
Group I Intron as a Potential Target for Antifungal Compounds: Development of a Trans-Splicing High-Throughput Screening Strategy
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
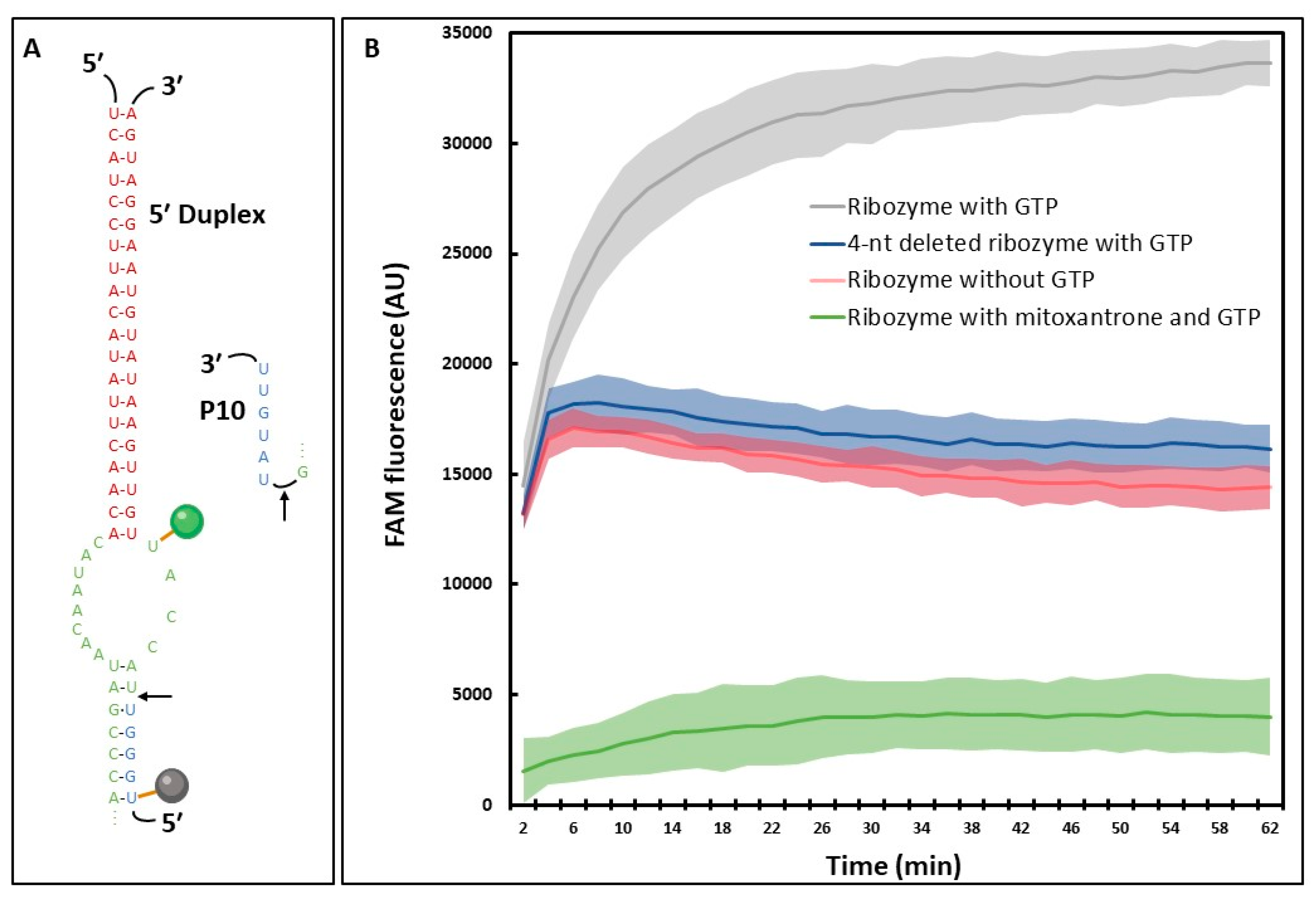
:1. Introduction
2. Results
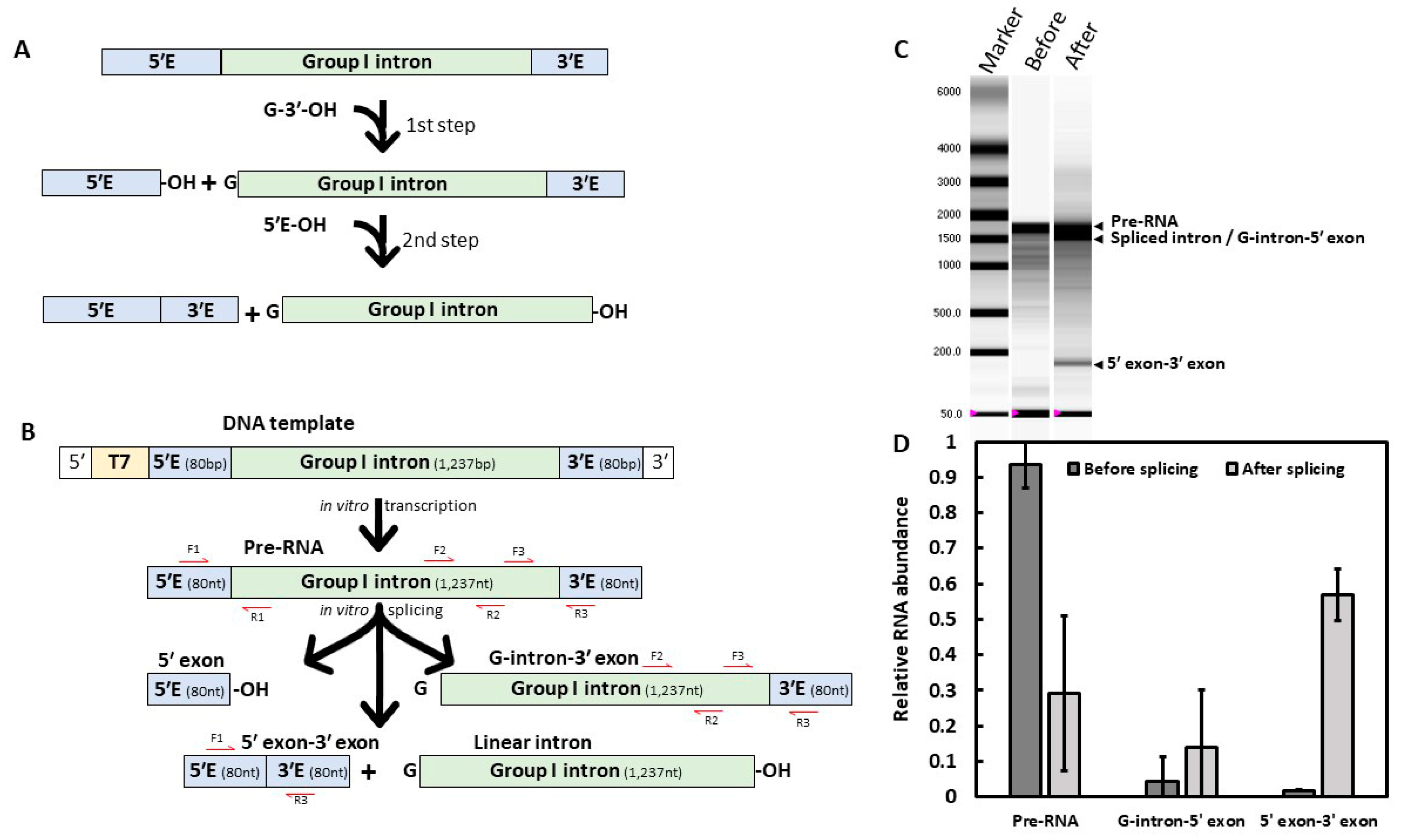
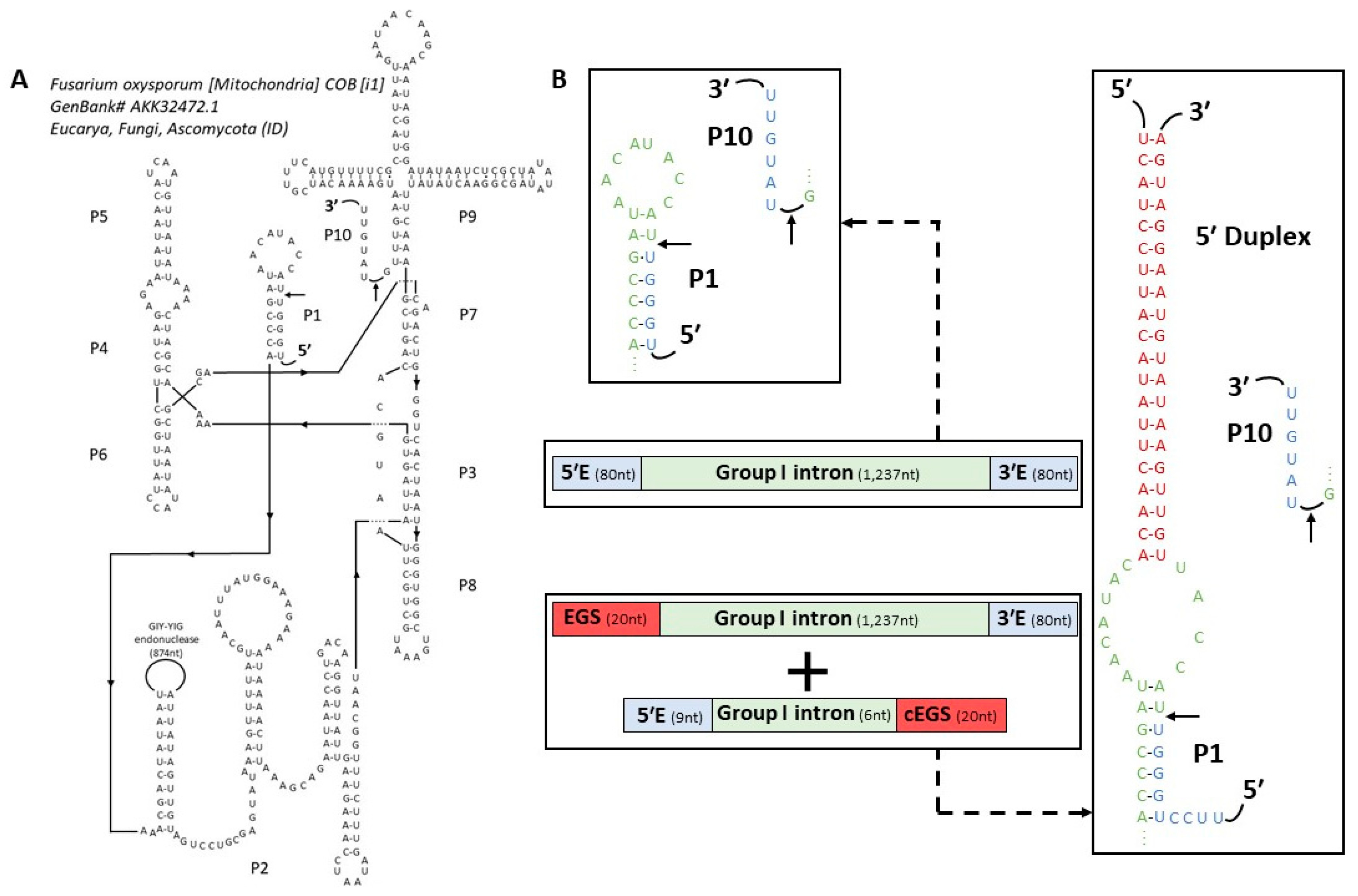
2.1. In Vitro Self-Splicing Activity of Fusarium Cob Intron
2.2. Trans-Splicing Ribozyme Design
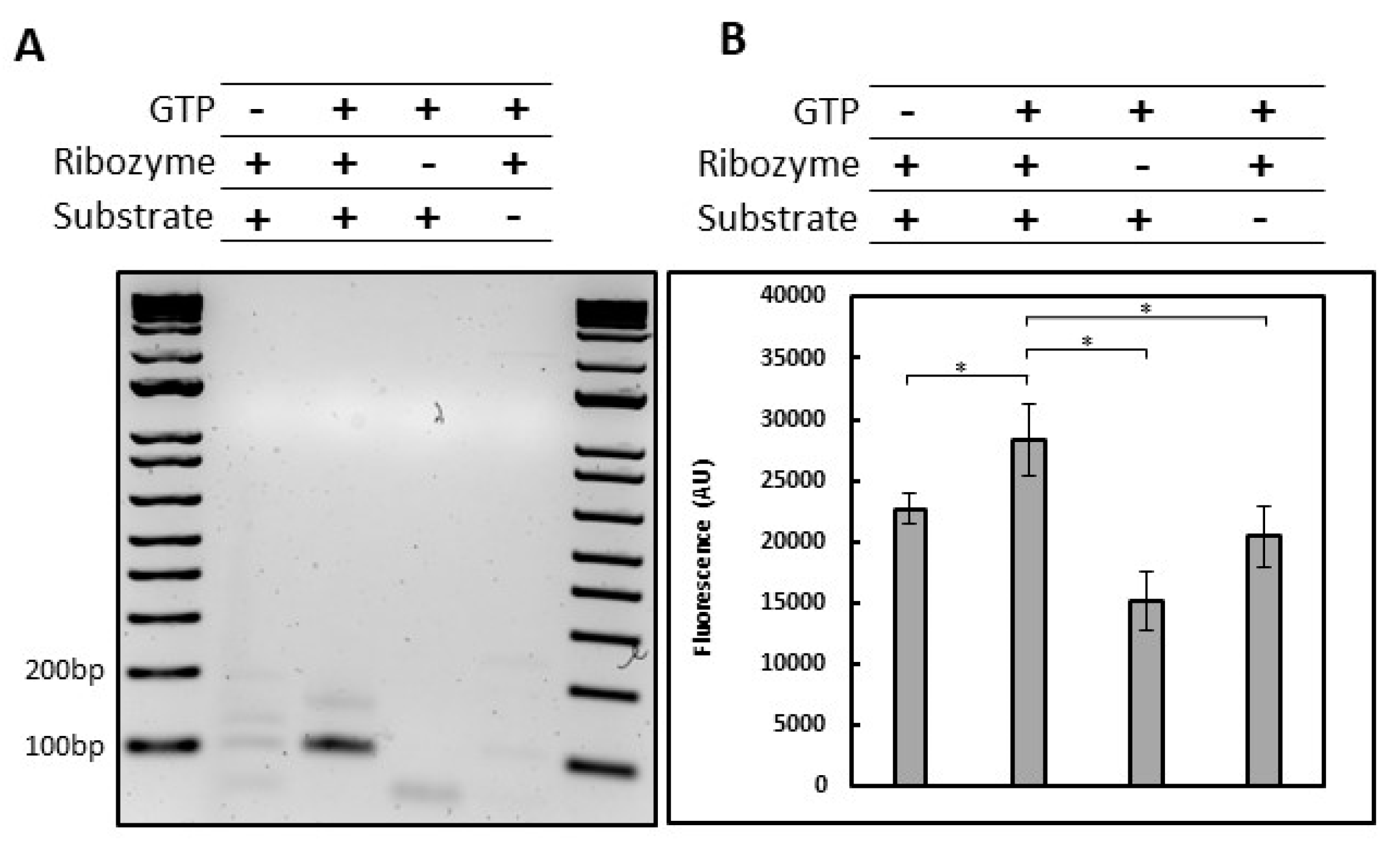
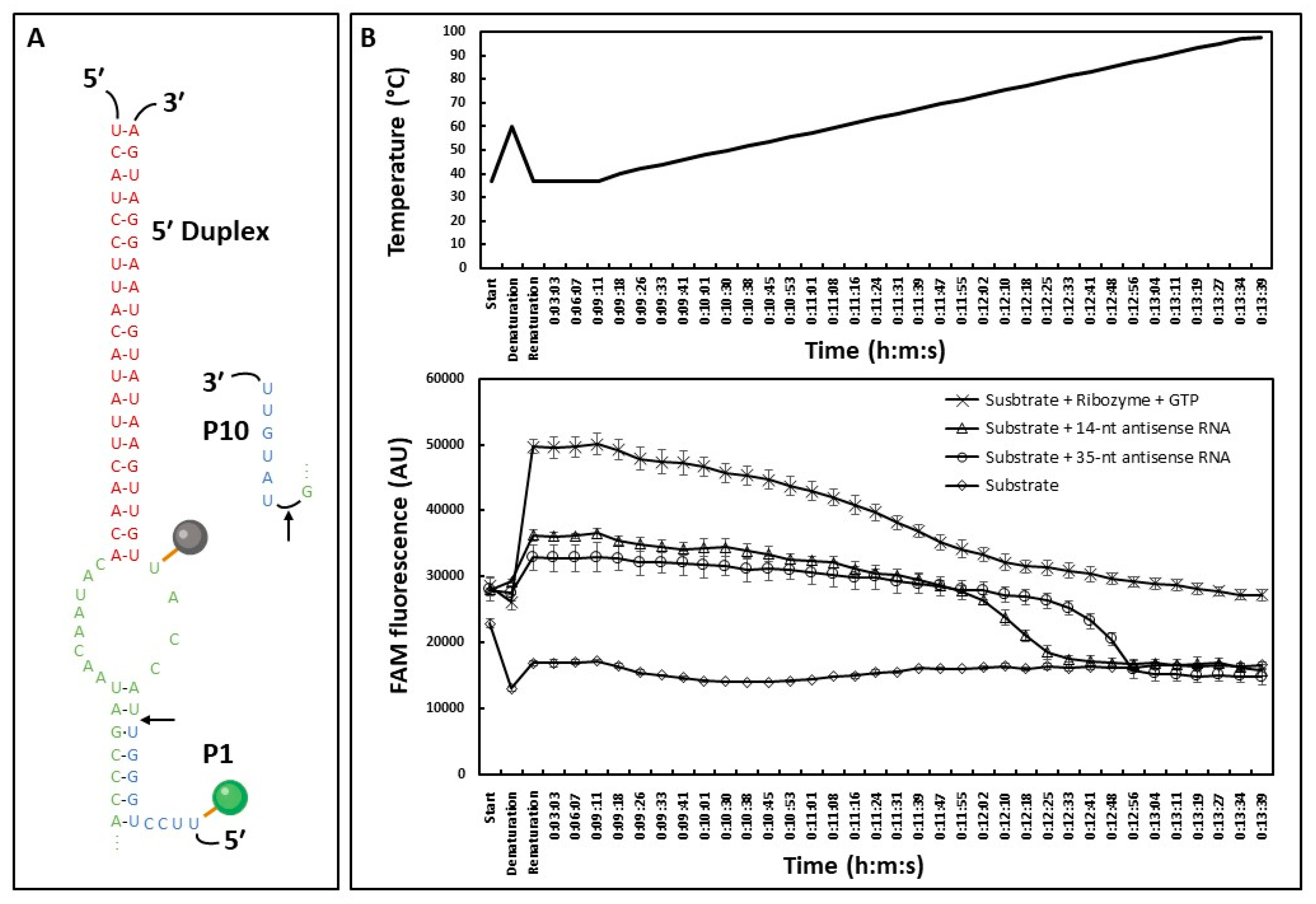
2.3. HTS-Compatible Fluorescent Reporting System
3. Discussion
3.1. A Low GC Content Group I Intron Able to Self-Splice In Vitro
3.2. Comparison between Fusarium Oxysporum and Neurospora Crassa cob (1) Intron
3.3. A high-Throughput Fluorescent Assay Based on Trans-Splicing Activity to Find New Antifungal Molecules
4. Materials and Methods
4.1. Intron Selection
4.2. RNA Production
4.2.1. Construct Preparation
4.2.2. In Vitro Transcription
4.3. Splicing Assays and Splicing Products Quantification
4.3.1. Splicing Assays
4.3.2. Primers Used for RT-PCR and RT-qPCR Analysis
4.3.3. Self-Splicing Direct Visualization and Relative Quantification
4.4. Secondary Structure Predictions and Ribozyme Construction
4.5. Trans-Splicing Validation
4.5.1. Trans-Splicing Fluorescent Assay
4.5.2. Beacon Assay
4.5.3. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Disney, M.D.; Yildirim, I.; Childs-Disney, J.L. Methods to Enable the Design of Bioactive Small Molecules Targeting RNA. Org. Biomol. Chem. 2014, 12, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Childs-Disney, J.L.; Disney, M.D. Approaches to Validate and Manipulate RNA Targets with Small Molecules in Cells. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Disney, M.D. Targeting RNA with Small Molecules To Capture Opportunities at the Intersection of Chemistry, Biology, and Medicine. J. Am. Chem. Soc. 2019, 141, 6776–6790. [Google Scholar] [CrossRef]
- Mei, H.Y.; Cui, M.; Lemrow, S.M.; Czarnik, A.W. Discovery of Selective, Small-Molecule Inhibitors of RNA Complexes—II. Self-Splicing Group I Intron Ribozyme. Bioorganic Med. Chem. 1997, 5, 1185–1195. [Google Scholar] [CrossRef]
- Seth, P.P.; Miyaji, A.; Jefferson, E.A.; Sannes-Lowery, K.A.; Osgood, S.A.; Propp, S.S.; Ranken, R.; Massire, C.; Sampath, R.; Ecker, D.J.; et al. SAR by MS: Discovery of a New Class of RNA-Binding Small Molecules for the Hepatitis C Virus: Internal Ribosome Entry Site IIA Subdomain. J. Med. Chem. 2005, 48, 7099–7102. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.K.; Hurley, B.; Kerrigan, R.; Shu, L.; Chin, D.N.; Shen, Y.; O’Brien, G.; Sung, M.J.; Hou, Y.; Axford, J.; et al. Discovery of Small Molecule Splicing Modulators of Survival Motor Neuron-2 (SMN2) for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 11021–11036. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O.; Jagdmann, G.E.; Adams, R.L.; Yuan, L.; Van Zandt, M.C.; Pyle, A.M. Small Molecules That Target Group II Introns Are Potent Antifungal Agents. Nat. Chem. Biol. 2018, 14, 1073–1078. [Google Scholar] [CrossRef]
- Hausner, G.; Hafez, M.; Edgell, D.R. Bacterial Group i Introns: Mobile RNA Catalysts. Mob. DNA 2014, 5, 8. [Google Scholar] [CrossRef]
- Vicens, Q.; Westhof, E. Brief Considerations on Targeting RNA with Small Molecules. Fac. Rev. 2022, 11, 39. [Google Scholar] [CrossRef]
- Cech, T.R. Self-Splicing Of Group I Introns. Annu. Rev. Biochem. 1990, 59, 543–568. [Google Scholar] [CrossRef]
- Inoue, T.; Cech, T.R. Secondary Structure of the Circular Form of the Tetrahymena RRNA Intervening Sequence: A Technique for RNA Structure Analysis Using Chemical Probes and Reverse Transcriptase. Proc. Natl. Acad. Sci. USA 1985, 82, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Köhler, U.; Ayre, B.G.; Goodman, H.M.; Haseloff, J. Trans-Splicing Ribozymes for Targeted Gene Delivery. J. Mol. Biol. 1999, 285, 1935–1950. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.; Lan, N.; Long, M.; Sullenger, B.A. Efficient and Specific Repair of Sickle β-Globin RNA by Trans -Splicing Ribozymes. RNA 2003, 9, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- Fiskaa, T.; Lundblad, E.W.; Henriksen, J.R.; Johansen, S.D.; Einvik, C. RNA Reprogramming of α-Mannosidase MRNA Sequences in Vitro by Myxomycete Group IC1 and IE Ribozymes. FEBS J. 2006, 273, 2789–2800. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.E.; Müller, U.F. An in Vivo Selection Method to Optimize Trans-Splicing Ribozymes. RNA 2012, 18, 581–589. [Google Scholar] [CrossRef]
- Michielse, C.B.; Rep, M. Pathogen Profile Update: Fusarium Oxysporum. Mol. Plant Pathol. 2009, 10, 311–324. [Google Scholar] [CrossRef]
- Brankovics, B.; van Dam, P.; Rep, M.; de Hoog, G.S.; van der Lee, T.A.J.; Waalwijk, C.; van Diepeningen, A.D. Mitochondrial Genomes Reveal Recombination in the Presumed Asexual Fusarium Oxysporum Species Complex. BMC Genom. 2017, 18, 735. [Google Scholar] [CrossRef]
- Kim, B.S.; Hwang, B.K. Microbial Fungicides in the Control of Plant Diseases. J. Phytopathol. 2007, 155, 641–653. [Google Scholar] [CrossRef]
- Zaug, A.J.; McEvoy, M.M.; Cech, T.R. Self-Splicing of the Group I Intron from Anabaena Pre-TRNA: Requirement for Base-Pairing of the Exons in the Anticodon Stem. Biochemistry 1993, 32, 7946–7953. [Google Scholar] [CrossRef]
- Vicens, Q.; Paukstelis, P.J.; Westhof, E.; Lambowitz, A.M.; Cech, T.R. Toward Predicting Self-Splicing and Protein-Facilitated Splicing of Group I Introns. RNA 2008, 14, 2013–2029. [Google Scholar] [CrossRef]
- Dolan, G.F.; Müller, U.F. Trans-Splicing with the Group I Intron Ribozyme from Azoarcus. RNA 2014, 20, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.F.; Laforest, M.J.; Burger, G. Mitochondrial Introns: A Critical View. Trends. Genet. 2007, 23, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Cinget, B.; Bélanger, R.R. Discovery of New Group I-D Introns Leads to Creation of Subtypes and Link to an Adaptive Response of the Mitochondrial Genome in Fungi. RNA Biol. 2020, 17, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Osawa, S.; Jukes, T.H.; Watanabe, K.; Muto, A. Recent Evidence for Evolution of the Genetic Code. Microbiol. Rev. 1992, 56, 229–264. [Google Scholar] [CrossRef] [PubMed]
- Meluzzi, D.; Olson, K.E.; Dolan, G.F.; Arya, G.; Müller, U.F. Computational Prediction of Efficient Splice Sites for Trans-Splicing Ribozymes. RNA 2012, 18, 590–602. [Google Scholar] [CrossRef]
- Legault, P.; Herschlag, D.; Celander, D.W.; Cech, T.R. Mutations at the Guanosine-Binding Site of the Tetrahymena Ribozyme Also Affect Site-Specific Hydrolysis. Nucleic Acids Res. 1992, 20, 6613–6619. [Google Scholar] [CrossRef]
- Tanaka, T.; Hirata, Y.; Tominaga, Y.; Furuta, H.; Matsumura, S.; Ikawa, Y. Heterodimerization of Group I Ribozymes Enabling Exon Recombination through Pairs of Cooperative Trans-Splicing Reactions. ChemBioChem 2017, 18, 1659–1667. [Google Scholar] [CrossRef]
- Omran, Q.Q.; Fedorova, O.; Liu, T.; Pyle, A.M. A Molecular Beacon Assay for Monitoring RNA Splicing. Nucleic Acids Res. 2022, 50, e74. [Google Scholar] [CrossRef]
- Donahue, C.P.; Ni, J.; Rozners, E.; Glicksman, M.A.; Wolfe, M.S. Identification of Tau Stem Loop RNA Stabilizers. J. Biomol. Screen. 2007, 12, 789–799. [Google Scholar] [CrossRef]
- Hagemeister, F.; Cabanillas, F.; Coleman, M.; Gregory, S.A.; Zinzani, P.L. The Role of Mitoxantrone in the Treatment of Indolent Lymphomas. Oncologist 2005, 10, 150–159. [Google Scholar] [CrossRef]
- Smith, D.R. Updating Our View of Organelle Genome Nucleotide Landscape. Front. Genet. 2012, 3, 175. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Liao, X.; Zhang, X.; Tembrock, L.R.; Broz, A. Genomic Architectural Variation of Plant Mitochondria—A Review of Multichromosomal Structuring. J. Syst. Evol. 2022, 60, 160–168. [Google Scholar] [CrossRef]
- Quiroga, C.; Kronstad, L.; Ritlop, C.; Filion, A.; Cousineau, B. Contribution of Base-Pairing Interactions between Group II Intron Fragments during Trans -Splicing in Vivo. RNA 2011, 17, 2212–2221. [Google Scholar] [CrossRef] [PubMed]
- Gampel, A.; Tzagoloff, A. In Vitro Splicing of the Terminal Intervening Sequence of Saccharomyces Cerevisiae Cytochrome b Pre-MRNA. Mol. Cell. Biol. 1987, 7, 2545–2551. [Google Scholar] [CrossRef]
- Kruger, K.; Grabowski, P.J.; Zaug, A.J.; Sands, J.; Gottschling, D.E.; Cech, T.R. Self-Splicing RNA: Autoexcision and Autocyclization of the Ribosomal RNA Intervening Sequence of Tetrahymena. Cell 1982, 31, 147–157. [Google Scholar] [CrossRef]
- Garriga, G.; Lambowitz, A.M. RNA Splicing in Neurospora Mitochondria: Self-Splicing of a Mitochondrial Intron in Vitro. Cell 1984, 39, 631–641. [Google Scholar] [CrossRef]
- Tabak, H.F.; van der Horst, G.; Osinga, K.A.; Arnberg, A.C. Splicing of Large Ribosomal Precursor RNA and Processing of Intron RNA in Yeast Mitochondria. Cell 1984, 39, 623–629. [Google Scholar] [CrossRef]
- Chu, F.K.; Maley, G.F.; West, D.K.; Belfort, M.; Maley, F. Characterization of the Intron in the Phage T4 Thymidylate Synthase Gene and Evidence for Its Self-Excision from the Primary Transcript. Cell 1986, 45, 157–166. [Google Scholar] [CrossRef]
- Sogin, M.L.; Edman, J.C. A Self-Splicing Intron in the Small Subunit RRNA Gene of Pneumocystis Carinii. Nucleic Acids Res. 1989, 17, 5349–5360. [Google Scholar] [CrossRef]
- Goodrich-Blair, H.; Scarlato, V.; Gott, J.M.; Xu, M.Q.; Shub, D.A. A Self-Splicing Group I Intron in the DNA Polymerase Gene of Bacillus Subtilis Bacteriophage SPO1. Cell 1990, 63, 417–424. [Google Scholar] [CrossRef]
- Xu, M.-Q.; Kathe, S.D.; Goodrich-Blair, H.; Nierzwicki-Bauer, S.A.; Shub, D.A. Bacterial Origin of a Chloroplast Intron: Conserved Self-Splicing Group I Introns in Cyanobacteria. Science 1990, 250, 1566–1570. [Google Scholar] [CrossRef]
- Gampel, A.; Cech, T.R. Binding of the CBP2 Protein to a Yeast Mitochondrial Group I Intron Requires the Catalytic Core of the RNA. Genes Dev. 1991, 5, 1870–1880. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, B.; Merlos-Lange, A.M.; Anderl, C.; Welser, F.; Zimmer, M.; Wolf, K. The Mitochondrial Genome of Fission Yeast: Inability of All Introns to Splice Autocatalytically, and Construction and Characterization of an Intronless Genome. Mol. Gen. Genet. MGG 1991, 225, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Reinhold-Hurek, B.; Shub, D.A. Self-Splicing Introns in TRNA Genes of Widely Divergent Bacteria. Nature 1992, 357, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Ruoff, B.; Johansen, S.; Vogt, V.M. Characterization of the Self-Splicing Products of a Mobile Intron from the Nuclear RDNA of Physarum Polycephalum. Nucleic Acids Res. 1992, 20, 5899–5906. [Google Scholar] [CrossRef]
- Wilcox, L.W.; Lewis, L.A.; Fuerst, P.A.; Floyd, G.L. Group I Introns within the Nuclear-Encoded Small-Subunit RRNA Gene of Three Green Algae. Mol. Biol. Evol. 1992, 9, 1103–1118. [Google Scholar] [CrossRef]
- Mercure, S.; Montplaisir, S.; Lernay, G. Correlation between the Presence of a Self-Splicing Intron in the 25S RDNA of C.Albicans and Strains Susceptibility to 5-Fluorocytosine. Nucleic Acids Res. 1993, 21, 6020–6027. [Google Scholar] [CrossRef]
- Sugita, M.; Luo, L.; Ohta, M.; Itadani, H.; Matsubayashi, T.; Sugiura, M. Genes Encoding the Group I Intron-Containing TRNALeu and Subunit L of NADH Dehydrogenase from the Cyanobacterium Synechococcus PCC 6301. DNA Res. 1995, 2, 71–76. [Google Scholar] [CrossRef]
- Hur, M.; Geese, W.J.; Waring, R.B. Self-Splicing Activity of the Mitochondrial Group-I Introns from Aspergillus Nidulans and Related Introns from Other Species. Curr. Genet. 1997, 32, 399–407. [Google Scholar] [CrossRef]
- Kapoor, M.; Nagai, T.; Wakasugi, T.; Yoshinaga, K.; Sugiura, M. Organization of Chloroplast Ribosomal RNA Genes and in Vitro Self-Splicing Activity of the Large Subunit RRNA Intron from the Green Alga Chlorella Vulgaris C-27. Curr. Genet. 1997, 31, 503–510. [Google Scholar] [CrossRef]
- Wallweber, G.J.; Mohr, S.; Rennard, R.; Caprara, M.G.; Lambowitz, A.M. Characterization of Neurospora Mitochondrial Group I Introns Reveals Different CYT-18 Dependent and Independent Splicing Strategies and an Alternative 3′ Splice Site for an Intron ORF. RNA 1997, 3, 114–131. [Google Scholar] [PubMed]
- Everett, K.D.E.; Kahane, S.; Bush, R.M.; Friedman, M.G. An Unspliced Group I Intron in 23S RRNA LinksChlamydiales, Chloroplasts, and Mitochondria. J. Bacteriol. 1999, 181, 4734–4740. [Google Scholar] [CrossRef]
- Landthaler, M.; Shub, D.A. Unexpected Abundance of Self-Splicing Introns in the Genome of Bacteriophage Twort: Introns in Multiple Genes, a Single Gene with Three Introns, and Exon Skipping by Group I Ribozymes. Proc. Natl. Acad. Sci. USA 1999, 96, 7005–7010. [Google Scholar] [CrossRef] [PubMed]
- Foley, S.; Bruttin, A.; Brüssow, H. Widespread Distribution of a Group I Intron and Its Three Deletion Derivatives in the Lysin Gene of Streptococcus Thermophilus Bacteriophages. J. Virol. 2000, 74, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Bonocora, R.P.; Shub, D.A. A Novel Group I Intron-Encoded Endonuclease Specific for the Anticodon Region of TRNAfMet Genes. Mol. Microbiol. 2001, 39, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.; Choi, H.; Park, C. Group I Self-Splicing Intron in the RecA Gene of Bacillus Anthracis. J. Bacteriol. 2002, 184, 3917–3922. [Google Scholar] [CrossRef]
- Gampel, A.; Nishikimi, M.; Tzagoloff, A. CBP2 Protein Promotes in Vitro Excision of a Yeast Mitochondrial Group I Intron. Mol. Cell. Biol. 1989, 9, 5424–5433. [Google Scholar] [CrossRef]
- Lambowitz, A.M.; Perlman, P.S. Involvement of Aminoacyl-TRNA Synthetases and Other Proteins in Group I and Group II Intron Splicing. Trends Biochem. Sci. 1990, 15, 440–444. [Google Scholar] [CrossRef]
- Weeks, K.M.; Cech, T.R. Assembly of a Ribonucleoprotein Catalyst by Tertiary Structure Capture. Science 1996, 271, 345–348. [Google Scholar] [CrossRef]
- Webb, A.E.; Weeks, K.M. A Collapsed State Functions to Self-Chaperone RNA Folding into a Native Ribonucleoprotein Complex. Nat. Struct. Biol. 2001, 8, 135–140. [Google Scholar] [CrossRef]
- Paukstelis, P.J.; Chen, J.H.; Chase, E.; Lambowitz, A.M.; Golden, B.L. Structure of a Tyrosyl-TRNA Synthetase Splicing Factor Bound to a Group I Intron RNA. Nature 2008, 451, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Mohr, S.; Stryker, J.M.; Lambowitz, A.M. A DEAD-Box Protein Functions as an ATP-Dependent RNA Chaperone in Group I Intron Splicing. Cell 2002, 109, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.; Schottler, M.A.; Karcher, D.; Thiele, W.; Bock, R. Elimination of a Group II Intron from a Plastid Gene Causes a Mutant Phenotype. Nucleic Acids Res. 2011, 39, 5181–5192. [Google Scholar] [CrossRef] [PubMed]
- Guilcher, M.; Liehrmann, A.; Seyman, C.; Blein, T.; Rigaill, G.; Castandet, B.; Delannoy, E. Full Length Transcriptome Highlights the Coordination of Plastid Transcript Processing. Int. J. Mol. Sci. 2021, 22, 11297. [Google Scholar] [CrossRef]
- Paukstelis, P.J.; Lambowitz, A.M. Identification and Evolution of Fungal Mitochondrial Tyrosyl-TRNA Synthetases with Group I Intron Splicing Activity. Proc. Natl. Acad. Sci. USA 2008, 105, 6010–6015. [Google Scholar] [CrossRef] [PubMed]
- Gribbon, P.; Sewing, A. Fluorescence Readouts in HTS: No Gain without Pain? Drug Discov. Today 2003, 8, 1035–1043. [Google Scholar] [CrossRef]
- Deng, J.; Shi, Y.; Peng, X.; He, Y.; Chen, X.; Li, M.; Lin, X.; Liao, W.; Huang, Y.; Jiang, T.; et al. Ribocentre: A Database of Ribozymes. Nucleic Acids Res. 2022, 51, D262–D2681. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Cannone, J.J.; Subramanian, S.; Schnare, M.N.; Collett, J.R.; D’Souza, L.M.; Du, Y.; Feng, B.; Lin, N.; Madabusi, L.V.; Müller, K.M.; et al. The Comparative RNA Web (CRW) Site: An Online Database of Comparative Sequence and Structure Information for Ribosomal, Intron, and Other RNAs. BMC Bioinform. 2002, 3, 2. [Google Scholar] [CrossRef]
- Mann, M.; Wright, P.R.; Backofen, R. IntaRNA 2.0: Enhanced and Customizable Prediction of RNA–RNA Interactions. Nucleic Acids Res. 2017, 45, W435–W439. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malbert, B.; Labaurie, V.; Dorme, C.; Paget, E. Group I Intron as a Potential Target for Antifungal Compounds: Development of a Trans-Splicing High-Throughput Screening Strategy. Molecules 2023, 28, 4460. https://doi.org/10.3390/molecules28114460
Malbert B, Labaurie V, Dorme C, Paget E. Group I Intron as a Potential Target for Antifungal Compounds: Development of a Trans-Splicing High-Throughput Screening Strategy. Molecules. 2023; 28(11):4460. https://doi.org/10.3390/molecules28114460
Chicago/Turabian StyleMalbert, Bastien, Virginie Labaurie, Cécile Dorme, and Eric Paget. 2023. "Group I Intron as a Potential Target for Antifungal Compounds: Development of a Trans-Splicing High-Throughput Screening Strategy" Molecules 28, no. 11: 4460. https://doi.org/10.3390/molecules28114460