
Synthesis and Structural Characterization of Phosphanide Gold(III)/Gold(I) Complexes and Their Thallium(III) and Gold(III) Precursors

Abstract
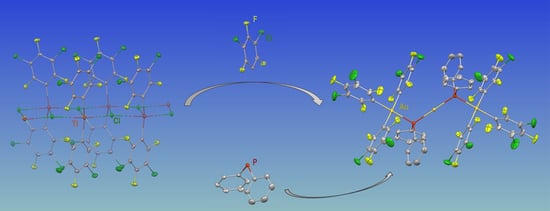
:
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization of the Thallium(III) Complex [Tl(3,5-C6Cl2F3)2Cl]n (1)
2.1.1. Synthesis and Spectroscopic Characterization of [Tl(3,5-C6Cl2F3)2Cl]n (1)
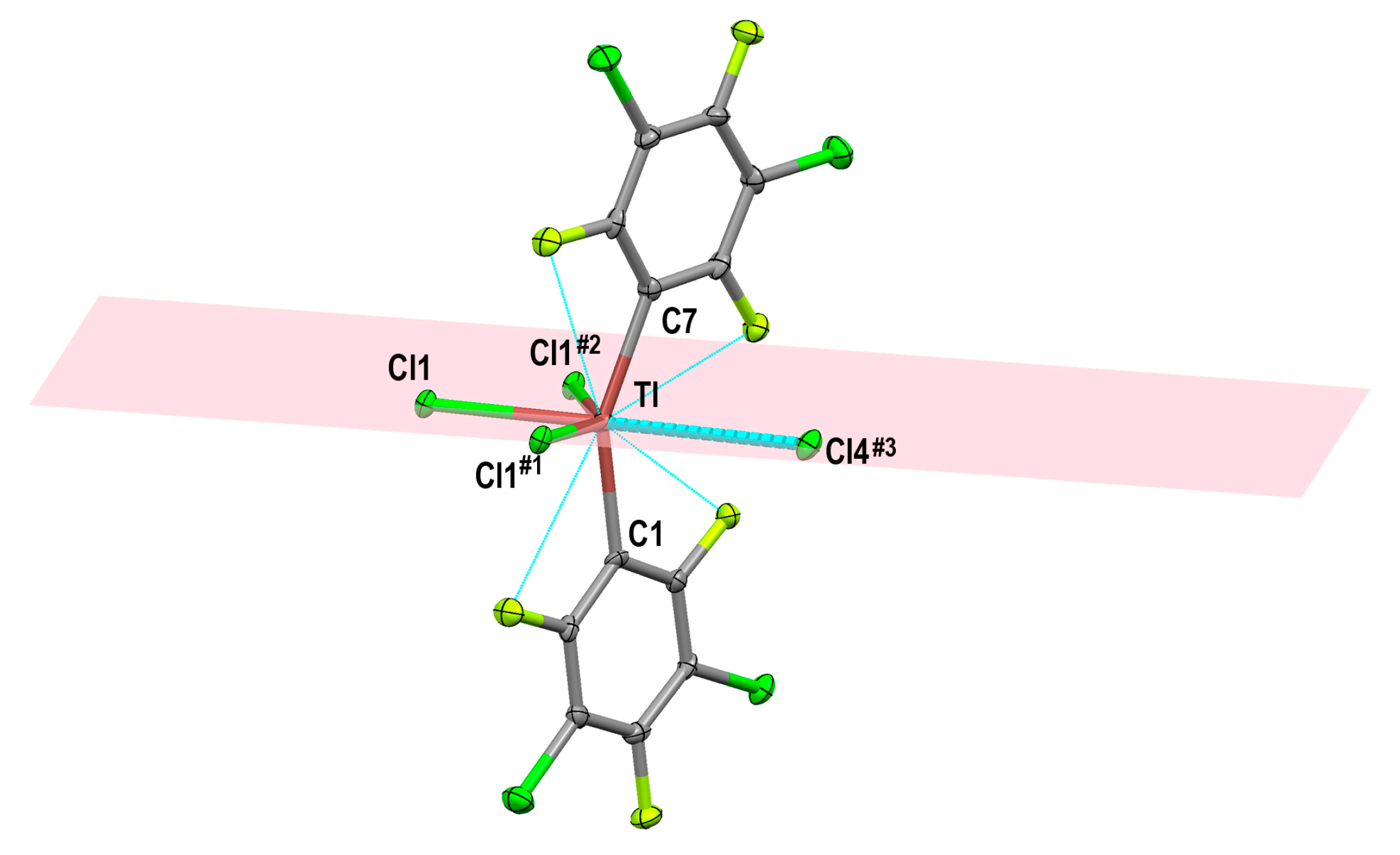
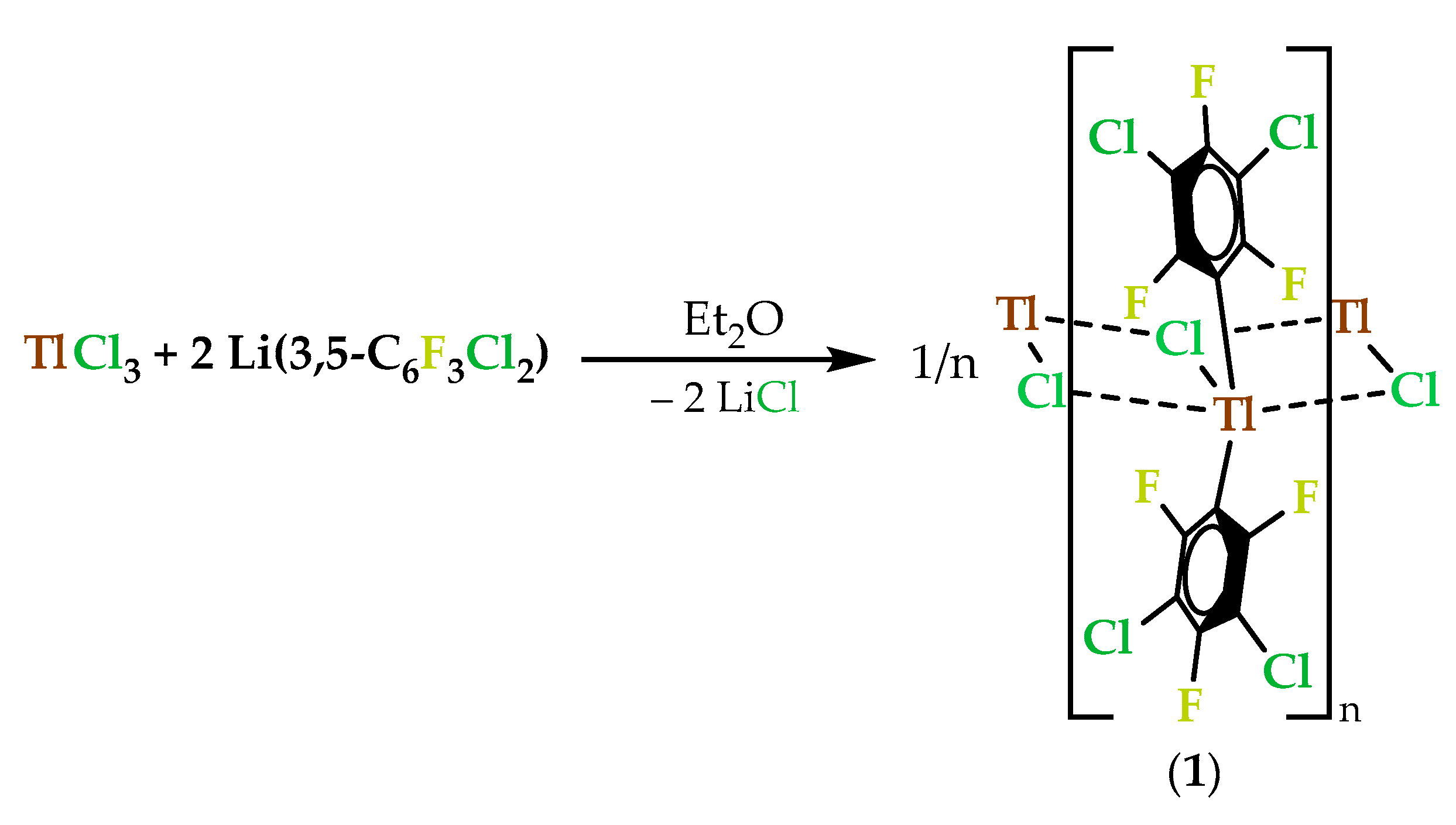
2.1.2. Crystal Structure of [Tl(3,5-C6Cl2F3)2Cl]n (1)
2.2. Synthesis and Characterization of Gold(III) Complexes
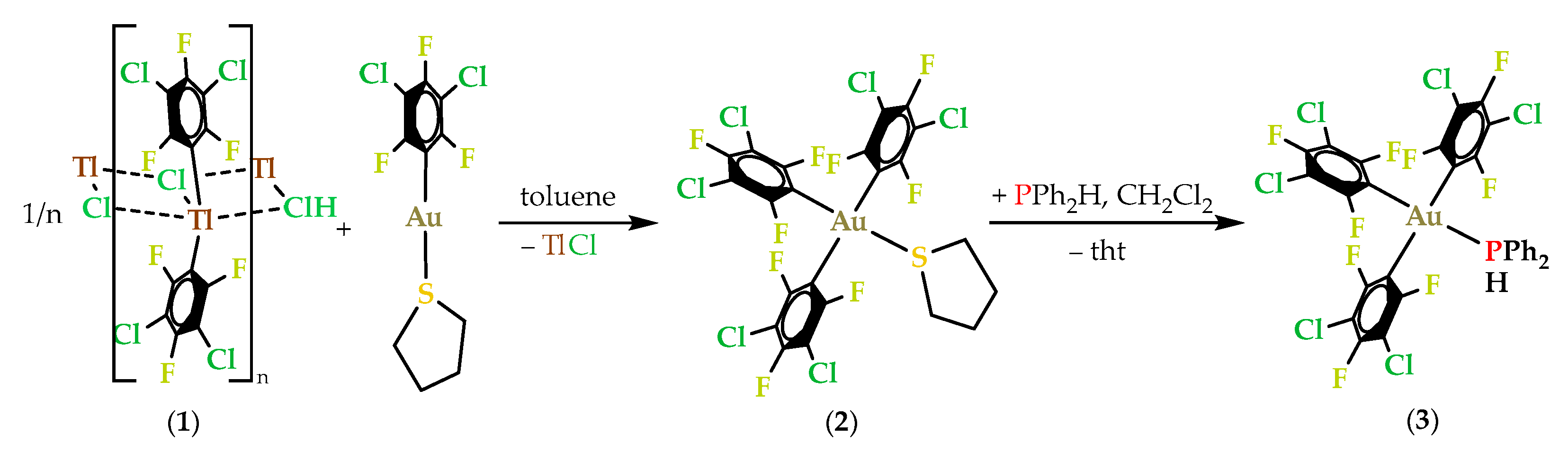
2.2.1. Synthesis and Spectroscopic Characterization of Gold(III) Complexes
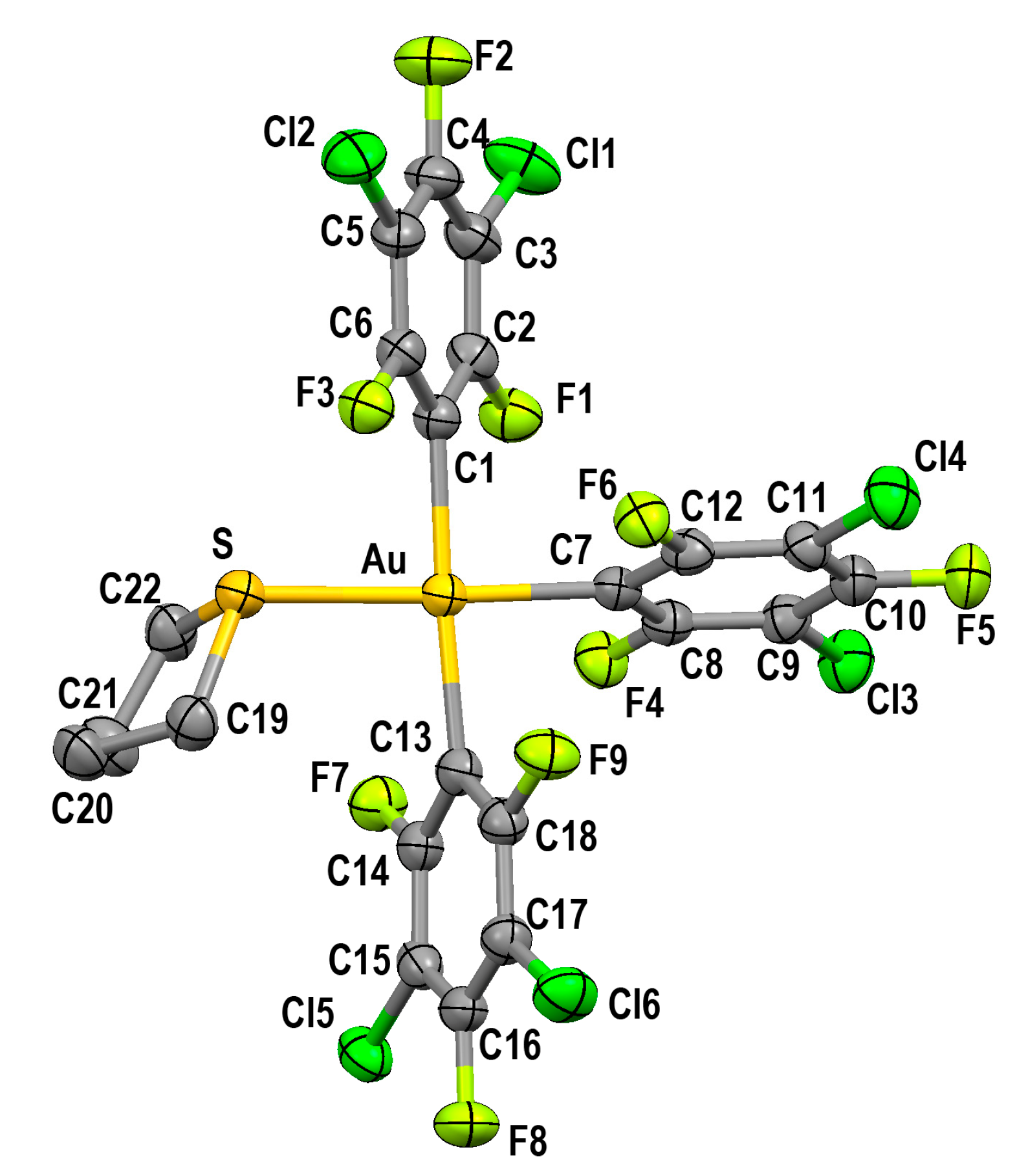
2.2.2. Crystal Structure of [Au(3,5-C6F5)3(tht)] (2)
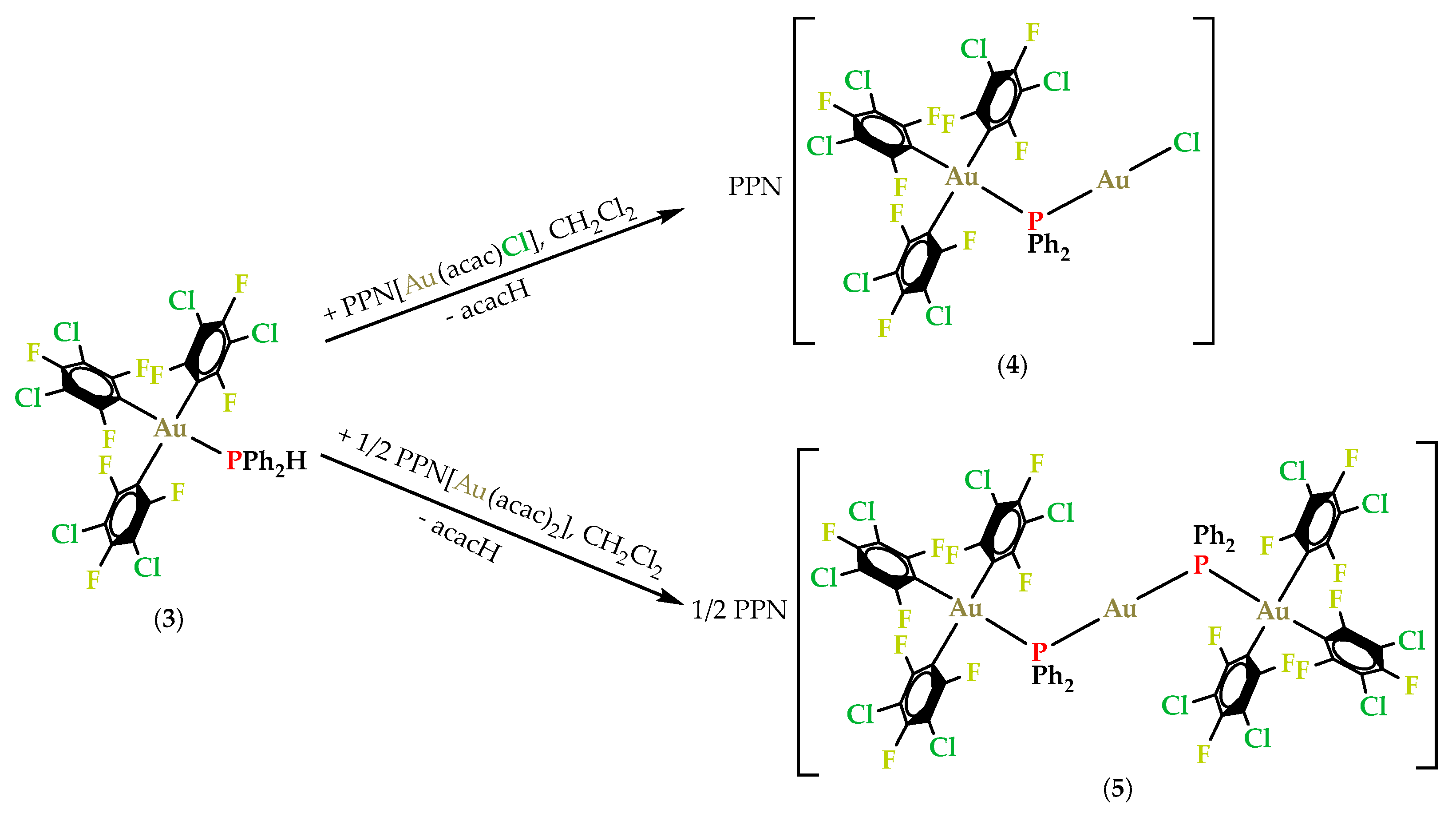
2.3. Synthesis and Characterization of Gold(III)/Gold(I) Phosphanido Complexes
2.3.1. Synthesis and Spectroscopic Characterization of Gold(III)/Gold(I) Phosphanido Complexes
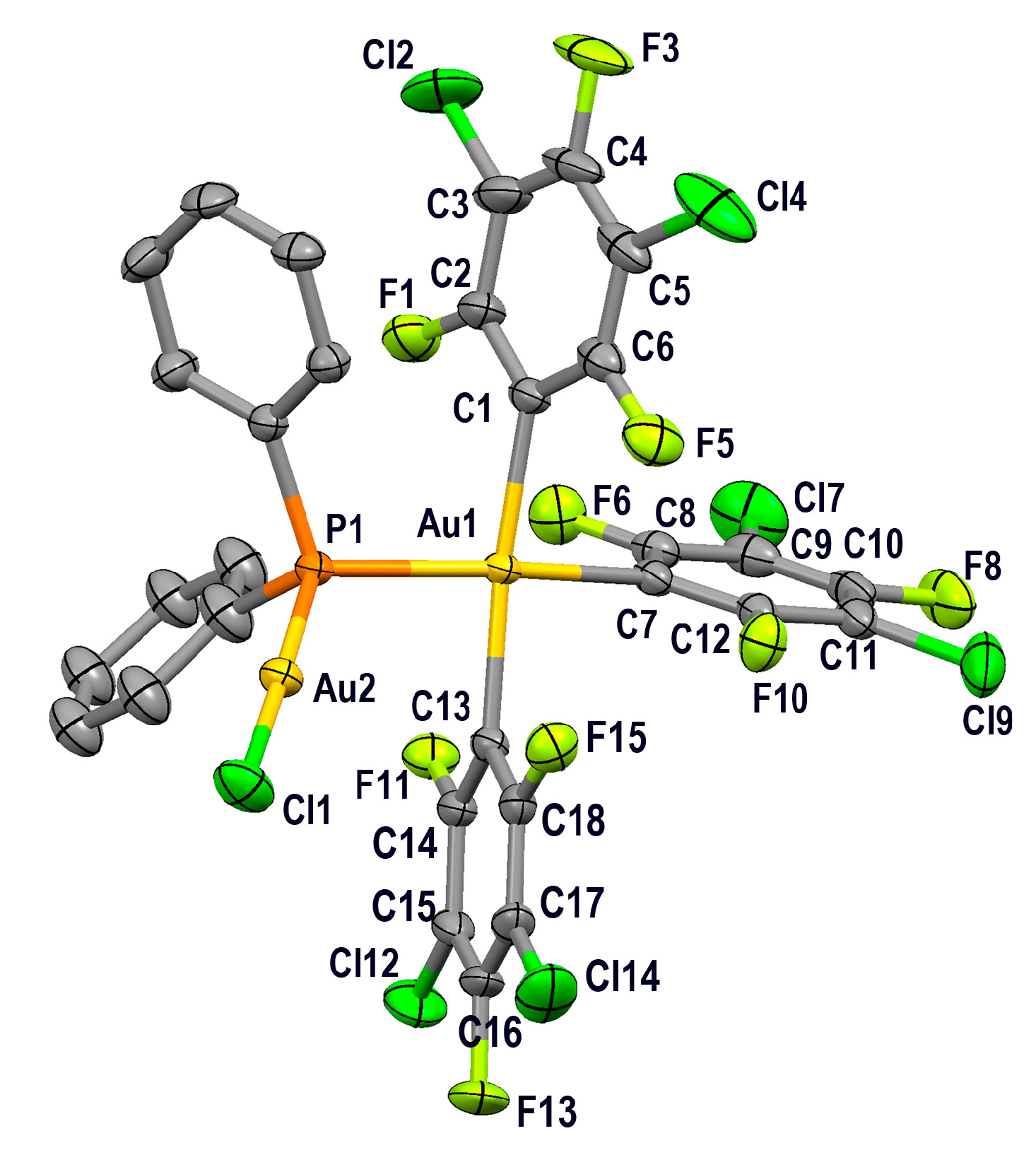
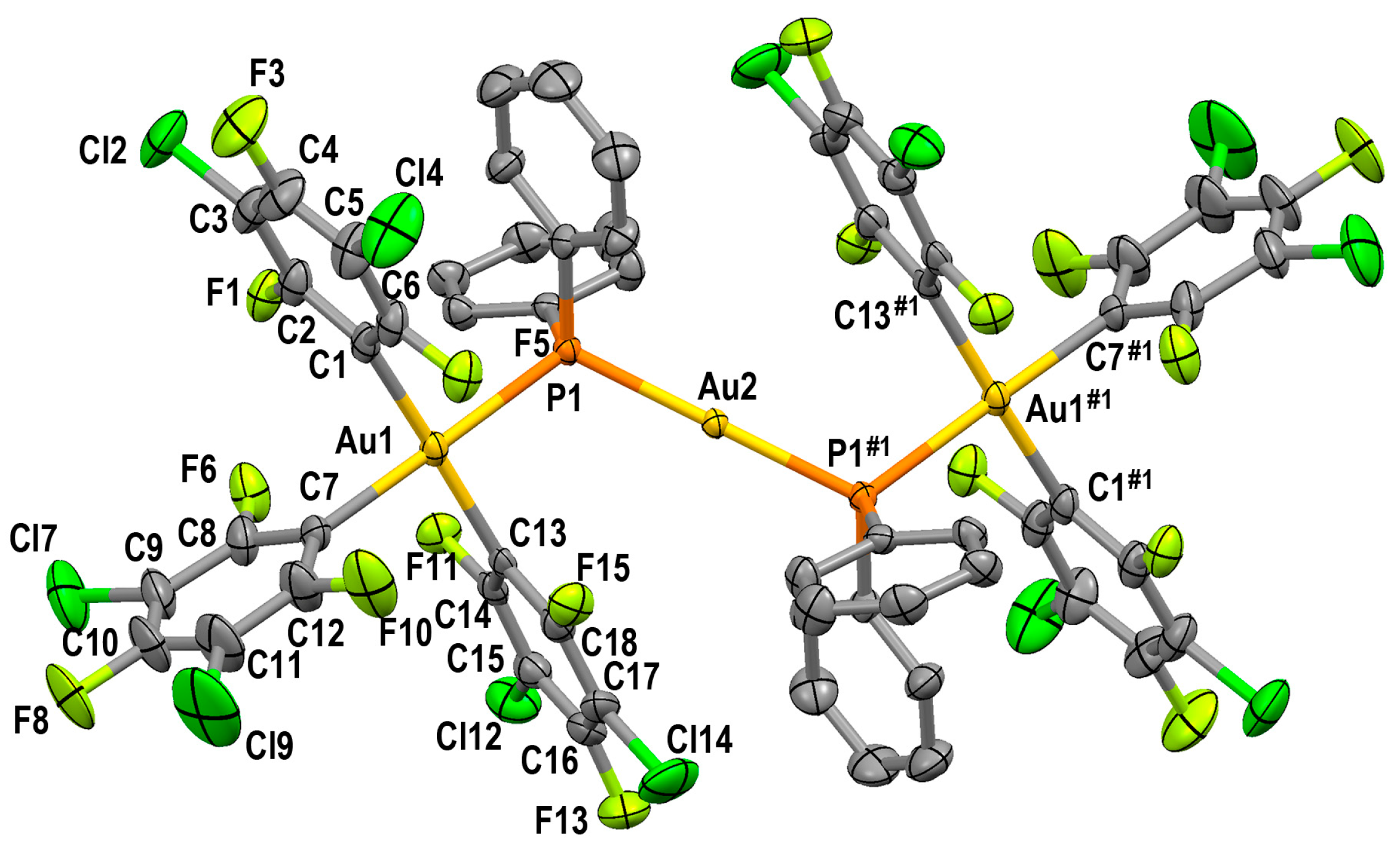
2.3.2. Crystal Structures of PPN[Au(3,5-C6Cl2F3)3(µ-PPh2)AuCl] (4) and PPN[{(3,5-C6Cl2F3)3Au(µ-PPh2)}2Au] (5)
3. Experimental Section
3.1. General
3.2. Instrumentation
3.3. Synthesis
3.3.1. Synthesis of [Tl(3,5-C6Cl2F3)2Cl]n (1)
3.3.2. Synthesis of [Au(3,5-C6Cl2F3)3(tht)] (2)
3.3.3. Synthesis of [Au(3,5-C6Cl2F3)3(PPh2H)] (3)
3.3.4. Synthesis of [PPN][Au(3,5-C6Cl2F3)3(µ-PPh2)AuCl] (4)
3.3.5. Synthesis of [PPN][{(3,5-C6Cl2F3)3Au(µ-PPh2)}2Au] (5)
3.4. Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sidgwick, N.V. The Electronic Theory of Valency; Clarendon Press: Oxford, UK, 1927; pp. 178–181. [Google Scholar]
- Fernández, E.J.; Laguna, A.; Olmos, M.E. Perfluoroarylgold complexes. Coord. Chem. Rev. 2008, 252, 1630–1667. [Google Scholar] [CrossRef]
- Starowieyski, K.B. Chemistry of Aluminium, Gallium, Indium and Thallium; Downs, A.J., Ed.; Blackie Academic and Professional: London, UK, 1993; pp. 272–322. [Google Scholar]
- Casas, J.S.; García-Tasende, M.S.; Sordo, J. Structural aspects of the coordination chemistry of organothallium(III) and organomercury(II) derivatives. Coord. Chem. Rev. 1999, 193–195, 286–359. [Google Scholar] [CrossRef]
- Ara, I.; Forniés, J.; García-Monforte, M.A.; Martín, A.; Menjón, B. Synthesis and characterization of pentachlorophenyl-metal derivatives with d0 and d10 electron configurations. Chem. Eur. J. 2004, 10, 4186–4197. [Google Scholar] [CrossRef] [PubMed]
- Usón, R.; Laguna, A.; Laguna, M.; Fernández, E.J.; Jones, P.G.; Sheldrick, G.M. Tris(pentafluorophenyl)gold(III) complexes. Dalton Trans. 1982, 10, 1971–1976. [Google Scholar] [CrossRef]
- Mastrorilli, P.; Fortuño, C. Platinum and palladium prompted couplings od bridging PPh2− with terminal bonded ligands. Inorg. Chim. Acta. 2020, 513, 119947. [Google Scholar] [CrossRef]
- Mastrorilli, P. Bridging and terminal (phosphanido)platinum complexes. Eur. J. Inorg. Chem. 2008, 4835–4850. [Google Scholar] [CrossRef]
- Harley, A.D.; Guskey, G.J.; Geoffroy, G.L. Interconversion of phosphido-bridged polynuclear cobalt carbonyl complexes. Cleavage of the phosphido bridge during hydroformylation catalysis. Organometallics 1983, 2, 53–59. [Google Scholar] [CrossRef]
- Patel, V.D.; Taylor, N.J.; Carty, A.J. Phosphido bridged iron carbonyl hydrides: Synthesis and X-ray structure of (µ-H)2Fe3(CO)8(µ-PPh2)2. J. Chem. Soc. Chem. Commun. 1984, 2, 99–100. [Google Scholar] [CrossRef]
- Carty, A.J.; Hogarth, G.; Enright, G.D.; Steed, J.W.; Georganopoulou, D. Transformations of alkynyl ligands at iron centres: Alkenyl formation via addition of PPh3 and enyne generation via head-to-head coupling and addition of benzene. Chem. Commun. 1999, 16, 1499–1500. [Google Scholar] [CrossRef]
- Fernández, E.J.; Laguna, A.; Olmos, M.E. Gold phosphide complexes. J. Chil. Chem. Soc. 2007, 52, 1200–1205. [Google Scholar] [CrossRef]
- Puddephatt, R.J.; Thompson, P.J. Some reactions of methylplatinum and methylgold compounds with phenylselenol, diphenylphosphine, diphenylarsine, N-bromosuccinimide and 2-nitrophenylsulphenyl chloride. J. Organomet. Chem. 1976, 117, 395–403. [Google Scholar] [CrossRef]
- Li, X.-S.; Mo, J.; Zhang, S.-M.; Yuan, L.; Liu, J.-H. (μ-Diphenylphosphanido-κ2P:P′)bis[2,2′-(pyridine-2,6-diyl)diphenyl-κ3C1,N,C1′)gold(III)] perchlorate acetonitrile solvate. Acta Crystallogr. Sect. E 2008, 64, m1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, M.C.; Fernández, E.J.; Jones, P.G.; Laguna, A.; López-de-Luzuriaga, J.M.; Olmos, M.E. [Au(C6F5)3(PPh2H)]: A precursor for the synthesis of gold(III) phosphide complexes. Angew. Chem. Int. Ed. 1998, 37, 3042–3043. [Google Scholar] [CrossRef]
- Blanco, M.C.; Fernández, E.J.; López-de-Luzuriaga, J.M.; Olmos, M.E.; Crespo, O.; Gimeno, M.C.; Laguna, A.; Jones, P.G. Heteropolynuclear phosphide complexes: Phosphorus as unique atom bridging coinage metal centres. Chem. Eur. J. 2000, 6, 4116–4123. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.C.; Fernández, E.J.; Fischer, A.K.; Jones, P.G.; Laguna, A.; Olmos, M.E.; Villacampa, M.D. NBu4[{Au(C6F5)3}2(μ-PPh2)]: A gold(III) phosphide with a single atom bridging the metallic centers. Inorg. Chem. Commun. 2000, 3, 163–165. [Google Scholar] [CrossRef]
- Blanco, M.C.; Fernández, E.J.; Olmos, M.E.; Crespo, O.; Laguna, A.; Jones, P.G. A pentanuclear mixed gold(III)–silver(I) phosphide with an unusual T-frame μ3-Cl bridge. Organometallics 2002, 21, 2426–2429. [Google Scholar] [CrossRef]
- Blanco, M.C.; Fernández, E.J.; Olmos, M.E.; Pérez, J.; Crespo, O.; Laguna, A.; Jones, P.G. Gold(III) phenylphosphides and -phosphodiides. Organometallics 2004, 23, 4373–4381. [Google Scholar] [CrossRef]
- Blanco, M.C.; Fernández, E.J.; Olmos, M.E.; Pérez, J.; Laguna, A. From diphosphane to diphosphodiide gold(III) derivatives of 1,2-diphosphinobenzene. Chem. Eur. J. 2006, 12, 3379–3388. [Google Scholar] [CrossRef]
- Usón, R.; Laguna, A.; García, J.; Laguna, M. Anionic perfluorophenyl complexes of gold(I) and gold(III). Inorg. Chim. Acta 1979, 37, 201–207. [Google Scholar] [CrossRef]
- Usón, R.; Laguna, A.; Laguna, M.; Abad, M. Synthesis and reactions of di-μ-halo- or -pseudohalotetrakis(pentafluorophenyl) digold(III). J. Organomet. Chem. 1983, 249, 437–443. [Google Scholar] [CrossRef]
- Fernández-Moyano, S.; Peñas-Defrutos, M.N.; Bartolomé, C.; Espinet, P. Striking ligand-disproportionative Cl/aryl scrambling in a simple Au(III) system. Solvent role, driving forces and mechanisms. Chem. Commun. 2021, 57, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Vicente, J.; Chicote, M.T. The ‘acac method’ for the synthesis of coordination and organometallic compounds: Synthesis of gold complexes. Coord. Chem. Rev. 1999, 193–195, 1143–1161. [Google Scholar] [CrossRef]
- Fenton, D.E.; Gillies, D.G.; Massey, A.G.; Randall, E.W. Fluorine-19 Nuclear Magnetic Resonance of pentafluorophenyl derivatives of thallium and mercury. Nature 1964, 201, 818. [Google Scholar] [CrossRef]
- Royo, P.; Serrano, R. bis(pentachlorophenyl)thallium(III) compounds. J. Organometal. Chem. 1977, 136, 309–313. [Google Scholar] [CrossRef] [Green Version]
- Usón, R.; Laguna, A.; Cuenca, T. Chlorobis(polyfluorophenyl)thallium(III) complexes and their reactions with gold(I) and tin(II) compounds. J. Organomet. Chem. 1980, 194, 271–275. [Google Scholar] [CrossRef]
- Laguna, A.; Fernández, E.; Mendía, A.; Ruiz-Romero, M.; Jones, P.G. Reactions of thallium(III) chloride with (aryl) silver(I) complexes. Crystal structure of [Tl(mes)2][TlCl3(mes)]. J. Organometal. Chem. 1989, 365, 201–206. [Google Scholar] [CrossRef]
- Deacon, G.B.; Phillips, R.J.; Henrick, K.; McPartlin, M. The structure of bromobis(2,3,5,6-tetrafluorophenyl) thallium(III). Inorg. Chim. Acta. 1979, 35, L335–L336. [Google Scholar] [CrossRef]
- Jefs, S.E.; Small, R.W.H.; Worral, I.J. Structure of the 4:1 complexes formed by pyridine and the Group III halides InCl3 and TlCl3: Mer-trichlorotris(pyridine)indium(III)–pyridine (1/1), [InCl3(C5H5N)3].C5H5N, and mer-trichlorotris(pyridine)thallium(III)–pyridine (1/1), [TlCl3(C5H5N)3].C5H5N. Acta Crystallogr. Sect. C 1984, 40, 1329–1331. [Google Scholar] [CrossRef]
- Jacob, K.; Scholz, J.; Merzweiler, K.; Pietzsch, C. Bis [2-(dimethylaminomethyl) ferrocenyl]-thalliumchlorid heterobimetallischer Chelatkomplex. J, Organomet. Chem. 1997, 527, 109–115. [Google Scholar] [CrossRef]
- Alvarez, B.; Fernández, E.J.; Gimeno, M.C.; Jones, P.G.; Laguna, A.; Laguna, M.; López-de-Luzuriaga, J.M. Synthesis and structure of [Tl(C6F5)2Cl{Au(C6F5)3(PPh2CH2PPh2(O))}2]. J. Organomet. Chem. 1996, 525, 109–113. [Google Scholar] [CrossRef]
- Mendia, A.; Cerrada, E.; Fernández, E.J.; Laguna, A.; Laguna, M. Four-, five- and six-co-ordinated pentafluorophenylthallium(III) complexes. Crystal structures of [Tl(C6F5)3(OPPh3)], [Tl(C6F5)2Cl(OPPh3)]2 and [Tl(C6F5)2(OdppmO)2][Tl(C6F5)2Cl2]. J. Organomet. Chem. 2002, 663, 289–296. [Google Scholar] [CrossRef]
- Powell, H.M.; Crowfoot, D.M. The crystal structures of dimethyl thallium halides. Z. Kristallogr. 1934, 87, 370–378. [Google Scholar] [CrossRef]
- Hausen, H.D.; Veigle, E.; Guder, H.J. Die Kristallstruktur von Dimethylthalliumchlorid. Z. Naturforsch. B 1974, 29, 269–270. [Google Scholar] [CrossRef]
- Henrick, K.; McPartlin, M.; Matthews, R.W.; Deacon, G.B.; Phillips, R.J. The Structure of di-μ-chlorobis[bis(2,3,5,6-tetraflourophenyl)(triphenylphosphine oxide)thallium(III)]. J. Organomet. Chem. 1980, 193, 13–20. [Google Scholar] [CrossRef]
- Ahmad, S.U.; Beckmann, J. Intramolecularly coordinated diarylindium and diarylthallium chlorides [(8-Me2NC10H6)2E]Cl (E = In, Tl) with essentially ionic structures. Main Group Met. Chem. 2012, 35, 29–33. [Google Scholar] [CrossRef]
- Kuzu, I.; Neumuller, B. Organothalliumhalogenide. Z. Anorg. Allg. Chem. 2006, 632, 1524–1530. [Google Scholar] [CrossRef]
- Bhandari, S.; Mahon, M.F.; Molloy, K.C.; Palmer, J.S.; Sayers, S.F. Thallium(I)- and organothallium(III)-substituted mono-, bis- and tris-tetrazoles: Synthesis and supramolecular structures. J. Chem. Soc. Dalton Trans. 2000, 7, 1053–1060. [Google Scholar] [CrossRef]
- Brady, F.; Henrick, K.; Matthews, R.W.; Gilles, D.G. Carbon-13 and proton NMR parameters of neopentyl-and trimethylsilylmethyl-thallium(III) derivatives and the x-ray crystal structure of bis(trimethylsilylmethyl)chlorothallium(III). J. Organomet. Chem. 1980, 193, 21–29. [Google Scholar] [CrossRef]
- Coetzee, J.; Gabrielli, W.F.; Coetzee, K.; Schuster, O.; Nogai, S.D.; Cronje, S.; Raubenheimer, H.G. Structural studies of gold(I, II, and III) compounds with pentafluorophenyl and tetrahydrothiophene ligands. Angew. Chem. Int. Ed. 2007, 46, 2497–2500. [Google Scholar] [CrossRef]
- Fernández, E.J.; Jones, P.G.; Laguna, A.; López-de-Luzuriaga, J.M.; Monge, M.; Olmos, M.E.; Puelles, R.C. Perhalophenyl(tetrahydrothiophene)gold(I) Complexes as Lewis Bases in Acid-Base Reactions with Silver Trifluoroacetate. Organometallics 2007, 26, 5931–5939. [Google Scholar] [CrossRef]
- Stefanescu, D.M.; Yuen, H.F.; Glueck, D.S.; Golen, J.A. Gold(I) phosphido complexes: synthesis, structure, and reactivity. Inorg. Chem. 2003, 42, 8891–8901. [Google Scholar] [CrossRef] [PubMed]
- Casado, A.L.; Espinet, P. A novel reversible aryl exchange involving two organometallics: Mechanism of the gold(I)-catalyzed isomerization of trans-[PdR2L2] complexes (R = aryl, L = SC4H8). Organometallics 1998, 17, 3677–3683. [Google Scholar] [CrossRef]
- Fernández, E.J.; Gimeno, M.C.; Jones, P.G.; Laguna, A.; Laguna, M.; López-de-Luzuriaga, J.M. Synthesis and structural characterization of polynuclear complexes containing the eight-electron donor bis(diphenylphosphino)methanediide ligand. J. Chem. Soc. Dalton Trans. 1992, 23, 3365–3370. [Google Scholar] [CrossRef]
- Vicente, J.; Chicote, M.T.; Saura-Llamas, I.; Lagunas, M.C. A facile, novel way to prepare anionic, neutral and cationic gold(I) complexes. J. Chem. Soc. Chem. Commun. 1992, 12, 915–916. [Google Scholar] [CrossRef]
- Uson, R.; Laguna, A.; Spencer, J.L.; Turner, D.G. Chlorobis (Pentafluorophenyl) Thallium (III). Inorg. Synth. 1982, 21, 71–74. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. 2015, A71, 3. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. 2015, C71, 3. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refine-ment and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON: A Multipurpose Crystallographic Tool; Ultrecht University: Ultrecht, The Netherlands, 1998. [Google Scholar]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tl–Cl (Bridging) | Tl–Cl (Terminal) | Ref. | |
|---|---|---|---|
| [Tl(3,5-C6Cl2F3)2Cl]n (1) | 2.6893(11), 2.7397(13) and 2.9856(13) | ||
| TlCl3·3C5H5N | 2.520(6), 2.498(4) | [30] | |
| [Tl(FcN)2Cl] | 2.581(3) | [31] | |
| [Tl(C6F5)2Cl{Au(C6F5)3(OdppmO)}2] | 2.469(7) | [32] | |
| [Tl(C6F5)2(OdppmO)2][Tl(C6F5)2Cl2] | 2.497(27), 2.567(2) | [33] | |
| [TlMe2Cl]n | 3.029 | [34,35] | |
| [Tl(p-C6F4H)2Cl(OPPh3)]2 | 2.936(3) | [36] | |
| {[Tl(8-Me2NC10H6)2]Cl}n | 2.929(2), 2.933(2) | [37] | |
| [TlPh2Cl(py)2]n | 2.907(4), 2.9285(5), 2.951(4) | [38] | |
| {1,2-(Ph2TlN4C)2C6H4·Ph2TlCl·2MeOH·2H2O}n | 2.777(1) | [39] | |
| [Tl(Me3SiCH2)2Cl]2 | 2.76(1) and 2.99(1) | [40] |
| AuIII–C (trans to aryl) | AuIII–C (trans to S/P) | AuIII–S | AuI–S | Ref. | |
|---|---|---|---|---|---|
| [Au(3,5-C6Cl2F3)3(tht)] (2) | 2.073(3) 2.068(3) | 2.035(3) | 2.3897(8) | ||
| [Au(3,5-C6Cl2F3)3(OH2)] | 2.056(7) | [23] | |||
| [Au(3,5-C6Cl2F3)4]− | 2.059(5) | [23] | |||
| [Au(C6F5)3(tht)] | 2.056(5)- -2.068(2) | 2.030(5) 2.032(3) | 2.3623(13) 2.37792(9) | [41] | |
| [Au(C6F5)(tht)] | 2.317(3) 2.320(3) | [41] | |||
| [Ag2Au(3,5-C6Cl2F3)(CF3CO2)2(tht)]n | 2.3158(16) | [42] |
| AuIII–C (trans to aryl) | AuIII–C (trans to S/P) | AuIII–P | AuI–P | C–AuIII–C/S/P (trans) | P–AuI–P/Cl (trans) | |
|---|---|---|---|---|---|---|
| 2 | 2.073(3) | 2.035(3) | 176.79(11) (C) 177.27(7) (S) | |||
| 2.068(3) | ||||||
| 4 | 2.058(5) | 2.076(5) | 2.3707(13) | 2.2436(13) | 172.4(2) (C) | 178.66(6) (Cl) |
| 2.068(5) | 171.13(15) (P) | |||||
| 5 | 2.044(5) 2.066(5) | 2.059(5) | 2.3645(11) | 2.3079(13) | 175.42(17) (C) 177.71(14) (P) | 180.0 (P) |
| Compound | 1 | 2 | 4 | 5 |
|---|---|---|---|---|
| CCD number | 2221966 | 2221967 | 2221968 | 2221968 |
| Empirical formula | C12Cl5F6Tl | C22H8AuCl6F9S | C66H40Au2Cl7F9NP3 | C96H50Au3Cl12F18NP4 |
| Formula weight | 639.74 | 885.01 | 1752.98 | 2699.55 |
| Temperature/K | 100(2) | 100(2) | 273(2) | 100(2) |
| Crystal system | Monoclinic | Monoclinic | Triclinic | Triclinic |
| Space group | P21/c | P21/n | P-1 | P-1 |
| a/Å | 14.3123(18) | 10.105(2) | 13.4927(10) | 13.0360(5) |
| b/Å | 5.5515(7) | 23.044(5) | 14.4362(13) | 14.5245(6) |
| c/Å | 20.123(2) | 12.100(2) | 19.1129(17) | 17.2981(8) |
| α/º | 90 | 90 | 73.616(3) | 92.010(2) |
| β/º | 103.725(5) | 110.680(7) | 73.465(3) | 109.185(2) |
| γ/º | 90 | 90 | 83.852(3) | 108.171(2) |
| Volume/Å3 | 1553.2(3) | 2635.9(9) | 3422.5(5) | 2904.3(2) |
| Z | 4 | 4 | 2 | 1 |
| ρcalcd/g·cm−3 | 2.736 | 2.230 | 1.701 | 1.543 |
| μ/mm−1 | 11.315 | 6.346 | 4.689 | 4.178 |
| F(000) | 1168 | 1672 | 1692 | 1296 |
| Crystal size/mm3 | 0.114 × 0.051 × 0.025 | 0.315 × 0.263 × 0.187 | 0.167 × 0.142 × 0.096 | 0.200 × 0.189 × 0.059 |
| Radiation | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) |
| 2θ range for data collection/° | 6.33 to 55.80 | 6.41 to 55.81 | 4.77 to 55.89 | 5.97 to 53.46 |
| Index rages | −18 ≤ h ≤ 18 −5 ≤ k ≤ 7 −26 ≤ l ≤ 26 | −13 ≤ h ≤ 13 −30 ≤ k ≤ 30 −14 ≤ l < 15 | −15 ≤ h ≤ 17 −19 ≤ k ≤ 19 −25 ≤ l < 25 | −15 ≤ h ≤ 16 −18 ≤ k ≤ 18 −21 ≤ l < 21 |
| Reflections collected | 23,964 | 62,875 | 93,776 | 58,913 |
| Independent reflections | 3,702 [R(int) = 0.0586] | 6,247 [R(int) = 0.0370] | 16,345 [R(int) = 0.0479] | 12,292 [R(int) = 0.0453] |
| Completeness to theta = 25.242° | 99.6 % | 99.4 % | 99.5 % | 99.5 % |
| Data/restraints/parameters | 3702/0/217 | 6247/68/352 | 16,345/36/763 | 12,292/87/522 |
| Goodness-of-fit in F2 | 1.057 | 1.101 | 1.104 | 1.017 |
| Final R indexes [I > 2sigma(I)] | R1 = 0.0297 wR2 = 0.0527 | R1 = 0.0217 wR2 = 0.0545 | R1 = 0.0384 wR2 = 0.0731 | R1 = 0.0339 wR2 = 0.0816 |
| R indices (all data) | R1 = 0.0401 wR2 = 0.0553 | R1 = 0.0231 wR2 = 0.0553 | R1 = 0.0591 wR2 = 0.0846 | R1 = 0.0446 wR2 = 0.0875 |
| Largest diff. peak and hole/e·Å−3 | 1.483 and −1.250 | 1.187 and −0.393 | 3.093 and −1.493 | 1.804 and −1.810 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coconubo-Guio, L.; López-de-Luzuriaga, J.M.; Moreno, S.; Olmos, M.E. Synthesis and Structural Characterization of Phosphanide Gold(III)/Gold(I) Complexes and Their Thallium(III) and Gold(III) Precursors. Molecules 2023, 28, 447. https://doi.org/10.3390/molecules28010447
Coconubo-Guio L, López-de-Luzuriaga JM, Moreno S, Olmos ME. Synthesis and Structural Characterization of Phosphanide Gold(III)/Gold(I) Complexes and Their Thallium(III) and Gold(III) Precursors. Molecules. 2023; 28(1):447. https://doi.org/10.3390/molecules28010447
Chicago/Turabian StyleCoconubo-Guio, Laura, José M. López-de-Luzuriaga, Sonia Moreno, and M. Elena Olmos. 2023. "Synthesis and Structural Characterization of Phosphanide Gold(III)/Gold(I) Complexes and Their Thallium(III) and Gold(III) Precursors" Molecules 28, no. 1: 447. https://doi.org/10.3390/molecules28010447