at δ = 8.9, 14.2 (2CH3), 117.0–156.7 (19 Ar-C and C=N), and 163.0, 171.3 (2C=O) ppm. In addition, its IR spectrum showed the expected characteristic stretching bands at υ = 3413, 3289 (2NH) and 1724, 1692 (2C=O) cm−1 (see Section 3 experimental section, supporting information).
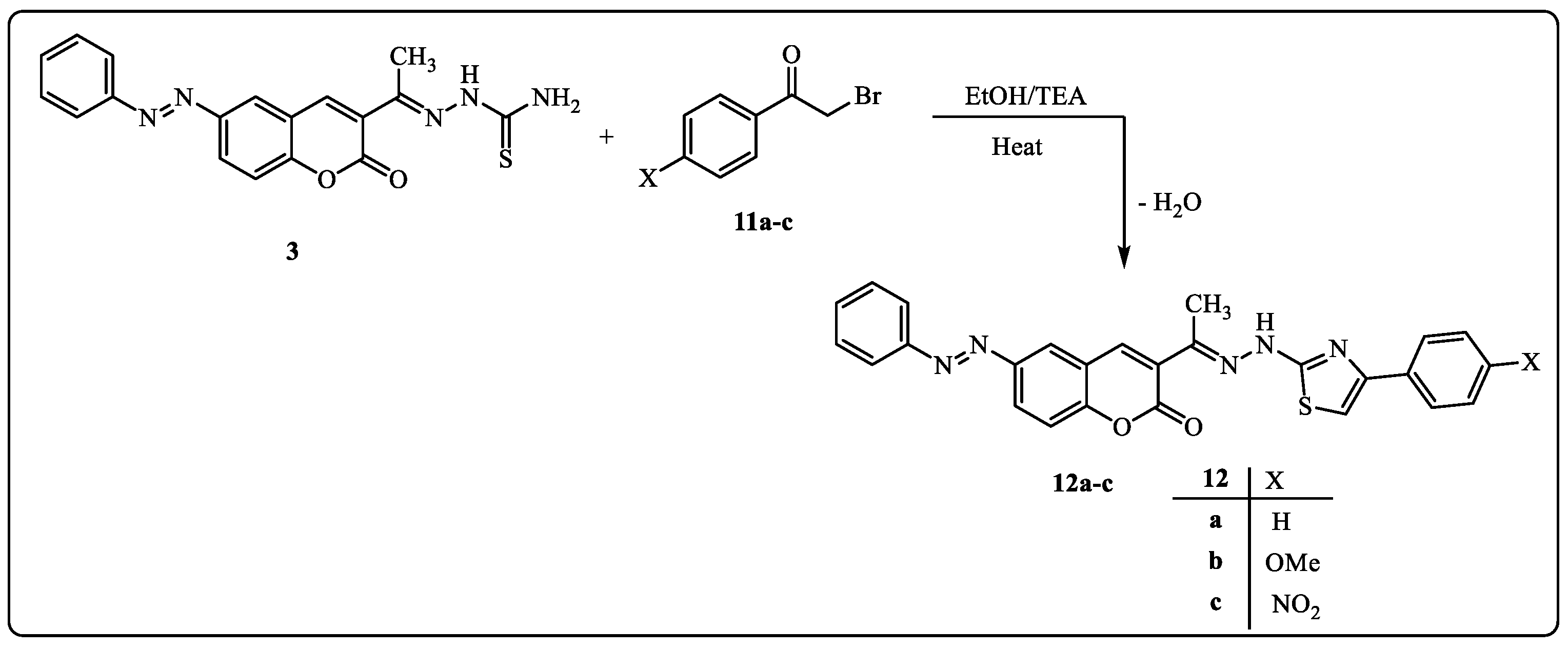
Furthermore, the reaction of compound 3 with α-bromoketones was used to investigate the potential of compound 3 as a building block for the production of another series of predicted physiologically active thiazoles. Thus, the reaction of compound 3 with substituted phenacyl bromides 11a–c in EtOH afforded thiazoles 12a–c (Scheme 2). The 1H-NMR spectrum of product 12a, considered an example of product 12, revealed the predicted signals assigned for the CH3, fourteen aromatic protons, and NH at δ = 2.31, 6.94–8.42, and 11.32 ppm, respectively. In addition, its 13C-NMR spectrum showed δ 10.1(CH3), 106.1–159.6 (20Ar-C and C=N) and 164.5 (C=O) ppm. The mass spectra of compounds 12a–c exhibited a peak that matched their molecular ions in each case. Infrared spectra showed four bands, each corresponding to the carbonyl and NH groups, at υ = 1722 and 3327 cm−1.
2.2. Molecular Docking
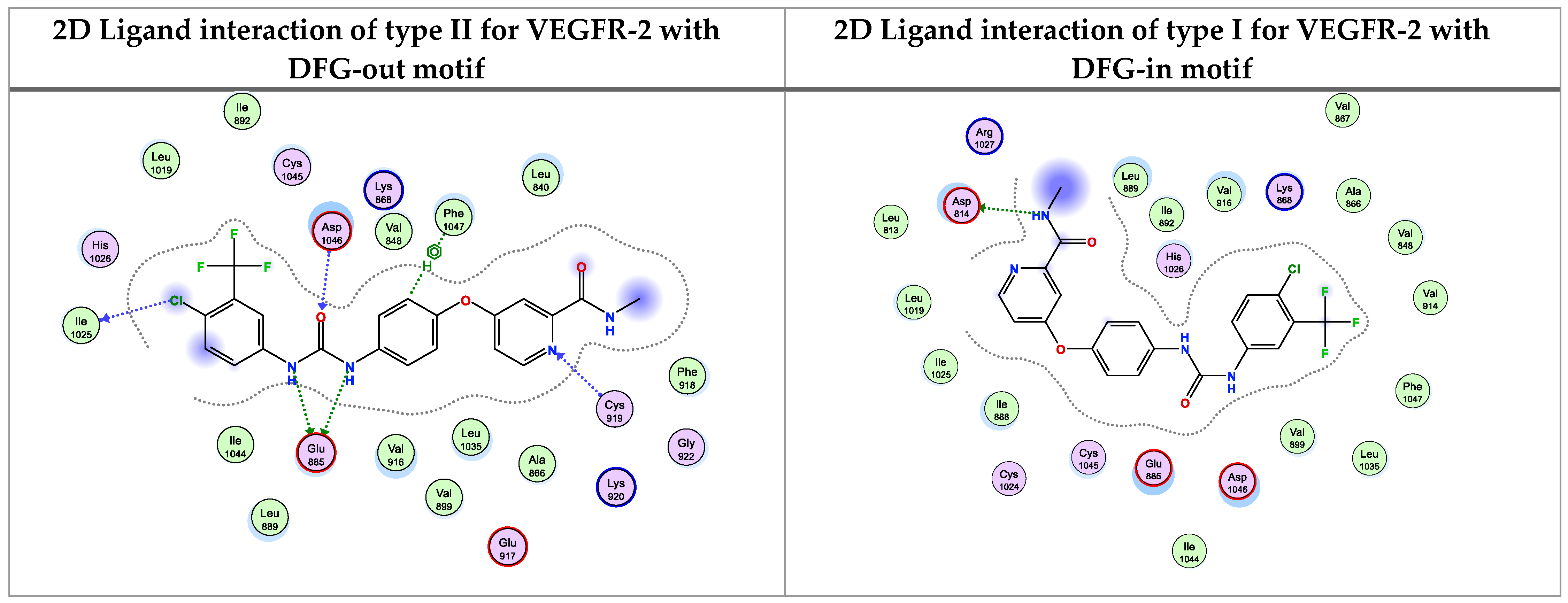
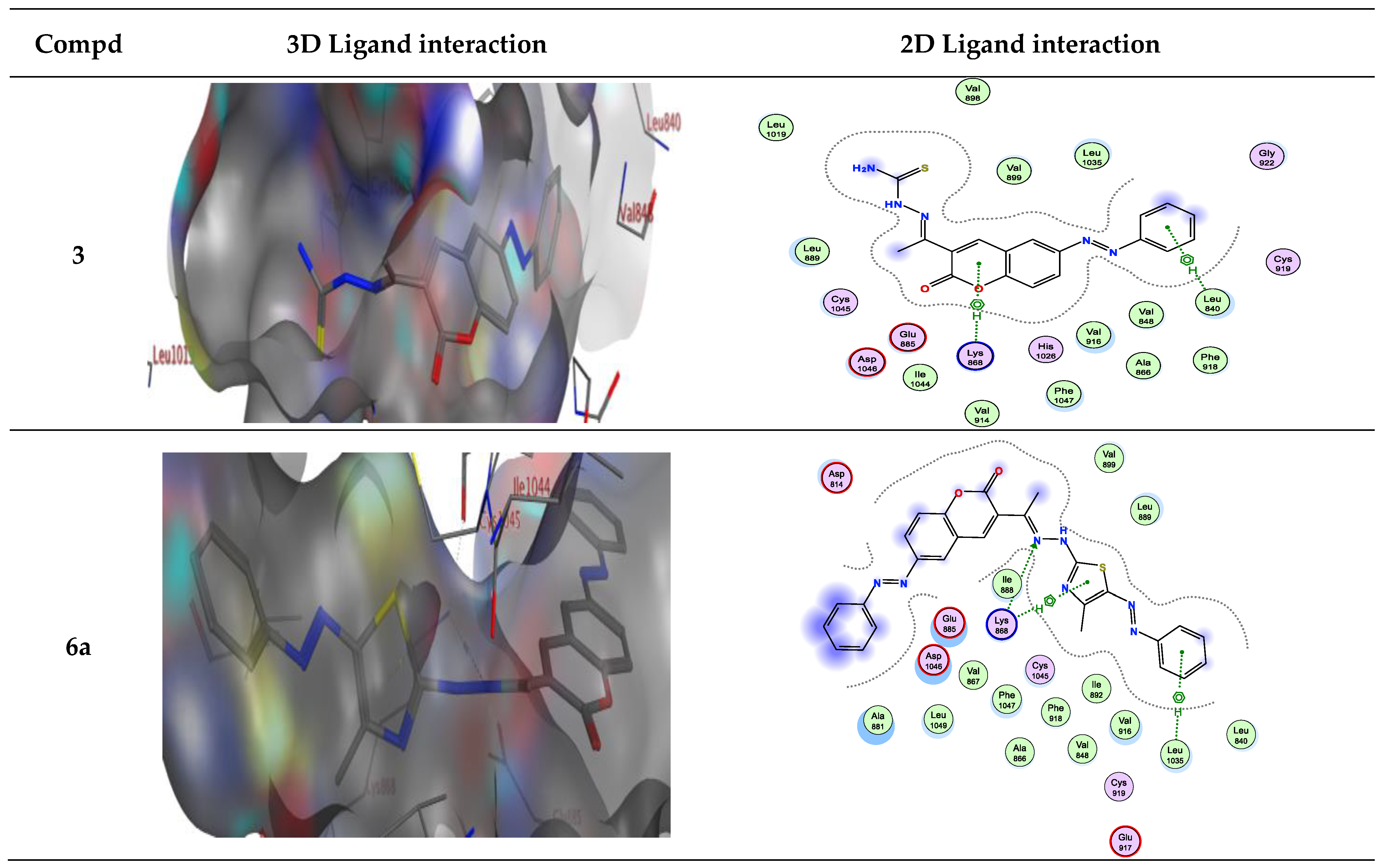
The molecular docking was performed using MOE (Molecular Operating Environment) software version 2015.10 and was confirmed by re-docking the native ligand Sorafenib in the VEGFR-2 active regions giving a docking score of −9.284 kcal/mol. The modes of Sorafenib’s interactions with VEGFR-2’s active site residues are demonstrated in Figure 3. It is evident that the urea linker of Sorafenib plays a significant role in its binding ability for the enzyme VEGFR2 as it fits into the enzyme’s allosteric site to form four significant hydrogen bonds with four essential residues (one H-bond acceptor between chloro atom with Ile1025, one H-bond acceptor between the oxygen of urea moiety and Asp1046, and finally two H-bonds donors between two NH of urea side chain with Glu885). The hydrophobic 4-chloro-3-trifluoromethylphenyl is directed toward the hydrophobic back pocket by the urea linker’s binding mechanism. In addition, one H-bond acceptor between N atom of pyridine with essential amino acid Cys919 (inserted in Figure 3 as a 2D view).
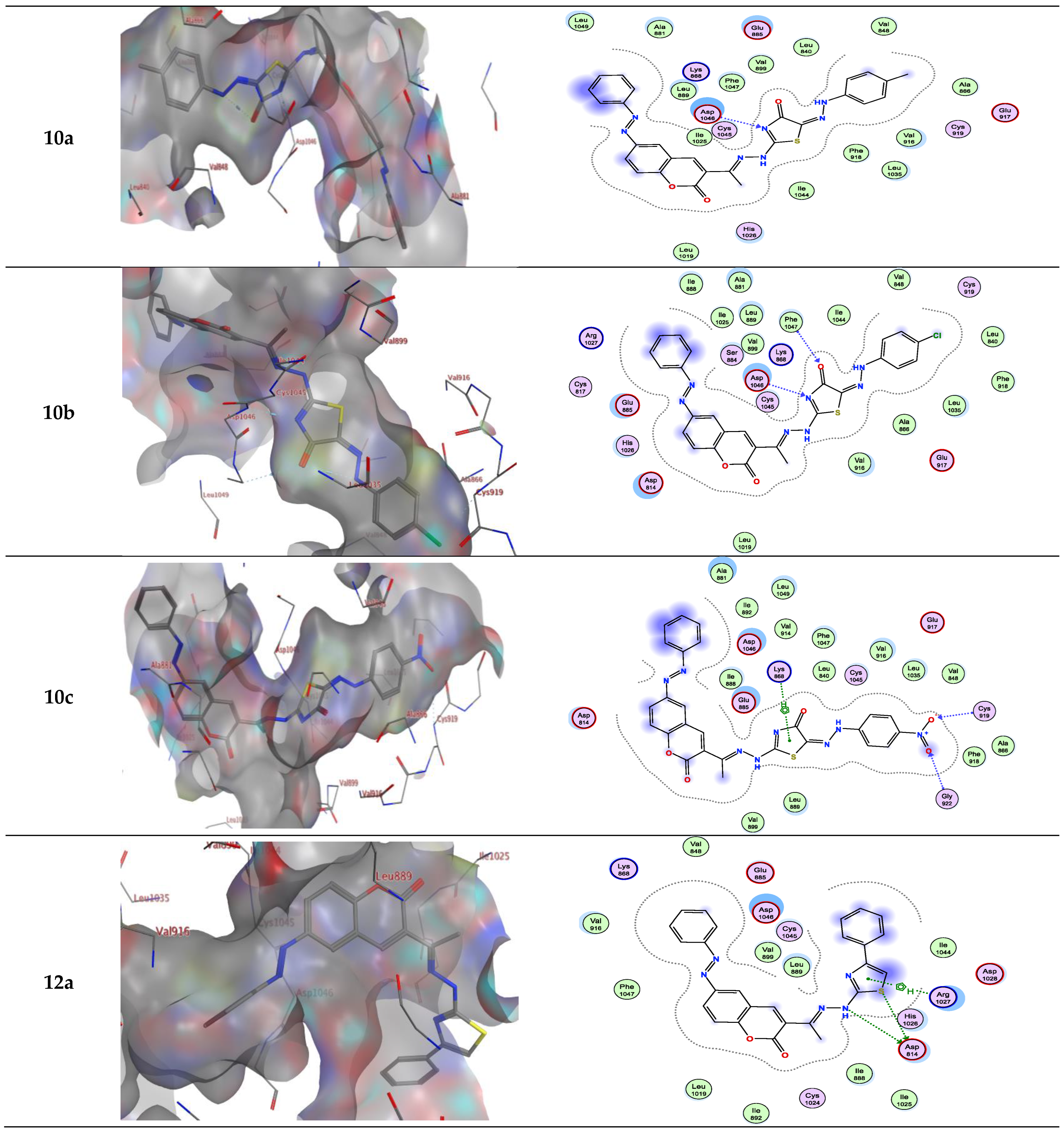
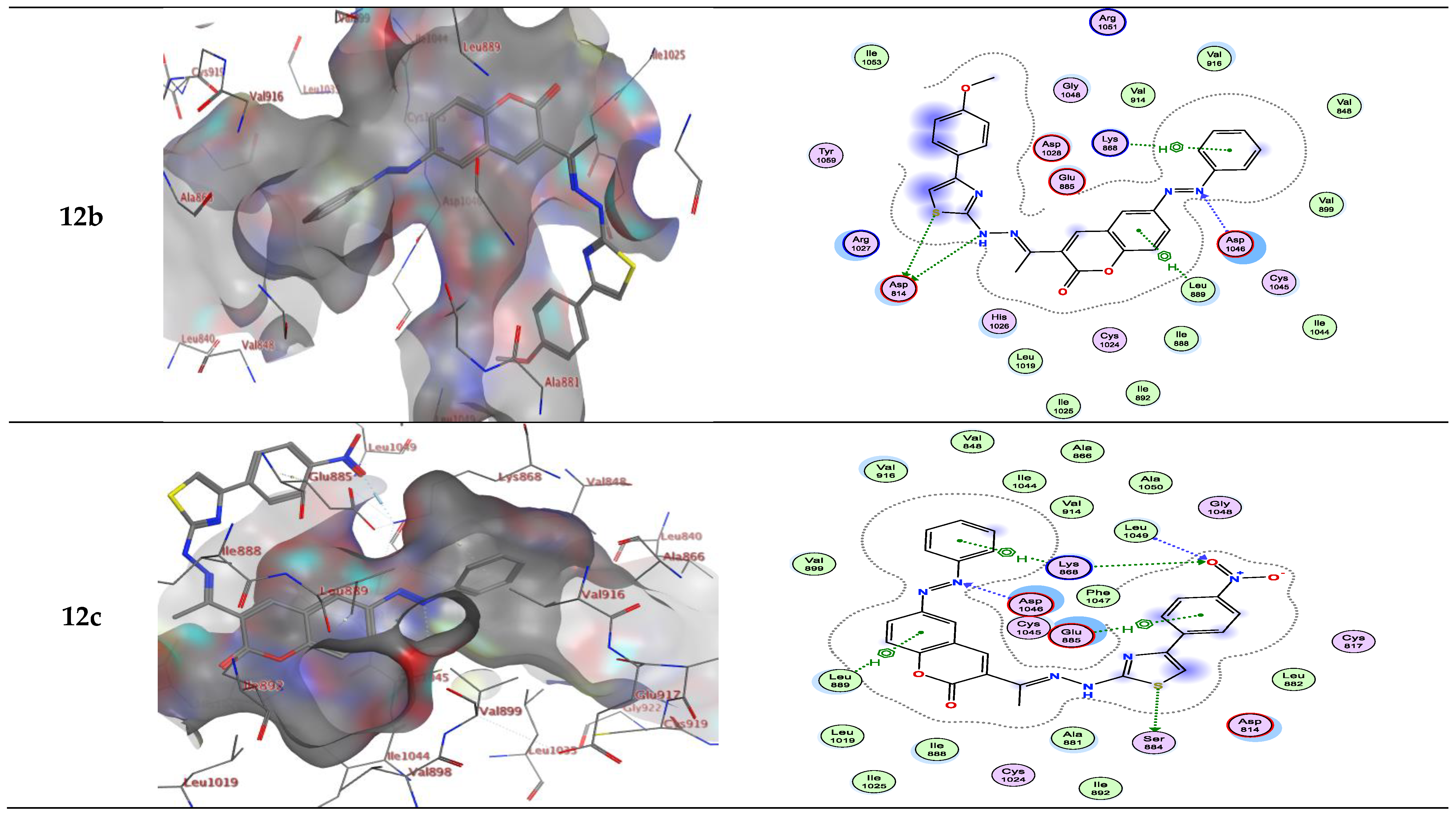
Molecular docking studies demonstrated that the synthesized coumarin-based derivatives (3, 6a–e, 10a–c and 12a–c) interact with the VEGFR-2 enzyme active site in similar ways to those of Sorafenib with binding energy values between −7.723 to −9.900 kcal/mol. The energy scores and receptor interactions of type II VEGFR-2 inhibitor for the synthesized compounds compared to the native ligand (Sorafenib) are summarized in Table 1 and Table 2.
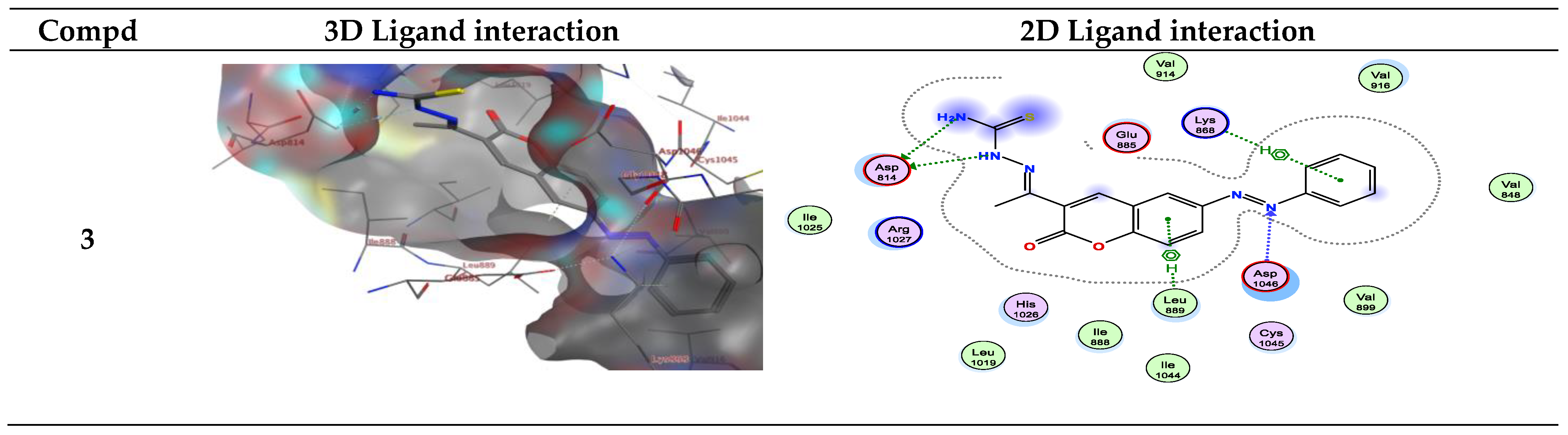
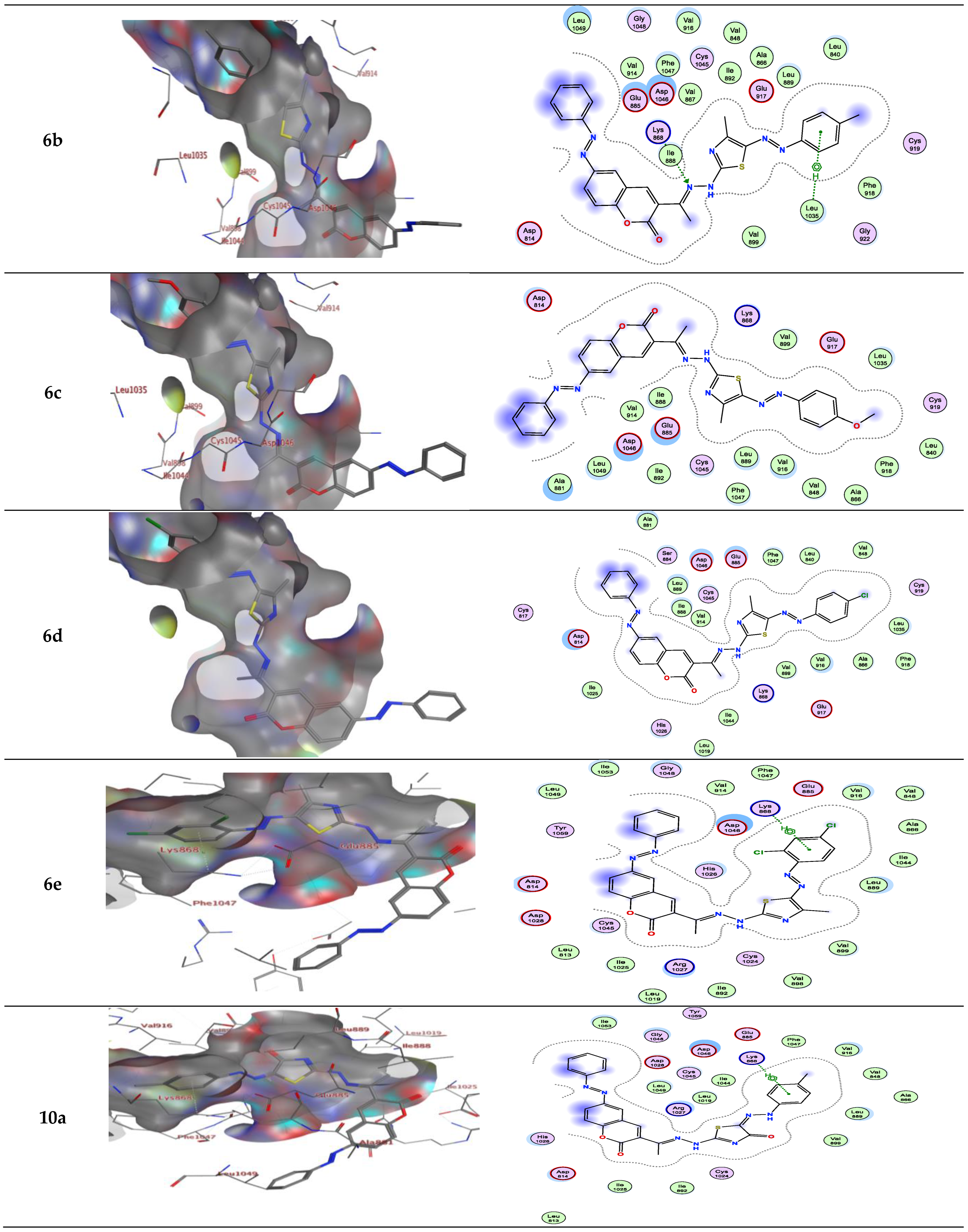
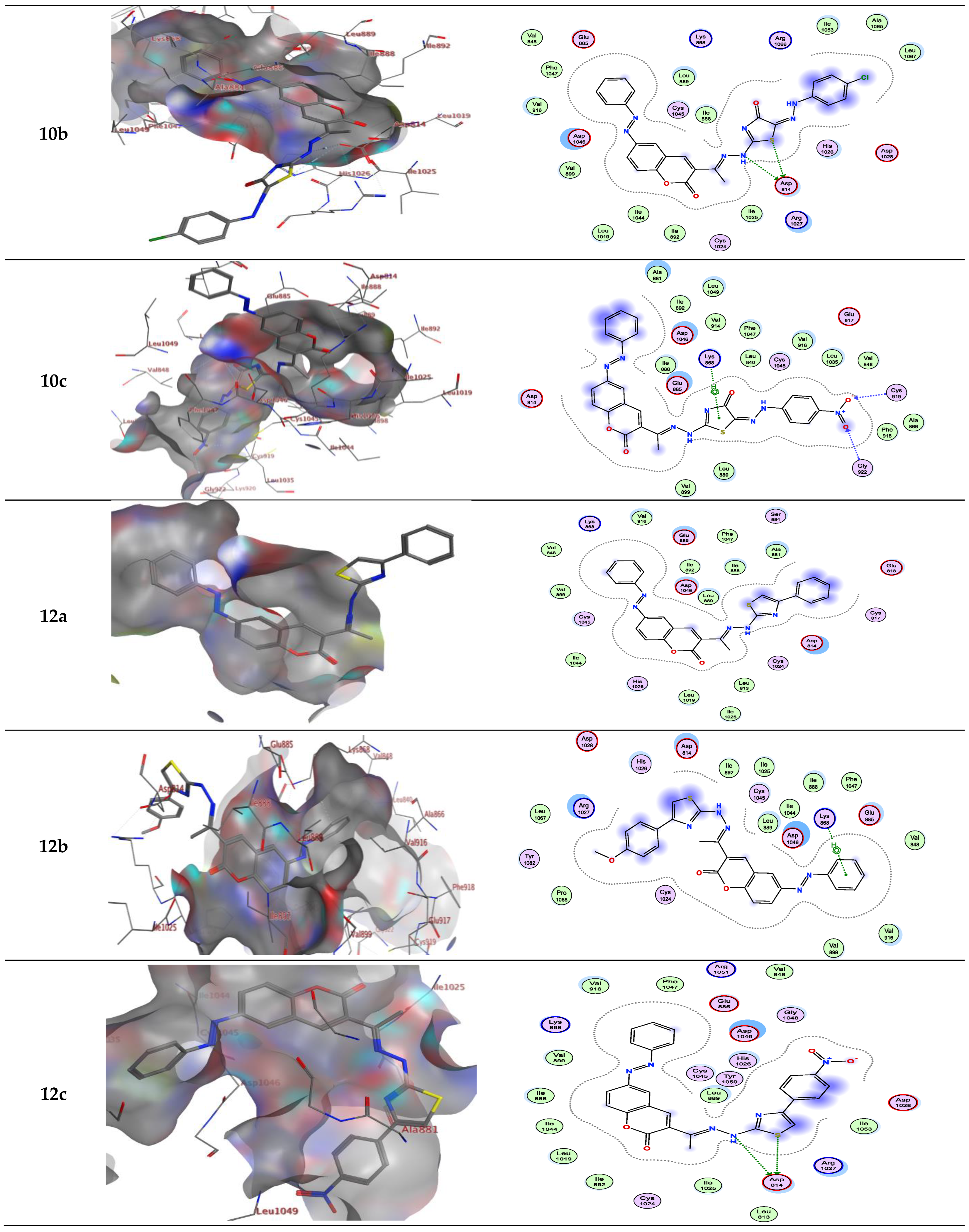
For example, compound 6a in type II VEGFR-2 inhibitor showed two H-bond; one H-bond donor between the carbon phenyl of 2H-chromen-2-one skeleton with Asp814, and another H-bond acceptor between the nitrogen of thiazole ring with Asp1046, respectively, but compounds 6a, b in type I VEGFR-2 inhibitor showed H-bond acceptor between the nitrogen atom with Lys868 (inserted in Figure 4 and Figure 5 as a 2D view).
Compound 6b in type II VEGFR-2 inhibitor form two H-bond acceptors; one between the nitrogen atom of thiazole moiety and Asp1046 and another with Arg1027. In addition, π-H bond interaction with Lys868. Compound 6c forms an H-bond acceptor with the Lys868. As depicted in Figure 4, compounds 6d and 6e form one H-bond acceptor between the nitrogen atom of the azo moiety and the side chain of Asp1046. Moreover, there was π-H interaction between the phenyl scaffold and Lys868. Moreover, compound 6e showed an additional H- bond donor between the sulfur atom of the thiazole ring and Asp1046.
On the other hand, compounds 10a and b form an H-bond acceptor with the Asp1046 via the nitrogen atom of the thiazole ring. Moreover, compound 10b in type II VEGFR-2 inhibitor showed an additional H-bond acceptor between the carbonyl group of the thiazole ring and Phe1047. However, this compound in type I VEGFR-2 inhibitor showed two H-bond donors between the nitrogen atom and the sulfur atom of the thiazole ring with Asp814 (inserted in Figure 5 as a 2D view). Compound 10c exhibits two H-bond acceptors between the two oxygen atoms of the nitro group with the side chain of Cys919 and Gly922, respectively. Moreover, there was one π-H interaction between the thiazole ring and Lys868 (inserted in Figure 4 and Figure 5 as a 3D view).
Compounds 12a and b form two H-bond donors with Asp814 via NH and the sulfur atom of thiazole moiety. Moreover, compound 12a exhibits an additional π-H interaction between the thiazole ring and Arg1027. Compound 12b shows an additional H-bond acceptor between the nitrogen atom of the azo group and Asp1046. In addition, two π-H interactions, one between the phenyl ring and Lys868, and another π-H interaction between the phenyl of 2H-chromen-2-one skeleton and Leu889. These latter π-H interactions of 12b are present in compound 3 in addition to two H-bond donors with Asp814 and one H-bond acceptor with Asp1046. Compound 12c in type II VEGFR-2 inhibitor showed three H-bonds; one H-bond donor between the sulfur atom of the thiazole ring and Ser884, and two H-bond acceptors between the oxygen atom of the nitro group between Lys868 with Leu1049, respectively. In addition, three π-H interactions; one between the phenyl ring with Lys868, another π-H interaction between the phenyl of 2H-chromen-2-one skeleton with Leu889, and finally, π-H interaction between the phenyl of the 4-nitrophenyl skeleton with Glu885. However, this compound in type I VEGFR-2 inhibitor exhibits two H-bond donors between the nitrogen atom and the sulfur atom of the thiazole ring with the side chain of Asp814 (inserted in Figure 5 as a 3D view). The results implied that the ligands under investigation occupy similar positions and orientations within Sorafenib’s hypothesized binding sites.
2.3. Cytotoxic Potential
Using the MTT test and Sorafenib as a reference medication, the cytotoxicity of the most active synthesized thiazole derivatives 6b, 6c, 6d, 10a, and 10c for their human breast cancer (MCF-7) cell line and normal cell line LLC-Mk2 was examined. Afterward, the determination of the tested sample concentrations that were sufficient to kill 50 percent of the cell population (IC50) was done by using the cytotoxicity results in plotting a dose-response curve.
In addition, cytotoxic activities were reported as the average IC50 from three separate tests. Table 3 and Figure 6 demonstrate that most of the evaluated compounds had very varied activity when compared to the reference drug.
Examination of the SAR leads to the following conclusions:
The 1,3-thiazoles 6d and 6b (IC50 = 10.5 ± 0.71 and 11.2 ± 0.80 μM, respectively) demonstrated promising anticancer activity against MCF-7 and outperformed the reference drug (IC50 = 5.10 ± 0.69 μM).
For 1,3-thiazoles 6: Substitution of the phenyl group at position 5 in the 1,3-thiazole ring with Cl atom (electron-withdrawing atom) enhances the anticancer activity (6d > 6b, 6c).
It was observed that the chloro 6d derivative is more active than its methyl 6b counterpart, which may be owing to the influence of substituent lipophilicity and the atomic size; hence, 6d is the most potent derivative. Thus, the orientation of compound 6d in the pocket allowed effective hydrophobic interactions as compared to compound 6b (see the 3D models of both compounds, Figure 3). Additionally, the electron-withdrawing effect of the chloro substituent of 6d has a greater stimulatory effect on activity than the electron-donating methyl substituent of 6b. On the other hand, 2,4-dichloro derivative 6e was more active than the unsubstituted one 6a.
For 1,3-thiazolones 10: The introduction of an electron-withdrawing group (e.g., NO2) at the para-position of the phenyl group at position 5 in the 1,3-thiazole ring enhances the antitumor activity (10c > 10a).
The introduction of the p-substitution of the electron-withdrawing group at derivative 12 has a similar activity enhancement effect. Thus, compound 12c was the most reactive in this series, and the unsubstituted derivative 12a exhibited the lowest activity. In addition, derivatives with thiazole moiety exhibited higher activity as compared to compound 3, thus highlighting the positive effect of the thiazole ring on the activity of the reported compounds.
The 1,3-thiazole derivatives 6 have higher anticancer activity towards MCF-7 cell lines as compared to thiazolone derivatives 10.
The cytotoxic activity of the Sorafenib standard drug and most active compounds 6b, 6c, 6d, 10a and 10c were also estimated on LLC-Mk2 (rhesus monkey kidney epithelial normal cells). The outcomes of these measurements demonstrated the non-toxic effect of the tested derivatives because their CC50 toward normal cell lines is higher than 100 μM, as shown in Table 3.
3. Experimental Section
See the supporting information file S1.
Synthesis of 2-(1-(2-oxo-6-(phenyldiazenyl)-2H-chromen-3-yl)ethylidene) hydrazine-1-carbothioamide (3).
A mixture of 3-acetyl-6-(phenyldiazenyl)-2H-chromen-2-one (1) (10 mmol, 2.92 g) and hydrazinecarbothioamide (2) (10 mmol, 0.91 g) in EtOH (40 mL) containing a few drops of concentrated HCl was heated under reflux for 4 h. The formed precipitate was recrystallized from EtOH to give product 3 as a yellowish-white solid in 73% yield; m.p. 177–179 °C; 1H-NMR (DMSO-d6) δ: 2.41 (s, 3H, CH3), 7.15–8.32 (m, 11H, Ar-H and NH2), 10.39 (s, br, 1H, NH) ppm; IR (KBr) ν cm−1: 3419, 3372, 3284 (NH2 and NH), 1725 (C=O), 1606 (C=N); MS m/z (%): 365 (M+, 37). Anal. Calcd: for C18H15N5O2S (365.41): C, 59.17; H, 4.14; N, 19.17. Found: C, 59.04; H, 4.05; N, 19.00%.
General procedure for the synthesis of 1,3-thiazole derivatives 6a–e, 10a–c and 12a–c.
Catalytic amounts of TEA were added into a solution of compound 3 (1 mmol, 0.365 g) and the appropriate hydrazonoyl chlorides 4a–e or 8a–c or α-bromoketones 11a–c (1 mmol for each) in EtOH (20 mL), and the reaction mixture was refluxed for 4–6 h (monitored by TLC). Finally, the formed precipitate was recrystallized to give thiazoles 6a–e or 10a–c, or 12a–c, respectively.
Alternate synthesis of 6a.
A cold aqueous solution of benzenediazonium salt was added portion wise to a cold solution of compound 7 (1 mmol, 0.403 g) in pyridine (15 mL) under stirring then the precipitated product was recrystallized from DMF to give compound 6a in 72% yield.
The physical properties and spectral data of the isolated products are listed in the supporting information file S1.
3.1. Molecular Docking
The most active compounds were sketched using Chemdraw 12.0, and their molecular modeling was performed using molecular operating environment software. The results were refined using the London DG force and force field energy. All minimizations were performed until a root mean square deviation (RMSD) gradient 0.1 kcal·mol−1Å−1 using MMFF 94× (Merck molecular force field 94×), and the partial charges were determined automatically. The binding affinity of the ligand was evaluated using the scoring function and dock function (S, Kcal/mol) created by the MOE software. The enzyme’s X-ray crystal structure (PDB ID: 4ASD, resolution: 2.03 Å) was downloaded in PDB format according to the protein data bank [51,52]. The enzyme was ready for studies using docking: (i) the water was eliminated from the protein; (ii) hydrogen atoms were added to the structure in their characteristic geometries, then reconnected the bonds broken and fixing the potential; (iii) as the large site, dummy atoms were used to execute a site search using MOE Alpha Site Finder upon this enzyme structure [53]; (iv) analyzing the ligand’s interaction with the active site’s amino acids. The best docking score is obtained as the most negative value for the active ligands. All docking procedures and scoring were recorded according to established protocols [54,55,56]. Triangle Matcher placement method and London dG score tool were used for docking.
3.2. Cytotoxic Assay
The cytotoxicity of the investigated substances was assessed and determined by MTT assay, and the detailed cytotoxicity assay is included in the Supplementary File S1 [57,58].
4. Conclusions
This study disclosed the design and synthesis of novel 3-thiazolhydrazinyl coumarins utilizing 3-acetyl-6-methyl-2H-chromen-2-one. Spectroscopy and elemental analyses were utilized to confirm the hypothesized product’s structures. Moreover, a molecular docking study of synthesized 2H-chromen-2-one derivatives was performed to investigate their interactions with VEGFR-2’s active region. In addition, the most active derivatives of the designed compounds were tested in vitro against the MCF-7 and LLC-Mk2 cell lines using MTT assay and Sorafenib as a reference drug. The results demonstrated the potential anti-tumor activities of compounds 6d and 6b (IC50 = 10.5 ± 0.71 and 11.2 ± 0.80 μM, respectively). Therefore, the present study demonstrated that the reported thiazolyl coumarins are potential (VEGFR-2) inhibitors and pave the way for the synthesis of additional libraries based on the reported scaffold, which could eventually lead to the development of efficient treatment for breast cancer
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28020689/s1. Supplementary File S1: Physical and spectral data of the synthesized compounds and their 1H- and 13C-NMR spectra.
Author Contributions
T.Z.A., M.F., B.F., M.E.A.Z. and S.M.G.: Supervision, Investigation, Methodology, Resources, Formal analysis, Data curation, Funding acquisition, Writing-original draft, Writing-review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request.
Acknowledgments
The authors express their recognition to Islamic University of Madinah, Saudi Arabia, for supporting and providing Labs and chemicals for the research.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds 3, 6a–e, 10a–c and 12a–c are available from the authors.
References
- Stewart, B.W.; Wild, C.P. World Cancer Report 2014; World Health Organization, International Agency for Research on Cancer [IARC]: Lyon, France, 2014. [Google Scholar]
- Lee, N.C.; Wong, F.L.; Jamison, P.M.; Jones, S.F.; Galaska, L.; Brady, K.T.; Wethers, B.; Stokes-Townsend, G.A. Implementation of the National Breast and Cervical Cancer Early Detection Program. Cancer 2014, 120, 2540–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Cancer Institute. SEER Stat Fact Sheet: Breastcancer. 2014. Available online: http://seer.cancer.gov/statfacts/html/breast.html (accessed on 22 August 2014).
- National Cancer Institute. SEER Stat Fact Sheet: Cervix Uteri Cancer. 2014. Available online: http://seer.cancer.gov/statfacts/html/cervix.html (accessed on 22 August 2014).
- Fox, S.B.; Generali, D.G.; Harris, A.L. Breast tumour angiogenesis. Breast Cancer Res. 2007, 9, 216. [Google Scholar] [CrossRef] [PubMed]
- Ryden, L.; Stendahl, M.; Jonsson, H.; Emdin, S.; Bengtsson, N.O.; Landberg, G. Tumor-specific VEGF-A and VEGFR2 in postmenopausal breast cancer patients with long-term follow-up. Implication of a link between VEGF pathway and tamoxifen response. Breast Cancer Res. Treat. 2005, 89, 135–143. [Google Scholar] [CrossRef]
- Roskoski, R.J. VEGF receptor protein-tyrosine kinases: Structure and regulation. Biochem. Biophys. Res. Commun. 2008, 37, 287–291. [Google Scholar] [CrossRef]
- Zuccotto, F.; Ardini, E.; Casale, E.; Angiolini, M. Through the “gatekeeper door: Exploiting the active kinase conformation. J. Med. Chem. 2010, 53, 2681–2694. [Google Scholar] [CrossRef]
- Regan, J.; Pargellis, C.A.; Cirillo, P.F.; Gilmore, T.; Hickey, E.R.; Peet, G.W.; Proto, A.; Swinamer, A.; Moss, N. The kinetics of binding to p38MAP kinase by analogues of BIRB 796. Bioorg. Med. Chem. Lett. 2003, 13, 3101. [Google Scholar] [CrossRef]
- Patel, R.R.; Sengupta, S.; Kim, H.R.; Klein-Szanto, A.J.; Pyle, J.R.; Zhu, F.; Li, T.; Ross, E.A.; Oseni, S.; Fargnoli, J.; et al. Experimental treatment of oestrogen receptor (ER) positive breast cancer with tamoxifen and brivanib alaninate, a VEGFR-2/FGFR-1 kinase inhibitor: A potential clinical application of angiogenesis inhibitors. Eur. J. Cancer 2010, 46, 1537–1553. [Google Scholar] [CrossRef] [Green Version]
- Rawat, A.; Reddy, A.V.B. Recent advances on anticancer activity of coumarin derivative. Eur. J. Med. Chem. Rep. 2022, 5, 100038. [Google Scholar] [CrossRef]
- Morigi, R.; Locatelli, A.; Leoni, A.; Rambaldi, M. Recent patents on thiazole derivatives endowed with antitumor activity. Recent Pat. Anticancer. Drug Discov. 2015, 10, 280–297. [Google Scholar] [CrossRef]
- Batran, R.Z.; Dawood, D.H.; El-Seginy, S.A.; Maher, T.J.; Gugnani, K.S.; Rondon-Ortiz, A.N. Coumarinyl pyranopyrimidines as new neuropeptide S receptor antagonists; design, synthesis, homology and molecular docking. Bioorg. Chem. 2017, 75, 274–290. [Google Scholar] [CrossRef]
- Abdelhafez, O.M.; Amin, K.M.; Ali, H.I.; Abdalla, M.M.; Batran, R.Z. Synthesis of new 7-oxycoumarin derivatives as potent and selective monoamine oxidase A inhibitors. J. Med. Chem. 2012, 55, 10424–10436. [Google Scholar] [CrossRef]
- Batran, R.Z.; Kassem, A.F.; Abbas, E.M.H.; Elseginy, S.A.; Mounier, M.M. Design, synthesis and molecular modeling of new 4-phenylcoumarin derivatives as tubulin polymerization inhibitors targeting MCF-7 breast cancer cells. Bioorg. Med. Chem. 2018, 26, 3474–3490. [Google Scholar] [CrossRef] [PubMed]
- Alshabanah, L.A.; Al-Mutabagani, L.A.; Gomha, S.M.; Ahmed, H.A. Three-component synthesis of some new coumarin derivatives as anti-cancer agents. Front. Chem. 2022, 9, 762248. [Google Scholar] [CrossRef]
- Zhao, P.; Chen, L.; Li, L.; Wei, Z.; Tong, B.; Jia, Y.; Kong, L.; Xia, Y.; Dai, Y. SC-III3, a novel scopoletin derivative, induces cytotoxicity in hepatocellular cancer cells through oxidative DNA damage and ataxia telangiectasia-mutated nuclear protein kinase activa-tion. BMC Cancer 2014, 14, 987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batran, R.Z.; Dawood, D.H.; El-Seginy, S.A.; Ali, M.M.; Maher, T.J.; Gugnani, K.S.; Rondon-Ortiz, A.N. New Coumarin Derivatives as Anti-Breast and Anti-Cervical Cancer Agents Targeting VEGFR-2 and p38α MAPK. Arch. Pharm. (Weinheim) 2017, 350, e1700064. [Google Scholar] [CrossRef]
- Gomha, S.M.; Abdel-aziz, H.M. Synthesis and antitumor activity of 1,3,4-thiadiazole derivatives bearing coumarine ring. Heterocycles 2015, 91, 583–592. [Google Scholar] [CrossRef]
- Luo, G.; Li, X.; Zhang, G.; Wu, C.; Tang, Z.; Liu, L.; You, Q.; Xiang, H. Novel SERMs based on 3-aryl-4-aryloxy-2H-chromen-2-one skeleton—A possible way to dual ERα/VEGFR-2 ligands for treatment of breast cancer. Eur. J. Med. Chem. 2017, 140, 252–273. [Google Scholar] [CrossRef]
- Pan, R.; Dai, Y.; Gao, X.H.; Lu, D.; Xia, Y.F. Inhibition of vascular endothelial growth factorinduced angiogenesis by scopoletin through interrupting the autophosphorylation of VEGF receptor 2 and its downstream signaling pathways. Vascul. Pharmacol. 2011, 54, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Park, S.L.; Won, S.Y.; Song, J.H.; Lee, S.Y.; Kim, W.J.; Moon, S.K. Esculetin inhibits VEGF-Induced angiogenesis both in vitro and in vivo. Am. J. Chin. Med. 2016, 44, 61–76. [Google Scholar] [CrossRef]
- Franchin, M.; Rosalen, P.L.; da Cunha, M.G.; Silva, R.L.; Colon, D.F.; Bassi, G.S.; de Alenca, S.M.; Ikegaki, M.; Alves-Filho, J.C.; Cunha, F.Q.; et al. Cinnamoyloxy-mammeisin Isolated from Geopropolis Attenuates Inflammatory Process by Inhibiting Cytokine Production: Involvement of MAPK, AP-1, and NF-κB. J. Nat. Prod. 2016, 79, 1828–1833. [Google Scholar] [CrossRef]
- Arshad, M.F.; Alam, A.; Alshammari, A.A.; Alhazza, M.B.; Alzimam, I.M.; Alam, M.A.; Mustafa, G.; Ansari, M.S.; Alotaibi, A.M.; Alotaibi, A.A.; et al. Thiazole: A Versatile Standalone Moiety Contributing to the Development of Various Drugs and Biologically Active Agents. Molecules 2022, 27, 3994. [Google Scholar] [CrossRef] [PubMed]
- Raveesha, R.; Anusuya, A.M.; Raghu, A.V.; Kumar, K.Y.; Kumar, M.G.D.; Prasad, S.B.B.; Prashanth, M.K. Synthesis and characterization of novel thiazole derivatives as potential anticancer agents: Molecular docking and DFT studies. Comput. Toxicol. 2022, 21, 100202–100219. [Google Scholar] [CrossRef]
- Gomha, S.M.; Riyadh, S.M.; Huwaimel, B.; Zayed, M.E.M.; Abdellattif, M.H. Synthesis, molecular docking study and cytotoxic activity on MCF cells of some new thiazole clubbed thiophene scaffolds. Molecules 2022, 27, 4639. [Google Scholar] [CrossRef]
- Abdel-Aziz, S.A.; Taher, E.S.; Lan, P.; El-Koussi, N.A.; Salem, O.I.A.; Gomaa, H.A.M.; Youssif, B.G.M. New pyrimidine/thiazole hybrids endowed with analgesic, anti-inflammatory, and lower cardiotoxic activities: Design, synthesis, and COX-2/sEH dual inhibition. Arch. Pharm. 2022, 355, e2200024. [Google Scholar] [CrossRef]
- Altıntop, M.D.; Sever, B.; Çiftçi, G.A.; Özdemir, A. Design, Synthesis, and Evaluation of a New Series of Thiazole-Based Anticancer Agents as Potent Akt Inhibitors. Molecules 2018, 23, 1318. [Google Scholar] [CrossRef] [Green Version]
- Hassan, A.; Badr, M.; Hassan, H.A.; Abdelhamid, D.; Abuo-Rahma, G.E.D.A. Novel 4-(piperazin-1-yl)quinolin-2(1H)-one bearing thiazoles with antiproliferative activity through VEGFR-2-TK inhibition. Bioorg. Med. Chem. 2021, 40, 116168–116181. [Google Scholar] [CrossRef]
- Perrot-Applanat, M.; Di Benedetto, M. Autocrine functions of VEGF in breast tumor cells: Adhesion, survival, migration and invasion. Cell Adh. Migr. 2012, 6, 547–553. [Google Scholar] [CrossRef] [Green Version]
- Song, G.; Li, Y.; Jiang, G. Role of VEGF/VEGFR in the pathogenesis of leukemias and as treatment targets. Oncol. Rep. 2012, 28, 1935–1944. [Google Scholar] [CrossRef] [Green Version]
- Bilodeau, M.T.; Rodman, L.D.; McGaughey, G.B.; Coll, K.E.; Koester, T.J.; Hoffman, W.F.; Thomas, K.A. The discovery of N-(1,3-thiazol-2-yl) pyridin-2-amines as potent inhibitors of KDR kinase. Bioorg. Med. Chem. Lett. 2004, 14, 2941–2945. [Google Scholar] [CrossRef]
- Bilodeau, M.T.; Balitza, A.E.; Koester, T.J.; Manley, P.J.; Rodman, L.D.; Buser-Doepner, C.; Hartman, G.D. Potent N-(1,3-thiazol-2-yl)pyridin-2-amine vascular endothelial growth factor receptor tyrosine kinase inhibitors with excellent pharmacokinetics and low affinity for the hERG ion channel. J. Med. Chem. 2004, 47, 6363–6372. [Google Scholar] [CrossRef]
- Sisko, J.T.; Tucker, T.J.; Bilodeau, M.T.; Buser, C.A.; Ciecko, P.A.; Coll, K.E.; Hartman, G.D. Potent 2-[(pyrimidin-4-yl)amine}-1,3-thiazole-5-carbonitrile-based inhibitors of VEGFR-2 (KDR) kinase. Bioorg. Med. Chem. Lett. 2006, 16, 1146–1150. [Google Scholar] [CrossRef]
- Kiselyov, A.S.; Piatnitski, E.; Semenov, V.V. N-(aryl)-4-(azolylethyl)thiazole-5-carboxamides: Novel potent inhibitors of VEGF receptors I and II. Bioorg. Med. Chem. Lett. 2006, 16, 602–606. [Google Scholar] [CrossRef]
- Wickens, P.; Kluender, H.; Dixon, J.; Brennan, C.; Achebe, F.; Bacchiocchi, A.; Levy, J. SAR of a novel “anthranilamide like” series of VEGFR-2, multi protein kinase inhibitors for the treatment of cancer. Bioorg. Med. Chem. Lett. 2007, 17, 4378–4381. [Google Scholar] [CrossRef]
- Abou-Seri, S.M.; Eldehna, W.M.; Ali, M.M.; Abou El Ella, D.A. 1-piperazinylphthalazines as potential VEGFR-2 inhibitors and anticancer agents: Synthesis and in vitro biological evaluation. Eur. J. Med. Chem. 2016, 107, 165–179. [Google Scholar] [CrossRef]
- El-Miligy, M.M.; Abd El Razik, H.A.; Abu-Serie, M.M. Synthesis of piperazine-based thiazolidinones as VEGFR2 tyrosine kinase inhibitors inducing apoptosis. Fut. Med. Chem. 2017, 9, 1709–1729. [Google Scholar] [CrossRef] [PubMed]
- Gomha, S.M.; Abdelhady, H.A.; Hassain, D.Z.H.; Abdelmonsef, A.H.; El-Naggar, M.; Elaasser, M.M.; Mahmoud, H.K. Thiazole based thiosemicarbazones: Synthesis, cytotoxicity evaluation and molecular docking study. Drug Des. Dev. Ther. 2021, 15, 659–677. [Google Scholar] [CrossRef] [PubMed]
- Aljohani, G.F.; Abolibda, T.Z.; Alhilal, M.; Al-Humaidi, J.Y.; Alhilal, S.; Ahmed, H.A.; Gomha, S.M. Novel thiadiazole-thiazole hybrids: Synthesis, molecular docking, and cytotoxicity evaluation against liver cancer cell lines. J. Taibah Uni. Sci. 2022, 16, 1005–1015. [Google Scholar] [CrossRef]
- Abouzied, A.S.; Al-Humaidi, J.Y.; Bazaid, A.S.; Qanash, H.; Binsaleh, N.K.; Alamri, A.; Ibrahim, S.M.; Gomha, S.M. Synthesis, molecular docking study, and cytotoxicity evaluation of some novel 1,3,4-thiadiazole as well as 1,3-thiazole derivatives bearing a pyridine moiety. Molecules 2022, 27, 6368. [Google Scholar] [CrossRef]
- Nayl, A.A.; Arafa, W.A.A.; Ahmed, M.; Abd-Elhamid, A.I.; El-Fakharany, E.M.; Abdelgawad, M.A.; Gomha, S.M.; Ibrahim, H.M.; Aly, A.A.; Bräse, S.; et al. Novel pyridinium based ionic liquid promoter for aqueous knoevenagel condensation: Green and efficient synthesis of new derivatives with their anticancer evaluation. Molecules 2022, 27, 2940. [Google Scholar] [CrossRef] [PubMed]
- Gomha, S.M.; Edrees, M.M.; Muhammad, Z.A.; El-Reedy, A.A. 5-(Thiophen-2-yl)-1,3,4-thiadiazole derivatives: Synthesis, molecular docking and in-vitro cytotoxicity evaluation as potential anti-cancer agents. Drug Des. Devel. Ther. 2018, 12, 1511–1523. [Google Scholar] [CrossRef]
- Gomha, S.M.; Edrees, M.M.; Faty, R.A.M.; Muhammad, Z.A.; Mabkhot, Y.N. Microwave-assisted one pot three-component synthesis of some novel pyrazole scaffolds as potent anticancer agents. Chem. Central J. 2017, 11, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomha, S.M.; Abdelaziz, M.R.; Kheder, N.A.; Abdel-aziz, H.M.; Alterary, S.; Mabkhot, Y.N. A Facile access and evaluation of some novel thiazole and 1,3,4-thiadiazole derivatives incorporating thiazole moiety as potent anticancer agents. Chem. Central J. 2017, 11, 105. [Google Scholar] [CrossRef]
- Edrees, M.M.; Abu-Melha, S.; Saad, A.M.; Kheder, N.A.; Gomha, S.M.; Muhammad, Z.A. Eco-friendly synthesis, characterization and biological evaluation of some new pyrazolines containing thiazole moiety as potential anticancer and antimicrobial agents. Molecules 2018, 23, 2970. [Google Scholar] [CrossRef] [Green Version]
- Sivaguru, P.; Sandhiya, R.; Adhiyaman, M.; Lalitha, A. Synthesis and antioxidant properties of novel 2H-chromene-3-carboxylate and 3-acetyl-2H-chromene derivatives. Tetrahedron Lett. 2016, 57, 2496–2501. [Google Scholar] [CrossRef]
- Badrey, M.G.; Gomha, S.M. 3-Amino-8-hydroxy-4-imino-6-methyl-5-phenyl-4,5-dihydro-3H-chromeno[2,3-d]pyrimidine: An efficient key precursor for novel synthesis of some interesting triazines and triazepines as potential anti-tumor agents. Molecules 2012, 17, 11538–11553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomha, S.M.; Khalil, K.D. A Convenient ultrasound-promoted synthesis and cytotoxic activity of some new thiazole derivatives bearing a coumarin nucleus. Molecules 2012, 17, 9335–9347. [Google Scholar] [CrossRef]
- Gomha, S.M. A facile one-pot synthesis of 6,7,8,9-tetrahydrobenzo[4,5]thieno[2,3-d]-1,2,4-triazolo[4,5-a]pyrimidin-5-ones. Monatsh. Chem. 2009, 140, 213–220. [Google Scholar] [CrossRef]
- McTigue, M.; Murray, B.W.; Chen, J.H.; Deng, Y.; Solowiej, J.; Kania, R.S. Molecular Conformations, Interactions, and Properties Associated with Drug Efficiency and Clinical Performance Among Vegfr Tk Inhibitors molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among Vegfr Tk Inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 18281–18289. [Google Scholar]
- Othman, I.M.M.; Alamshany, Z.M.; Tashkandi, N.Y.; Gad-Elkareem, M.A.; Anwar, M.M.; Nossier, E.S. New pyrimidine and pyrazole-based compounds as potential EGFR inhibitors: Synthesis, anticancer, antimicrobial evaluation and computational studies. Bioorg. Chem. 2021, 114, 105078. [Google Scholar] [CrossRef]
- Labute, P. Protonate3D: Assignment of Ionization States and Hydrogen Coordinates to Macromolecular Structures. Proteins 2008, 75, 187–205. [Google Scholar] [CrossRef] [Green Version]
- Kattan, S.W.; Nafie, M.S.; Elmgeed, G.A.; Alelwani, W.; Badar, M.; Tantawy, M.A. Molecular docking, anti-proliferative activity and induction of apoptosis in human liver cancer cells treated with androstane derivatives: Implication of PI3K/AKT/mTOR pathway. J. Steroid Biochem. Mol. Biol. 2020, 198, 105604. [Google Scholar] [CrossRef]
- Tantawy, M.A.; Sroor, F.M.; Mohamed, M.F.; El-Naggar, M.E.; Saleh, F.M.; Hassaneen, H.M.; Abdelhamid, I.A. Molecular Docking Study, Cytotoxicity, Cell Cycle Arrest and Apoptotic Induction of Novel Chalcones Incorporating Thiadiazolyl Isoquinoline in Cervical Cancer. Anti-Cancer Agents Med. Chem. 2020, 20, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Nafie, M.S.; Tantawy, M.A.; Elmgeed, G.A. Screening of different drug design tools to predict the mode of action of steroidal derivatives as anti-cancer agents. Steroids 2019, 152, 108485. [Google Scholar] [CrossRef] [PubMed]
- Gomha, S.M.; Riyadh, S.M.; Mahmmoud, E.A.; Elaasser, M.M. Synthesis and anticancer activities of thiazoles, 1,3-thiazines, and thiazolidine using chitosan-grafted-poly(vinylpyridine) as basic catalyst. Heterocycles 2015, 91, 1227–1243. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Examples of some reported coumarins, thiazoles and thiazolylcoumarins as anticancer agents.
Figure 1.
Examples of some reported coumarins, thiazoles and thiazolylcoumarins as anticancer agents.

Figure 2.
Examples of some reported coumarins (I–V) and thiazoles (VI–VIIII) based VEGFR-2 inhibitors and the target compounds 6a–e, 10a–c and 12a–c.
Figure 2.
Examples of some reported coumarins (I–V) and thiazoles (VI–VIIII) based VEGFR-2 inhibitors and the target compounds 6a–e, 10a–c and 12a–c.

Scheme 1.
Synthesis of arylazothiazole derivatives 6a–e and arylhydrazothiazolone derivatives 10a–c.
Scheme 1.
Synthesis of arylazothiazole derivatives 6a–e and arylhydrazothiazolone derivatives 10a–c.

Scheme 2.
Synthesis of thiazole derivatives 12a–c.

Figure 3.
2D Binding of sorafenib (a reference ligand) with DFG-out and DFG-in motif of type II and I for VEGFR-2, respectively.
Figure 3.
2D Binding of sorafenib (a reference ligand) with DFG-out and DFG-in motif of type II and I for VEGFR-2, respectively.

Figure 4.
Interactions between ligands in three dimensions (left) and two dimensions (right) within a compound’s binding pocket of 4ASD (type II VEGFR-2 inhibitor) for compounds 3, 6a–e, 10a–c and 12a–c.
Figure 4.
Interactions between ligands in three dimensions (left) and two dimensions (right) within a compound’s binding pocket of 4ASD (type II VEGFR-2 inhibitor) for compounds 3, 6a–e, 10a–c and 12a–c.




Figure 5.
Interactions between ligands in three dimensions (left) and two dimensions (right) within a compound’s binding pocket of 4ASD (type I VEGFR-2 inhibitor) for compounds 3, 6a–e, 10a–c and 12a–c.
Figure 5.
Interactions between ligands in three dimensions (left) and two dimensions (right) within a compound’s binding pocket of 4ASD (type I VEGFR-2 inhibitor) for compounds 3, 6a–e, 10a–c and 12a–c.



Figure 6.
SAR of the synthesized thiazoles.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Energy scores and receptor interactions of type II VEGFR-2 inhibitors for the synthesized compounds (3, 6a–e, 10a–c and 12a–c), compared to Sorafenib.
Table 1.
Energy scores and receptor interactions of type II VEGFR-2 inhibitors for the synthesized compounds (3, 6a–e, 10a–c and 12a–c), compared to Sorafenib.
| Compound | Energy Score (S) (kcal/mol) | Interacting Residues |
|---|---|---|
| 3 | −7.72 | Asp814, Asp1046, Lys868, and Leu889 |
| 6a | −9.15 | Leu1035 and Lys868 |
| 6b | −9.81 | Asp1046, Arg1027 and Lys868 |
| 6c | −9.58 | Lys868 |
| 6d | −9.90 | Asp1046 and Lys868 |
| 6e | −9.37 | Asp1046 and Lys868 |
| 10a | −9.67 | Asp1046 |
| 10b | −9.42 | Asp1046 and Phe1047 |
| 10c | −9.69 | Cys919, Gly922 and Lys868 |
| 12a | −8.52 | Asp814 and Arg1027 |
| 12b | −8.90 | Asp814, Asp1046, Lys868 and Leu889 |
| 12c | −8.95 | Asp814 |
| Sorafenib | −8.65 | Cys1045, Asp1046, Glu885 and Cys919 |
Table 2.
Energy scores and receptor interactions of active VEGFR-2 with DFG-in motif for the synthesized compounds (3, 6a–e, 10a–c and 12a–c), compared to Sorafenib.
Table 2.
Energy scores and receptor interactions of active VEGFR-2 with DFG-in motif for the synthesized compounds (3, 6a–e, 10a–c and 12a–c), compared to Sorafenib.
| Compound | Energy Score (S) (kcal/mol) | Receptor Interactions |
|---|---|---|
| 3 | −7.319 | Leu840/π-H Lys868/π-H |
| 6a | −9.155 | Lys868/H-bond acceptor Lys868/π-H Leu1035/π-H |
| 6b | −9.089 | Lys868/H-bond acceptor Leu1035/π-H |
| 6c | −9.580 | - |
| 6d | −9.857 | - |
| 6e | −9.254 | Lys868/π-H |
| 10a | −9.205 | Lys868/π-H |
| 10b | −8.728 | Asp814/H-bond donor |
| 10c | −9.572 | Cyss919/H-bond acceptor Cly922/H-bond acceptor Lys868/π-H |
| 12a | −8.135 | - |
| 12b | −8.363 | Lys868/π-H |
| 12c | −8.735 | Asp814/H-bond donor |
| Sorafenib | −8.214 | Asp814 |
Table 3.
In vitro cytotoxic activity of thiazoles 6b, 6c, 6d, 10a and 10c against MCF-7 and LLC-MK2.
Table 3.
In vitro cytotoxic activity of thiazoles 6b, 6c, 6d, 10a and 10c against MCF-7 and LLC-MK2.
| Tested Compounds | MCF-7IC50 (μM) * | LLC-MK2 * CC50 (μM) |
|---|---|---|
| 6b | 11.2 ± 0.80 | 152.3 ± 9.30 |
| 6c | 14.1 ± 0.63 | 141.0 ± 5.15 |
| 6d | 10.5 ± 0.71 | 132.2 ± 8.26 |
| 10a | 28.6 ± 0.64 | 140.3 ± 10.29 |
| 10c | 15.7 ± 0.67 | 163.1 ± 9.12 |
| Sorafenib | 5.10 ± 0.49 | 135.3 ± 4.08 |
* The data are presented as mean, standard error (SE).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Abolibda, T.Z.; Fathalla, M.; Farag, B.; Zaki, M.E.A.; Gomha, S.M. Synthesis and Molecular Docking of Some Novel 3-Thiazolyl-Coumarins as Inhibitors of VEGFR-2 Kinase. Molecules 2023, 28, 689. https://doi.org/10.3390/molecules28020689
AMA Style
Abolibda TZ, Fathalla M, Farag B, Zaki MEA, Gomha SM. Synthesis and Molecular Docking of Some Novel 3-Thiazolyl-Coumarins as Inhibitors of VEGFR-2 Kinase. Molecules. 2023; 28(2):689. https://doi.org/10.3390/molecules28020689
Chicago/Turabian StyleAbolibda, Tariq Z., Maher Fathalla, Basant Farag, Magdi E. A. Zaki, and Sobhi M. Gomha. 2023. "Synthesis and Molecular Docking of Some Novel 3-Thiazolyl-Coumarins as Inhibitors of VEGFR-2 Kinase" Molecules 28, no. 2: 689. https://doi.org/10.3390/molecules28020689