
Synthesis and Applications of Nitrogen-Containing Heterocycles as Antiviral Agents


1
Department of Chemistry, Georgia State University, 100 Piedmont Avenue SE, Atlanta, GA 30303, USA
2
Center of Diagnostics and Therapeutics, Department of Chemistry, Georgia State University, 100 Piedmont Avenue SE, Atlanta, GA 30303, USA
*
Author to whom correspondence should be addressed.
Molecules 2022, 27(9), 2700; https://doi.org/10.3390/molecules27092700
Submission received: 15 March 2022
/
Revised: 10 April 2022
/
Accepted: 11 April 2022
/
Published: 22 April 2022
(This article belongs to the Special Issue Synthesis of Heteroaromatic Compounds)
Abstract
:Viruses have been a long-term source of infectious diseases that can lead to large-scale infections and massive deaths. Especially with the recent highly contagious coronavirus (COVID-19), antiviral drugs were developed nonstop to deal with the emergence of new viruses and subject to drug resistance. Nitrogen-containing heterocycles have compatible structures and properties with exceptional biological activity for the drug design of antiviral agents. They provided a broad spectrum of interference against viral infection at various stages, from blocking early viral entry to disrupting the viral genome replication process by targeting different enzymes and proteins of viruses. This review focused on the synthesis and application of antiviral agents derived from various nitrogen-containing heterocycles, such as indole, pyrrole, pyrimidine, pyrazole, and quinoline, within the last ten years. The synthesized scaffolds target HIV, HCV/HBV, VZV/HSV, SARS-CoV, COVID-19, and influenza viruses.
1. Introduction
In recent years, outbreaks of infectious viral diseases have been increasing unexpectedly and costing millions of human lives. Despite many developed vaccines and therapeutics for prevention and treatment, viruses have continuously evolved and re-emerged to threaten public health, social relations, and economic stability.
In the early 1980s, the human immunodeficiency virus/acquired immunodeficiency syndrome (HIV/AIDS) infected 79 million people, with 39 million deaths over three decades [1]. The influenza virus series also have a long-term effect on public health. The seasonal influenza viruses infect 2–5 million people and kill 250,000–500,000 people worldwide per year [2]. Other influenza viruses also have caused occasional pandemics throughout history. The H1N1 influenza pandemic of 1918 cost approximately 40 million lives. H2N2 caused another epidemic in 1957, while H3N2 struck in 1968, and H1N1 again in 2009 [2]. During the early 21st century, the severe acute respiratory syndrome coronavirus (SARS-CoV) from a zoonotic source started in Guangdong Province, China. It spread through the global community, causing about 8000 infections and 800 deaths worldwide [3]. Middle East respiratory syndrome coronavirus (MERS-CoV) emerged in 2012 and caused 2029 infections and 704 deaths in 27 countries [4]. The novel coronavirus (SARS-CoV-2) has caused a worldwide pandemic with more than 99 million infections and 2 million deaths within the first twelve months, starting in December 2019 in Wuhan, China. Although some antiviral drugs and vaccines are available for certain viruses, it is necessary to continuously develop new drugs and methods for drug-resistant viruses and viruses newly evolved from mutations.
The most promising antiviral drugs are small organic molecules that can target specific parts of the viruses and interfere with different stages of the viral life cycle [4]. Various strategies have been applied to target different viruses effectively. For example, for SARS-CoV, small molecules were used to target protease activity and inhibit viral replication. For anti-HIV/AIDS agents, viral glycoproteins were targeted to hinder their interaction with the receptors on cell surfaces, which then activate the virus’s endocytosis into the cell [4]. Among many chemical scaffolds, nitrogen-containing heterocyclic small compounds have been exploited extensively due to their broad range of applications in biological and pharmacological activities. The nitrogen-containing heterocyclic bases have high versatility in synthesis with different moieties. Those heterocyclic backbones have rigid aromatic structures that can be incorporated into the binding pockets and provide various molecular interactions, such as ionic bonding, hydrogen bonding, hydrophobic interaction, non-covalent bonding, etc., for ligand binding with receptor proteins [5,6]. The interaction can mimic specific properties that can effectively inhibit the activities of various biological enzymes and components that play vital roles in the development of viral infections [7,8]. Previous reviews focused on single types of nitrogen-containing heterocycles such as indole and/or imidazothiazole derivatives in designing antiviral agents [7,9]. Other reviews covered broader details on various biological applications such as antimicrobial, anti-inflammatory, anti-tubercular, anti-depressant, and anti-cancer activities [10,11,12]. Our current review focuses specifically on the common nitrogen-containing heterocycles including indoles, pyrroles, pyrimidines, pyrazoles, and quinolines that have been applied in drug design for antiviral purposes during the past ten years. These heterocycles are significant backbones of pharmaceutical products with exceptional biological activity to interfere with various viral infections [8]. While the indole cores are known for their wide existence in natural products with biological activity [7], the properties of pyrrole can be expanded for chemical design and are suitable for biological systems [13]. Pyrimidine derivatives have widespread therapeutic applications, as they are essential building blocks of nucleic acids in DNA and RNA [14]. Pyrazoles can be fused with other heterocycles to extend their active biological potential [15]. Quinoline derivatives have versatile chemical properties for synthesis and biological activities [16]. Our review discusses the synthesis and biological characteristics of nitrogen-containing heterocycle derivatives in detail. Further modifications and functions of nitrogen heterocycles are introduced along with various antiviral purposes.
2. Indole
Indole derivatives are one of the well-known scaffolds in drug discovery that can inhibit a wide diversity of enzymes by binding with potential ligands [7]. The first indole was prepared using Fischer indole synthesis, which was reported in 1866. Nowadays, most indole synthesis pathways start with incorporating a benzene ring with additional factors that stabilize the formation with the fused pyrrole ring. Indole is an aromatic ring with ten π electrons and a lone pair of nitrogens. The lone pair is not available for protonation but is involved in the delocalization of the indole conjugated system. The protonation and other electrophilic substitutions mostly occur at the C-3 position, which has the highest electron density and is most thermodynamically stable for modifying antiviral agents [17].
Hassam et al. designed new scaffolds of HIV non-nucleoside reverse transcriptase (RT) inhibitor with the addition of a cyclopropyl group at the C-3 of the indole base [18]. The RT enzyme plays a crucial role in the reverse transcription of viral RNA to single-stranded DNA in host cells. The binding of specific cyclopropyl chemical moiety to the RT pocket can significantly inhibit the enzyme’s function by improving electrostatic interaction with the hydrophobic pocket [18]. As shown in Scheme 1, the ester-substituted indole 1 reacted with benzoyl chloride for the electrophilic substitution at the C-3 position. Tert-butyloxycarbonyl (Boc) was added to intermediate 2 to prevent reactions at the amine before further modifying cyclopropyl moiety. Intermediate 3 underwent a Wittig reaction with methyl triphenylphosphonium ylide reagent to convert ketone to alkene 4, which then reacted with diiodomethane and zinc alloy for cyclopropyl addition to furnish product 5 [18].
As shown in Figure 1, in the in vitro phenotypic assay, compounds 5a–c showed high inhibitory activity (IC50), with low cytotoxicity (CC50) in 5a and 5c. In the modeling study, 5a was better at accommodating the binding site. The indole NH and ester moiety was incorporated for hydrogen bonding in the reverse transcriptase’s allosteric site at Lys 101. The ester group associates well in the binding site with the ethyl directed out of the site. Modification of the ethyl group decreased the potency of the compound. The cyclopropyl moiety was implemented to bind with Val 179 binding pocket, a small, hydrophobic cleft located near the catalytic site. The aromatic ring of R2 was favorable for the interaction with Tyr 188 and Trp 299. The modification at R2 from phenyl to 2-thiophenyl resulted in a slightly better inhibitory value but increased toxicity in cells. Increasing the size of the halogen at R3 also enhanced IC50 and CC50 values [18].
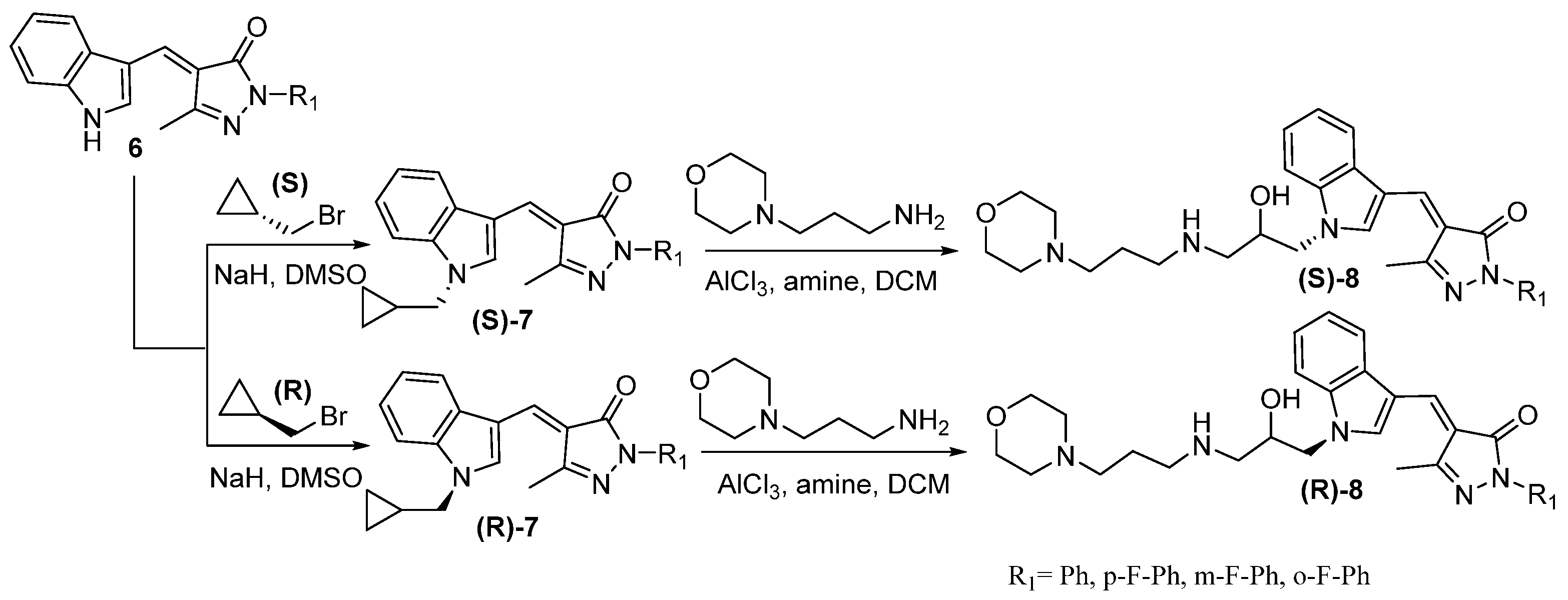
Another study of antiviral agents with substitution at C-3 of indole was utilized to target the hepatitis C virus (HCV). HCV infection can lead to acute or chronic liver diseases, such as hepatocellular carcinoma and liver cirrhosis. Specific treatments have been approved for clinical applications. However, those treatments possessed low effectiveness in the patient population with additional side effects [19]. Han et al. [20] introduced the small molecules of N-protected indole scaffolds (NINS) to inhibit HCV. As shown in Scheme 2, scaffold 6 reacted with the racemic epibromohydrin to form N-protection derivative 7 with two enantiomers. The substitution of NH was further extended by ring-opening of epoxide with the nucleophilic amine chain to afford 8.
In the structure–activity relationship (SAR) study, multiple modifications of the phenyl ring at R1 were analyzed. As shown in Table 1, p-F-Ph and m-F-Ph improved inhibitory potency, but o-F-Ph reduced anti-HCV activity. The m-F-Ph exhibited lower cytotoxicity compared to p-F-Ph. The (R)-enantiomer can possess better anti-HCV potency and less cytotoxicity than its corresponding (S)-enantiomer of both m-F-Ph and p-F-Ph. The results indicated that the position of substitution and chirality has a high impact on their inhibitory effect. The scaffold was also proved to inhibit viral entry into the viral cycle rather than interfere in any viral RNA replication in host cells with a mechanism of action (MoA) study.
The varicella-zoster virus (VZV) infection can cause acute varicella and herpes zoster, leading to various disease complications in the central nervous system from latent viruses [21]. Most approved drugs for VZV-associated treatments were nucleosides that competitively inhibit the viral DNA polymerase and interfere with DNA replication. However, those nucleosides implied multiple drug resistance and produced low efficacy in the anti-VZV virus. Mussela et al. [22] synthesized a family of indole derivatives as non-nucleoside antivirals to mimic deoxythymidine. The compounds can inhibit thymidine kinase (TK) by interfering with the series phosphorylation of thymidine triphosphates, which can be used as a complementary base in the replication process [23]. As shown in Scheme 3, the 3-(2-bromoethyl)-1H-indole 9 was reacted with methyl iodide for N-alkylation. The nucleophilic displacement of bromine in 10 was conducted with chain-linked amine and used palladium as a catalyst under microwave conditions to obtain 11, which was then treated with acetyl chloride under the basic condition to yield the final compound 12.
Compound 12 has low cytotoxicity in cells and good inhibitory activity at low EC50 due to the alkylation of NH and acylation of the amine group in C-3 substitution [22]. This tryptamine derivative has lower potency than reference drugs Acyclovir and Brivudine but displayed similar inhibiting activity against TK in anti-VZV mechanisms. Additionally, the derivative 12 selectively targeted VZV strains (OKA, 07-1, and YS-R) only when tested with other members in the same family of Herpesviridae (HMCV, HSV-1, and HSV-2) and various RNA virus strains such as HIV and influenza, as shown in Table 2. Compound 12 can be a leading compound for further investigation in inhibiting VZV specifically.
Other synthesized potential antiviral agents have substitution at C-2 of indole, the second most reactive site for electrophiles. For targeting SARS-CoV viruses, Thanigaimalai et al. [24] discovered compounds to target the chymotrypsin-like protease (CLpro), which plays a vital role in cleaving polyproteins to produce functional proteins directly involved in viral replication and transcription [25]. The analogs were peptidomimetic covalent inhibitors that can mimic the substrate of CoV 3CLpro. As shown in Scheme 4, to synthesize compound 17, the peptidomimetic chain was added to the commercially available indole derivative 13, through peptide coupling with the carboxyl group at C2 to form the peptide 14. For the other intermediate, γ-lactam acid 15 coupled with N, O-dimethylhydroxylamine to form substituted amide from the carboxylic end using Weinreb–Nahm synthesis. The amide continuously reacted with benzothiazole to create 16, which then deprotected and coupled with the peptide 14 in the presence of peptide coupling reagent to furnish compound 17 [24].
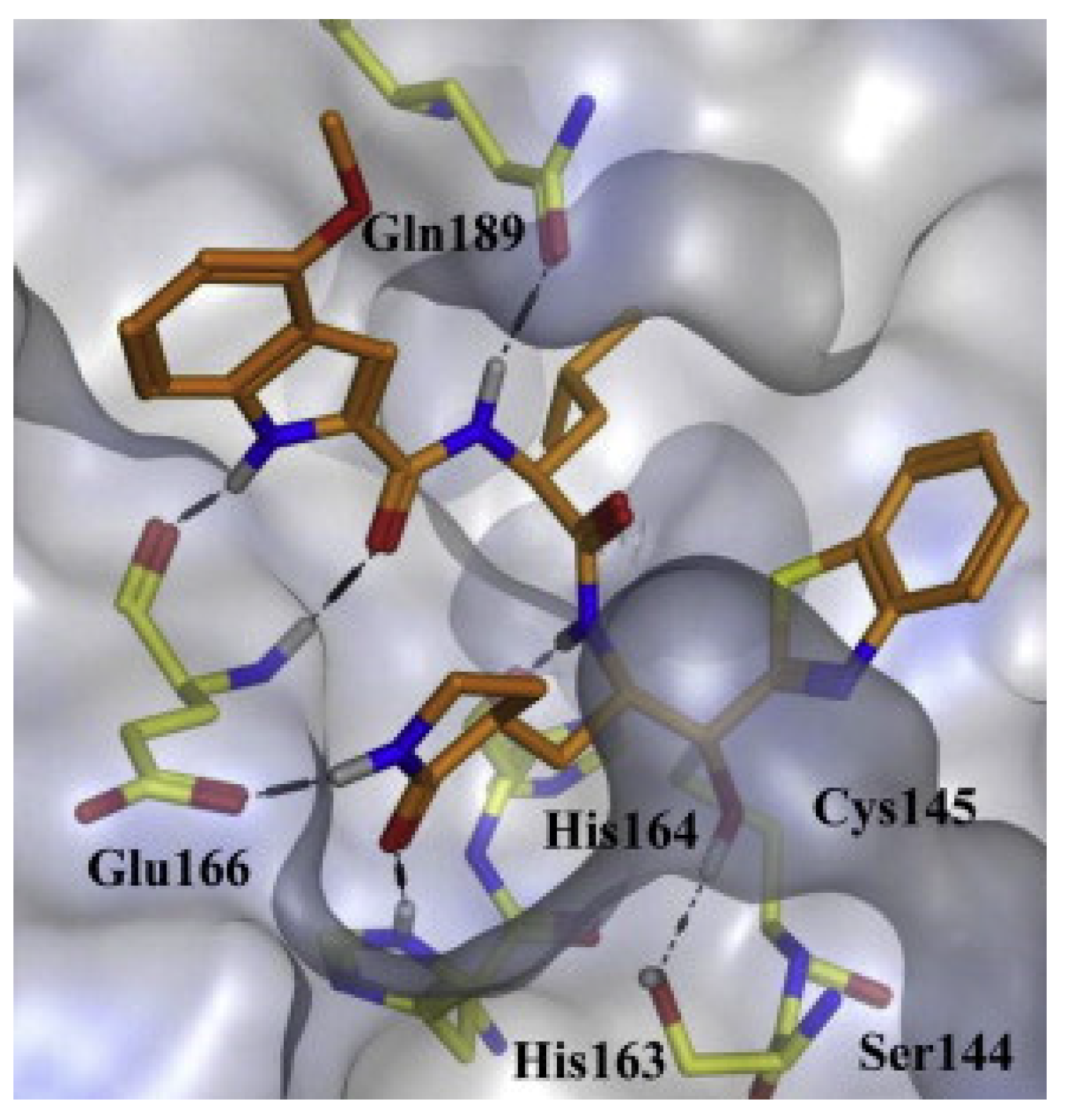
The synthetic inhibitor 17 has four main features that are suitable for different pockets in the active site of CoV CLpro. The compounds included ketone to target cysteine residue’s thiol (Cys 145) in the S1″ pocket and (S)-γ-lactam ring for the S1 pocket. The hydrophobic leucine was used for the S2 position [24,26]. In another study, the leucine was replaced by the π conjugated system and hydrophobic interaction of the aryl or cyclohexyl group to enhance access of the S2 pocket to target SARS-CoV-2 [27]. Based on its SAR study, compound 17 exhibited great inhibitory (Ki = 0.006 μM or IC50 = 0.74 μM) activity with the methoxy at C-4 of the indole. Indole possessed the best inhibitory effect among other heterocycles due to its NH, which can provide hydrogen bonding interaction with Gln 166, as shown in Figure 2 from the molecular docking study.
3. Pyrrole
Pyrrole is a heterocyclic aromatic five-membered ring that was first observed in coal tar and bone oil in 1834. Many investigations reported pyrrole as an integral part of different natural compounds. The delocalization of the lone pair of electrons from the nitrogen atom provided additional stabilization of the ring. In total, six π electrons delocalized over the five-membered ring formed the isoelectronic system and allowed electrophilic attack in different reactions [28].
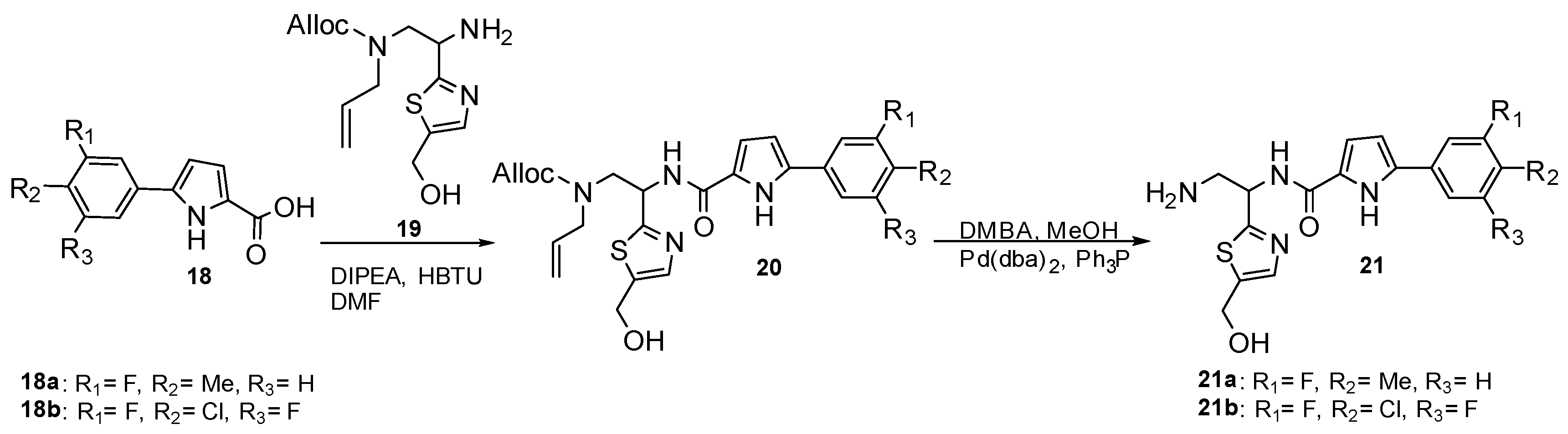
Curreli et al. [29] have developed drugs to block HIV-1 envelope glycoprotein (gp120) from binding the receptor CD4 of the host cells and prevent the entry of viral RNA [30]. The scaffolds were designed to mimic receptor CD4 and act as HIV-1 entry antagonists. Intermediates 18 and 19 were synthesized and coupled to yield compound 20, as shown in Scheme 5 [28,31]. The deprotonation of amine followed to afford product 21. Intermediate 19 can be synthesized from R- and S-isomer imines that derived isomers in compounds 20 and 21 [29].
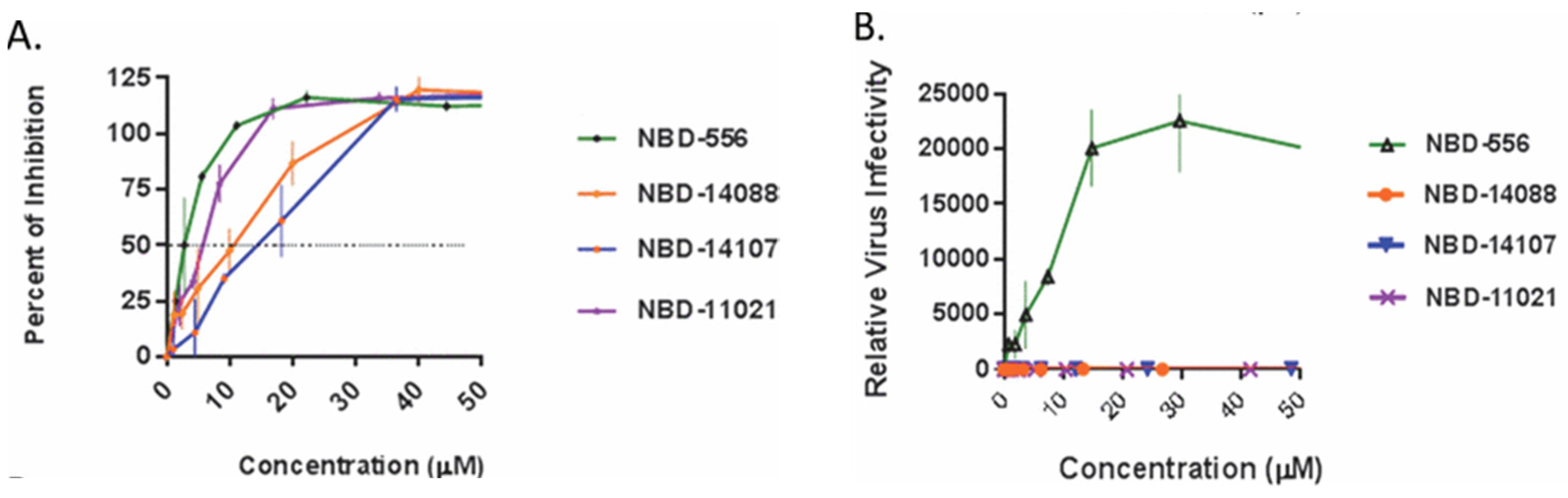
Based on X-ray crystal structure analysis, pyrrole allowed a potential hydrogen bond of NH with the residue Asn425 of gp120. Methylation of NH lost an H-bond donor atom and increased steric hindrance, which interferes with binding capability. The scaffold loses its antiviral activity when replacing the pyrrole ring with imidazole. According to Table 3, the compounds exhibited high antiviral potency with low cytotoxicity and good selectivity for HIV-1 gp120. The methyl substitution at R2 of 21a enhanced antiviral potency, while the addition of fluoro at R3 of 21b showed similar activity but improved metabolic stability. Because (R) 21a (NBD-14088) and (S) 21b (NBD-14107) have better selectivity, they were used to measure HIV-1 entry antagonist properties with cell-to-cell fusion inhibition assay and infectivity in cells, as shown in Figure 3. Even though (R) 21a and (S) 21b required higher IC50 compared to NBD-556 (HIV-1 entry agonist) and NBD-11021 (HIV-1 entry antagonist), negative and positive controls, respectively, two synthesized compounds still exhibited antagonist properties to reduce infections. The compounds also can inhibit HIV-1 reverse transcriptase from converting viral RNA into complementary DNA in hosts [29].
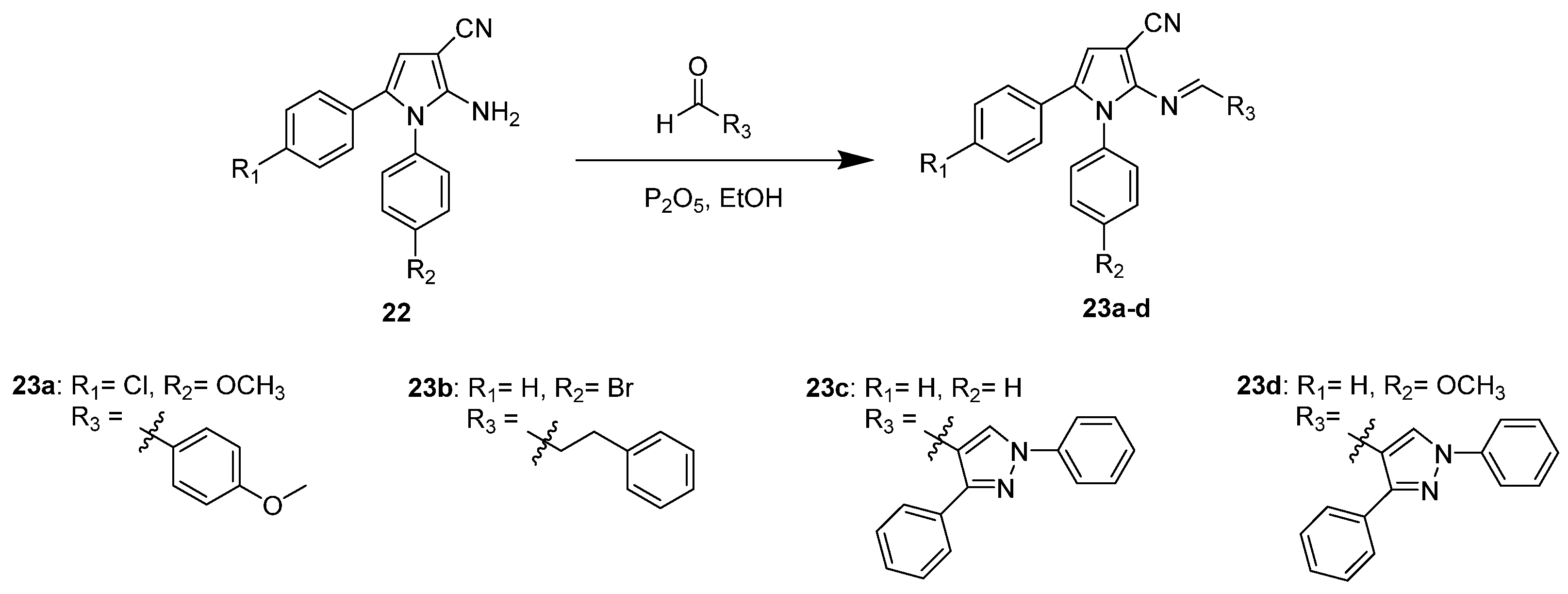
Herpes simplex virus (HSV) is another virus in the family of α-herpesviruses with VZV that can cause common, self-resolving diseases of skin or mucosa, such as herpes labialis and other infectious diseases. However, HSV and VZV possess differences in the route of infection, spread pathway, and range of hosts [32]. Due to the viral strains highly resistant to the nucleoside treatments, non-nucleoside compounds were synthesized to inhibit thymidine kinase (TK) from early DNA replication. According to Hilmy et al. [33], the pyrrole analogs 23a–d were synthesized using different substituted 2-amino-3-cyano-1,5-diarylpyrroles to react with various aryl aldehydes under the basic condition with phosphorous pentoxide, as shown in Scheme 6.
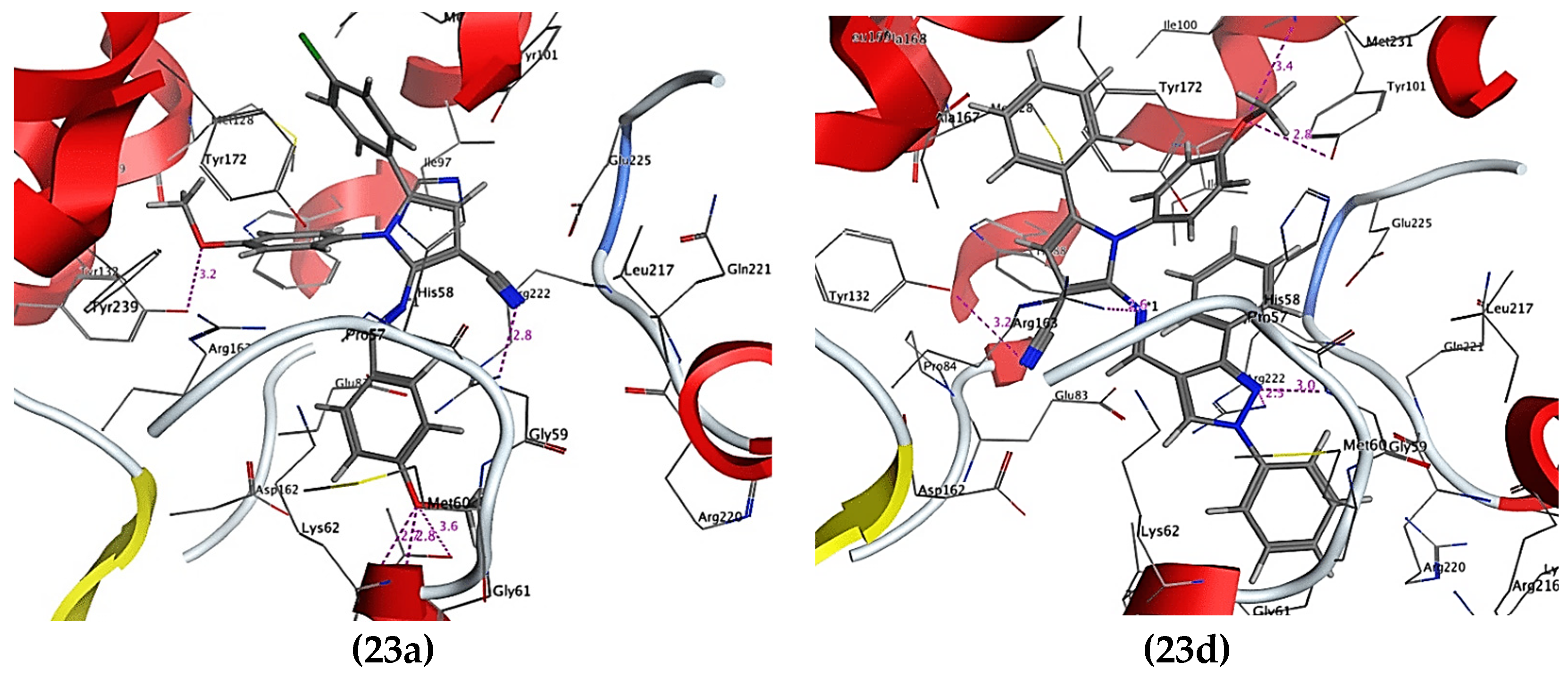
The analogs were compared with Acyclovir (ACV), a standard drug used to treat HSV, for anti-HSV activity and cytotoxicity. As shown in Table 4, all new compounds exhibited a high percentage of reduction (94–99%) in the number of virus plaques. Compounds 23a (99%) and 23d (97%) even had better results than ACV due to their similarity at substituted N of pyrrole with 4-methoxyphenyl. Compound 23a had the highest activity with the 4-methoxybenzylideneamino at position C-2 of pyrrole. The synthesized compounds had better docking scores than ACV, indicating that they had better ligand–receptor interaction in the TK active site. Comparing compounds 23a and 23d in Figure 4, they had different interactions in the binding pocket. The two oxygens of the methoxy group in 23a had hydrogen bonding with Lys62 and Tyr132. Another hydrogen bond was formed between the cyano nitrogen and Arg222. However, for 23d, the methoxy group formed two hydrogen bonds with Arg176 and Tyr101. Other hydrogen bonds were also established from the nitrogens of the compounds. Both also formed hydrophobic and van der Waals interactions with other amino acids. However, 23d interacted with crucial amino acids that actively contribute to the function of TK; hence, 23d showed better inhibitory effect compared to 23a. Additionally, compounds 27b and 27c might have different anti-HSV activity mechanisms rather than targeting TK [33].
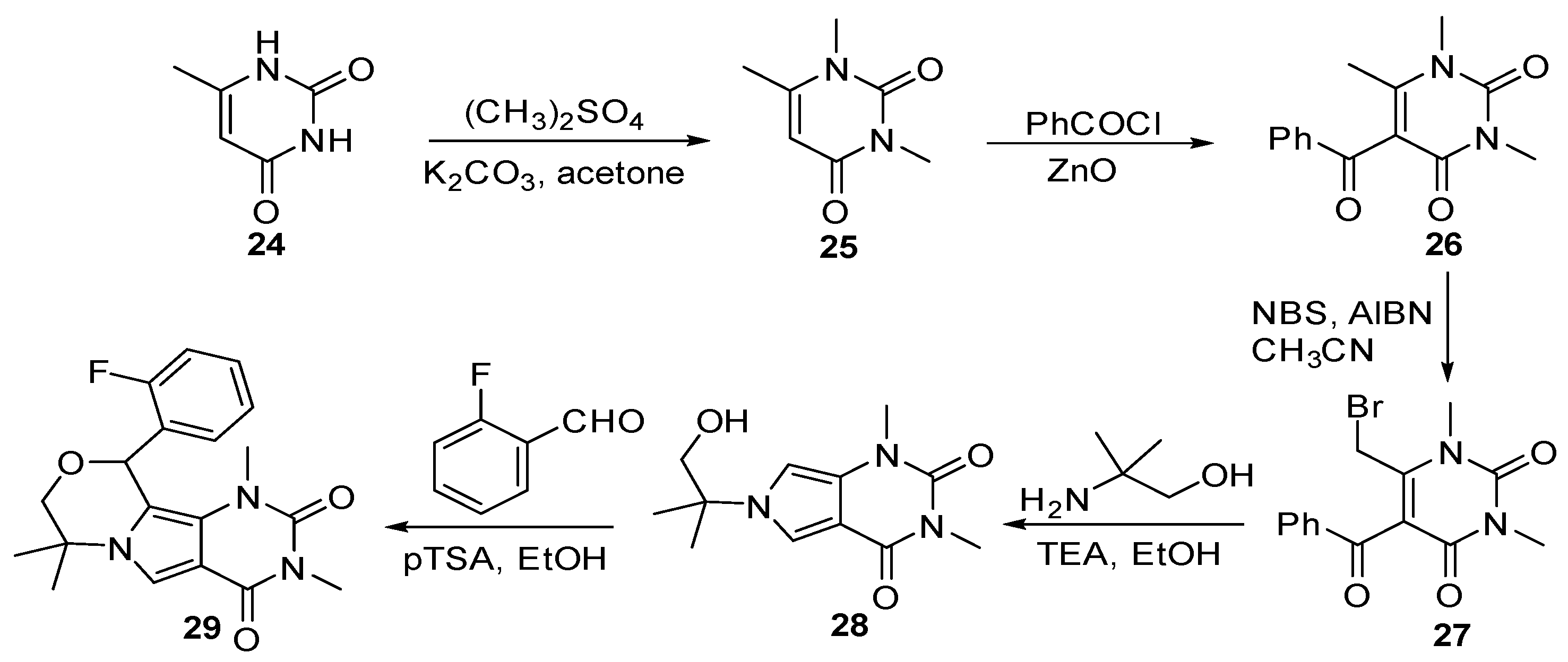
Lin et al. [34] introduced a class of anti-influenza agents targeting the viral nucleoprotein (NP), a binding protein that contributes to the transcription and packaging processes. The synthesized pyrimido-pyrrolo-quinoxalinedione analogs were aimed to inhibit the synthesis of NP and interrupt viral replication [35]. As shown in Scheme 7, substituted pyrimidinedione 24 was methylated at the two atoms of nitrogenusing dimethyl sulfate, followed by Friedel–Crafts acylation with benzyl chloride to furnish compound 26. The synthesis was continued with the addition of bromine and the formation of fused pyrrole 28 from the reaction of 27 with 2-amino-2-methylpropan-1-ol. The annulation of intermediate 28 with F- substituted aryl aldehyde produced the final product, compound 29.
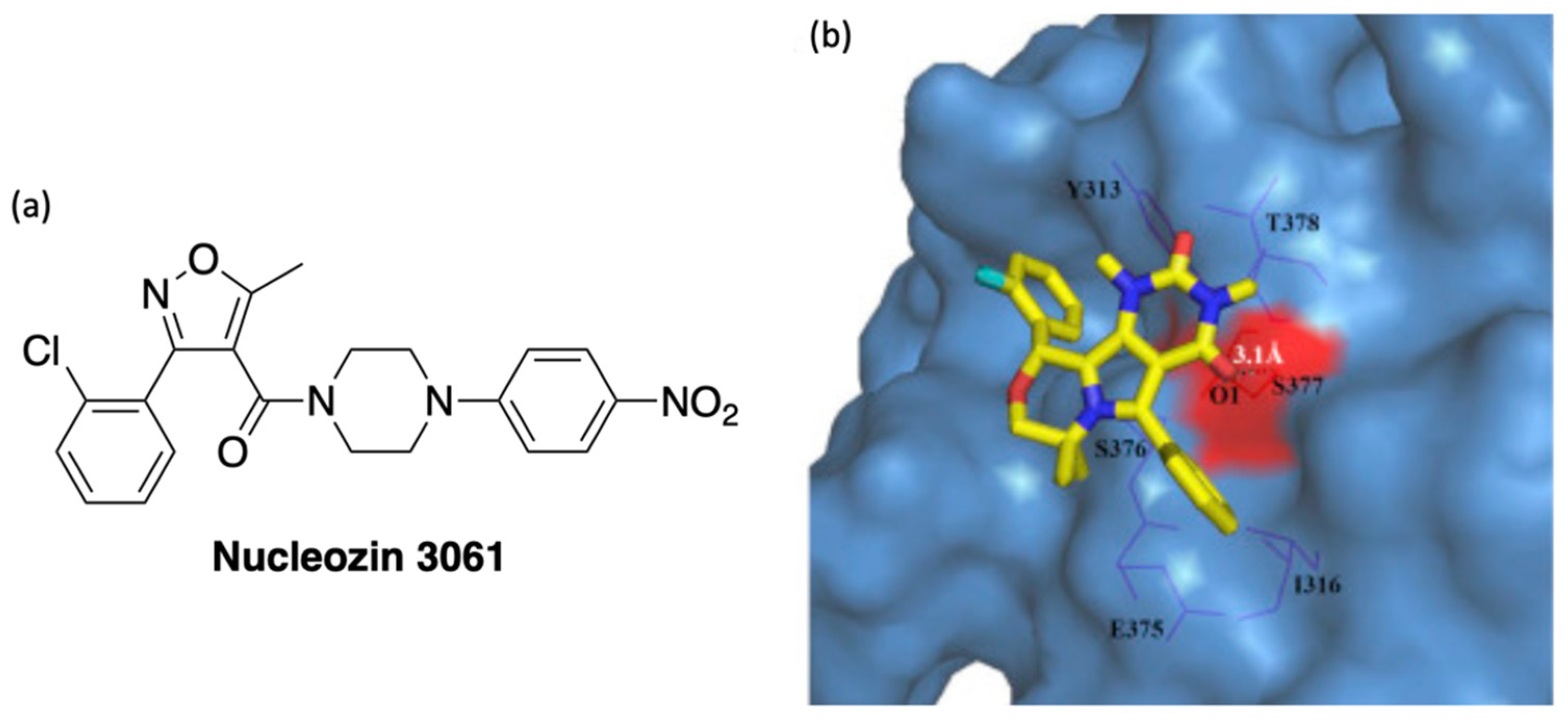
The inhibiting effect of compound 29 (PPQ-581) was compared with that of the nucleozin 3061, a potent antagonist of NP. Both have a similar trend of effectively inhibiting nucleoprotein (NP) synthesis shortly after infection. Treatment with 29 and nucleozin from 3–8 h post-infection partially inhibits NP synthesis. The docking study of compound 29 in the influenza A nucleoprotein is shown in Figure 5. The oxygen of ketone formed hydrogen bonding with S377, which was a crucial binding area for compound 29. The mutation of the S377 sidechain can result in the loss of the anti-influenza activity of compound 29. Compound 29 also inhibited the influenza RNA-dependent RNA polymerase (RdRP) activity of nucleozin-resistant influenza strains.
4. Pyrimidine
Pyrimidine is a heterocycle composed of a six-membered ring with two nitrogens at positions 1 and 3. The first pyrimidine derivative was synthesized in 1818 by Gaspare Brugnatelli. Pyrimidines were continuously developed for applications in biological systems, as their nitrogen bases were associated with DNA and RNA. The two nitrogens in pyrimidines are electron withdrawers, leaving specific carbons with electron deficiency [36]. Pyrimidine has also been fused with other heterocycles to improve its applicable spectrum of biological activity [37].
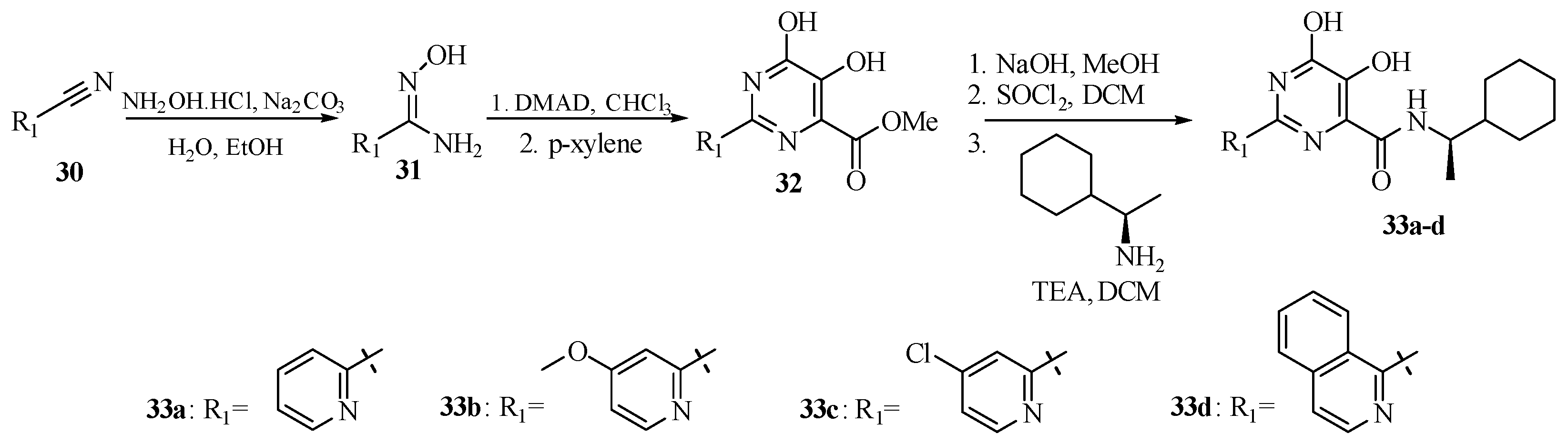
Currently, HIV/AIDS is still causing a major global health issue. The treatments for HIV/AIDS require a long-term commitment to target different stages of viral DNA replication. Malancona et al. [38] introduced the 5,6-dihydroxypyrimidine scaffolds to target the HIV nucleocapsid (NC), which is a multifunctional protein that plays crucial roles in binding amino acids for their sequence-specific binding in reverse transcription. NC is also a nucleic acid chaperone that can manipulate nucleic acid structures for their thermodynamically stable conformations [39]. As shown in Scheme 8, the central dihydroxypyrimidine cores were obtained by reacting substituted 2-pyridinecarbonitrile with hydroxylamine hydrochloride to form intermediate 31. The synthesis was continued with the reaction of compound 31 with dimethyl acetylenedicarboxylate (DMAD) for thermal cyclization to produce 32, which coupled with (R)-1-cyclohexylethanamine to furnish 33a–33d.
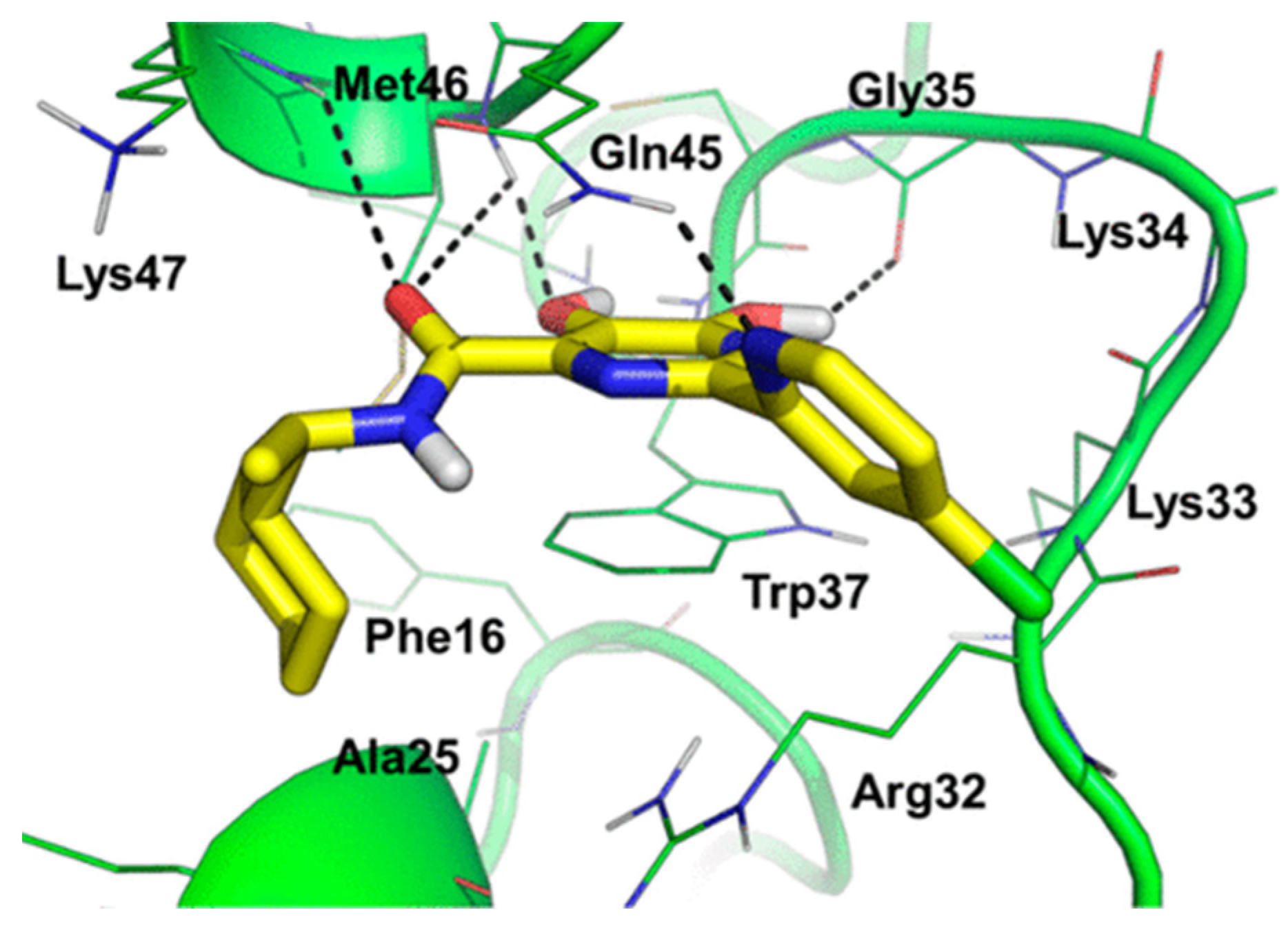
The 2-(pyridin-2-yl) substituent at R1 was essential for the inhibitory activity of the analogs. The amide moiety (N-cyclohexylmethyl) targeted the hydrophobic binding site of the protein. As shown in Table 5, further modification with 5-methoxy, 5-chlorine at the pyridine group, and quinoline resulted in 33b–33d with 40–80 folds of improved potency and higher SI values (25–75) than without any modification of 2-(pyridin-2-yl) substituent. As shown in Figure 6, the molecular docking of compound 33c in the NC demonstrated that 2-(pyridin-2-yl) substituent has π-stacking interaction with Trp37, while the dihydroxypyrimidine formed H-bonds with Gly35, Met46, and Gln45. The amide moiety interacted with the hydrophobic pocket, including Phe16, Ala25, Trp37, and Met46. Other in vitro and in vivo tests against multiple drug-resistant HIV-1 strains were also analyzed in the study. The analogs were also active in different HIV-1 resistant strains, with high oral bioavailability and excellent in vitro metabolic stability in rat and human samples and half-life in the in vivo study [38].
Mohamed et al. [40] reported the synthesis of non-nucleoside antiviral agents for hepatitis C virus (HCV), which has the highest infection rate in Egypt and causes 350,000 deaths worldwide annually. The HCV NS5B polymerase functioned as RNA-dependent RNA polymerase (RdRp) and enabled the catalyzation of viral genome synthesis [41]. The synthesis of pyrrolopyrimidine derivative 39 as a non-nucleoside purine scaffold to inhibit HCC NS5B polymerase is summarized in Scheme 9. The benzoin 34 was condensed with aryl amine to form intermediate 35, which then reacted with malononitrile to produce pyrrole derivative 36 via Dakin West reaction. The 2-aminopyrrole-3-carbonitrile 36 coupled with formic acid to furnish associated pyrrolopyrimidine derivative 37. The carbonyl was replaced by chlorine at C-4 pyrimidine, resulting in intermediate 38 for further modification at this position. Aryl amine was introduced to produce 4-aryl amino derivative 39.
Compound 39 exhibited the highest antiviral activity in the study. The arylamino group (‘‘) enhanced the toxicity of the scaffold in HCV genotype 4a cells. In the molecular docking, it was found that 39 bound strongly with Mg2+ in the docked site. Other measurements in the docking system proved that 39 was stable in the binding pocket and improved binding affinity. As shown in Figure 7, the carbonyl C=O of Gln446 in the HCV RdRp catalytic site can form a hydrogen bond with aniline NH. At the same time, the NH2 interacted with nitrogen in the pyrimidine ring to enhance the binding of compound 39 to the inhibition site.
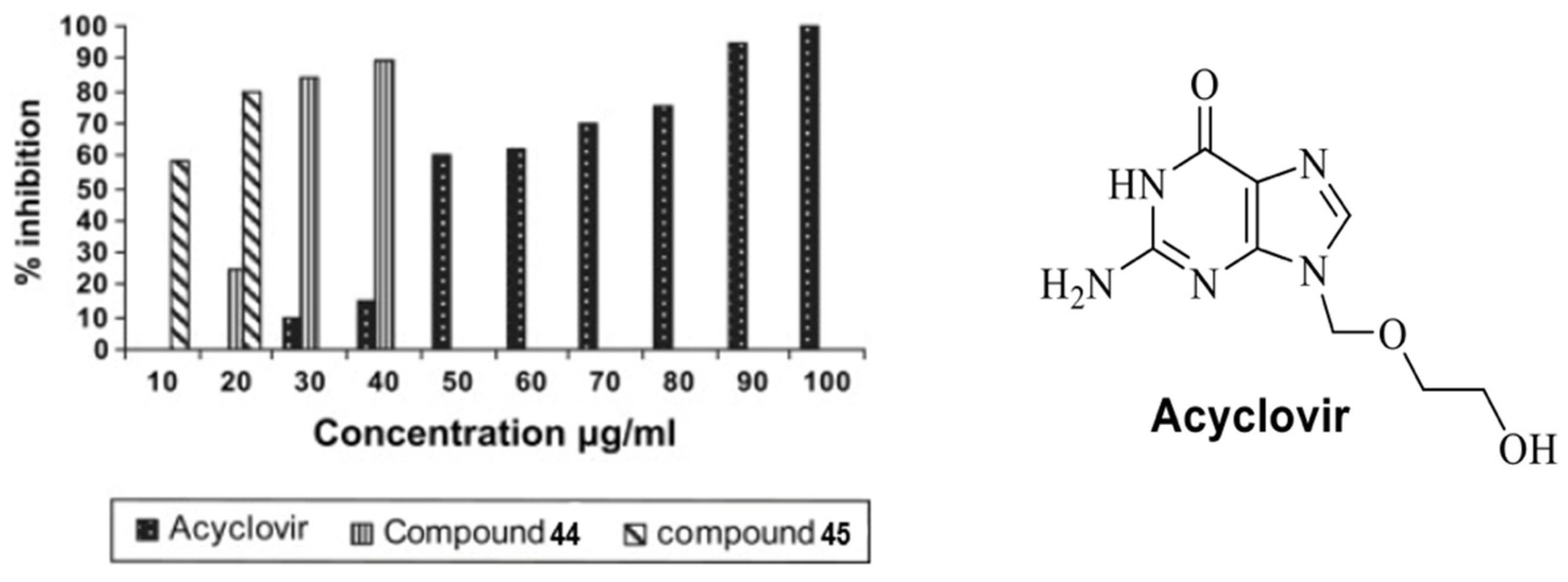
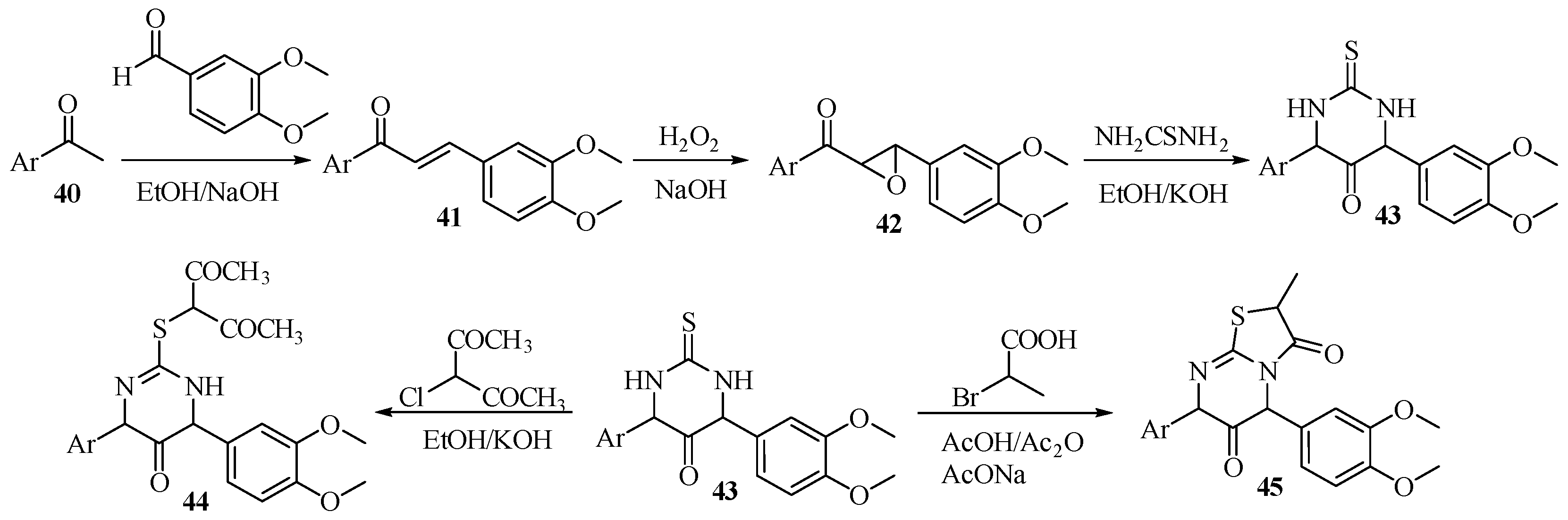
Pyrimidine derivatives were also employed in the study of anti-HSV activity. Currently, Acylclovir (Figure 8) is one of the drugs used to treat the herpes simplex virus (HSV). However, in some cases, the viruses can resist the drug; hence, new agents are continuously developed and tested for antiviral activity. Mohamed et al. [42] reported the synthesis of pyrimidine derivatives as anti-HSV agents. As shown in Scheme 10, acetophenone 40 was coupled with 3,4-dimethoxybenzaldehyde via aldol condensation to afford chalcone 41, which was reacted with hydrogen peroxide to form epoxide 42. The condensation of compound 42 with thiourea resulted in the cyclic formation of compound 43, which could either react with 3-chloroacetylacetone to produce compound 44 or be condensed with 2-bromopropionic acid to form methylthiazolo compound 45.
For the antiviral activity of compounds 44 and 45, a plaque reduction assay was conducted using concentrations of 2 and 5 μg/mL, and the viral count was recorded after adding the compounds. As shown in Table 6, compounds 44 and 45 showed a slightly higher percentage of inhibition with the 5 μg/mL concentration. Compound 44 was more effective than compound 45, achieving 100% inhibition with 5 μg/mL in the test sample. As shown in Figure 8, both compounds showed a higher percentage of inhibition at much lower concentrations compared to Acyclovir®. For the antiviral mechanism, 44 and 45 showed viricidal activity that possibly altered viral epitopes to inhibit binding to cells. Besides viricidal activity, compound 44 also has the potential to interfere with the replication processes of HSV. The two compounds are highly promising as potential new anti-HSV agents.
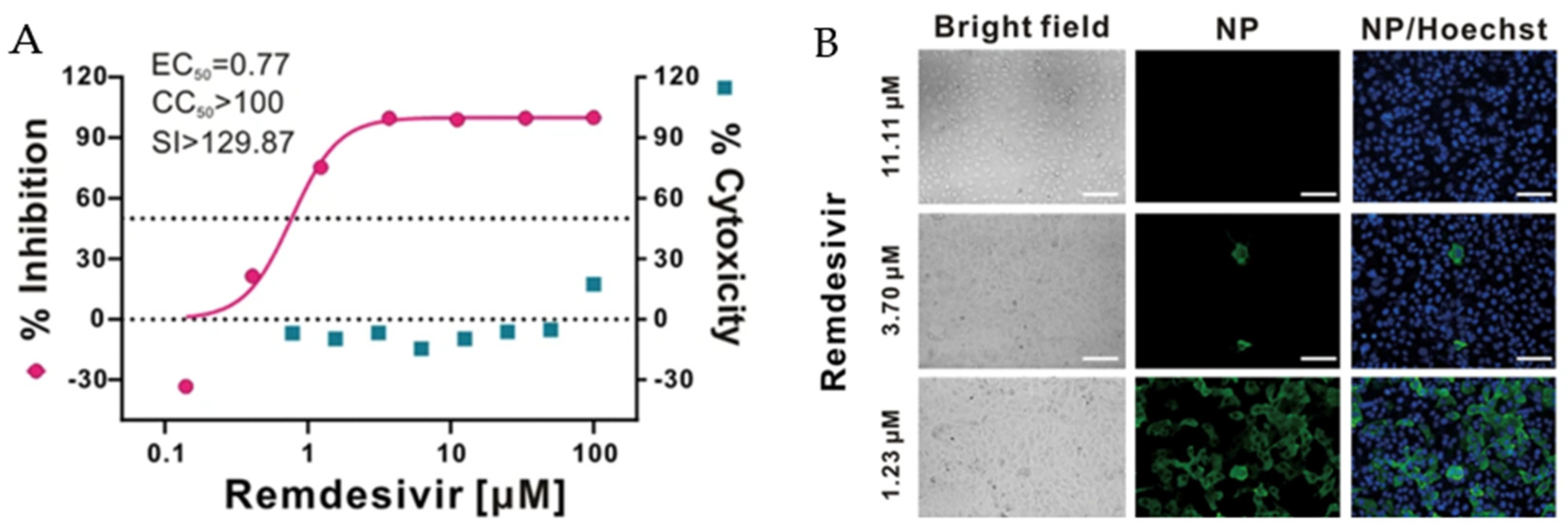
During the current COVID-19 pandemic, Remdesivir® (Figure 9) was approved for use in the treatment of patients with confirmed SARS-CoV-2 infection. Remdesivir® is an adenosine nucleoside prodrug that exhibits a broad antiviral spectrum. Due to the rapid emergence of coronavirus infections that threaten millions of people in the global community, a safe, approved drug like Remdesivir® was temporarily employed for treatment. It showed in vitro antiviral activity with animal and human coronaviruses, including SARS-CoV-2. As shown in Figure 10, Remdesivir® can effectively inhibit viral infection at a low concentration (EC50 = 0.77 μM) with a high SI value (129.87 μM). The antiviral activity of Remdesivir® was confirmed with an immunofluorescence assay. The results indicated complete viral reduction, as the viral nucleoproteins could not be observed with a 3.70 μM concentration at 48 h post-infection [43].
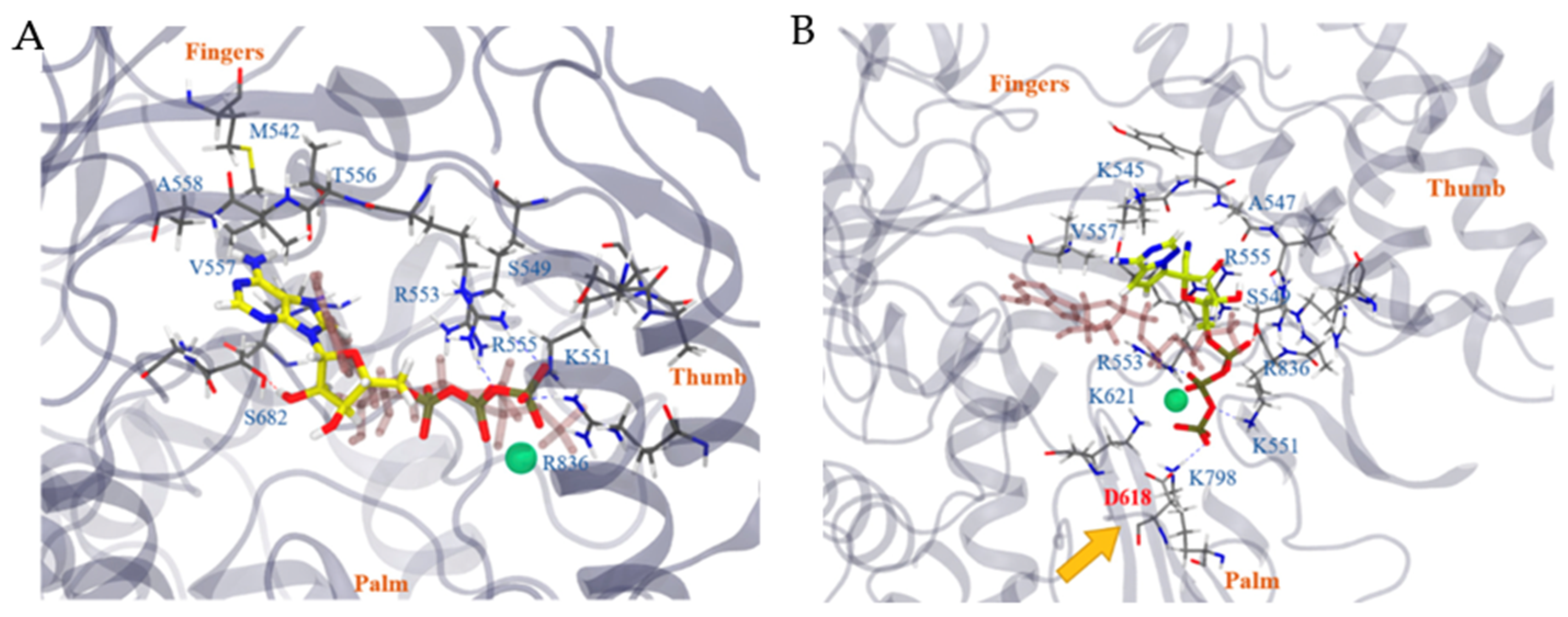
Remdesivir® was originally targeted to inhibit RNA-dependent RNA polymerase (RdRp), which has adenosine triphosphate (ATP) as the main substrate. Zhang et al. [44] reported the homology modeling of NSP12 (the RdRp complex with multiple nonstructural protein units) of SARS-CoV-2 RdRp (SARS-CoV-2 NSP12). When accumulated in cells, Remdesivir® was hydrolyzed and coupled with triphosphate (RemTP) to compete with ATP actively. As shown in Figure 11, the triphosphate of RemTP has other interactions with crucial residues compared to ATP interactions to inhibit NSP12. The adenosine of ATP and the Remdesivir® core also interacted differently from each other in the binding pocket. Additionally, RemTP affected D618, which is an essential residue for the function of SARS-CoV-2 RdRp by forming a hydrogen bond with K798. The binding free energies of ATP and RemTP were represented by FEP calculations, which showed that RemTP binds more strongly than ATP in the pocket of SARS-CoV-2 RdRp with approximately 800 folds in Kd value to effectively inhibit SARS-CoV-2 RdRp activity in RNA reproduction [44].
In addition, Grein et al. [45] further investigated Remdesivir® clinical application conducted within a cohort of hospitalized patients diagnosed with COVID-19. The data were collected from 53 patients across three continents. The majority of patients (34 out of 53) had severe symptoms and were under invasive oxygen support. The treatment plan was designed for a 10-day course with 200 mg administered intravenously on day 1 and 100 mg daily for the next nine days. Only 40 patients completed ten days of treatments, while others discontinued treatment due to serious adverse effects. After 18 days of day one treatment, 12 patients (100%) previously with no oxygen or low-flow oxygen support were discharged. Five out of seven patients (71%) previously under non-invasive ventilation were also discharged. Nineteen out of thirty-four showed improvement within the oxygen-support group, as they were discharged without oxygen aid and under non-invasive oxygen support. The other patients in this group either showed no improvement (9 out of 34) or died (6 out of 34). About 83% experienced various common to serious adverse effects. Overall, Remdesivir in this study showed improvement in the treatment of COVID-19 based on the oxygen support scale. Among the patients, 68% showed improvement in oxygen supporting status or were fully discharged, with a 13% mortality rate after treatment.
5. Pyrazole
Pyrazole is an aromatic heterocycle composed of a five-membered ring with two nitrogens. Pyrazole core structures have been employed in wide applications from natural components to pharmaceutical activities [46]. With high electron densities of nitrogen at positions 1 and 2, pyrazole is considered as a π-excessive heterocycle. The ring is available for various modifications with multiple reagents in synthetic reactions. Substituted pyrazole can form chelation with metal ions to inhibit enzymes [47].
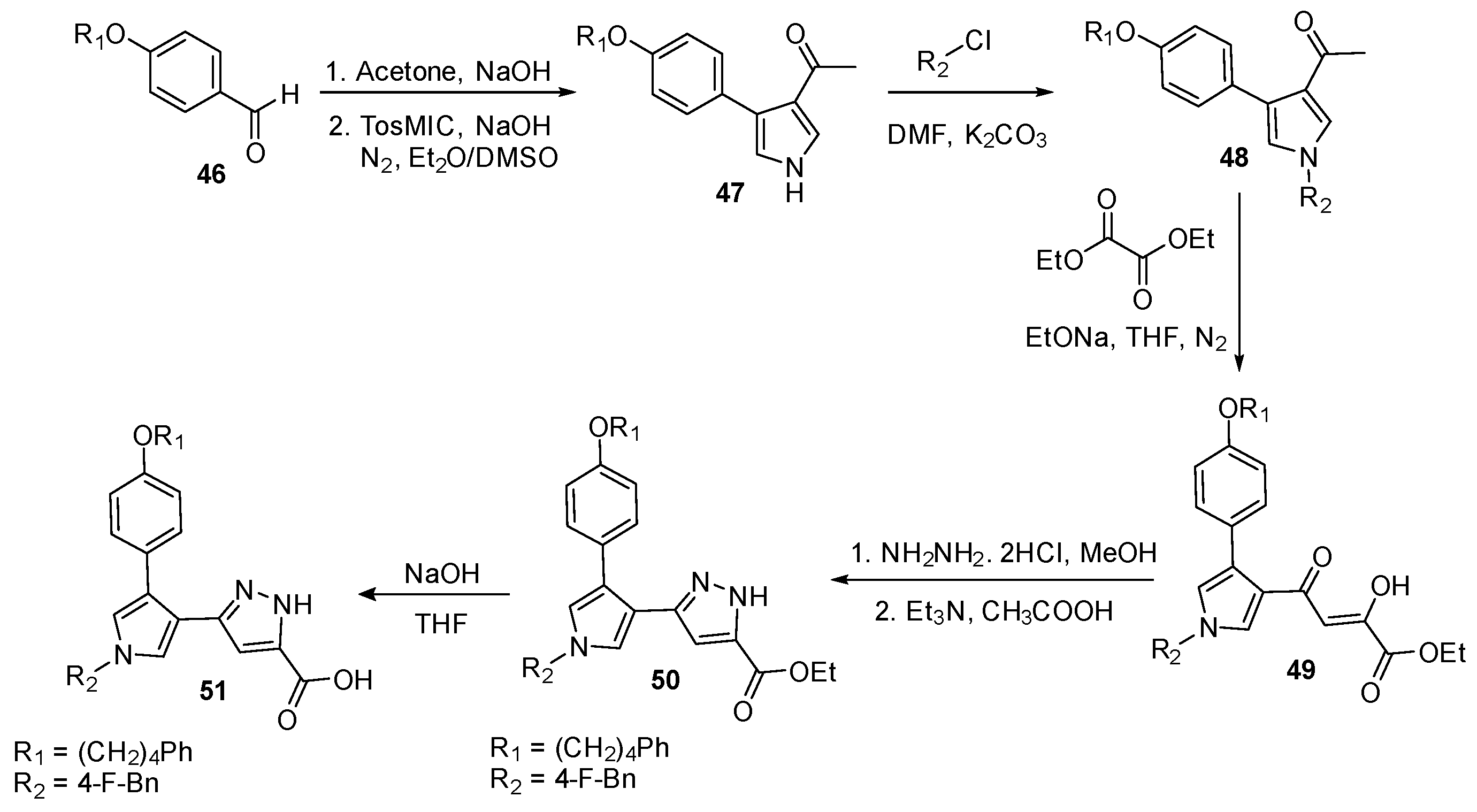
Currently, HIV-1 is still considered one of the most threatening infectious diseases to people with a high rate of infection. The crucial step of viral infection is the integration of viral RNA into host DNA by reverse transcriptase (RT). RT is composed of a DNA polymerase domain and a ribonuclease H domain (RNase H) [48]. Messore et al. [49] reported the synthesis of specific inhibitory analogs to target RNase H. As shown in Scheme 11, the synthesis started with the aldol condensation of substituted benzaldehyde 46 with acetone, then its reaction with TosMIC to furnish pyrrole derivative 47. Compound 47 coupled with substituted aryl or alkyl chloride for the N-substitution of pyrrole ring to form compound 48. Diethyl oxalate was coupled with intermediate 48 for aldol condensation product 49. Then, hydrazine reacted with compound 49 for the formation of the pyrazole ring of product 50. The ester of product 50 was hydrolyzed under a basic condition to yield carboxylic acid for a separate analysis of antiviral activity.
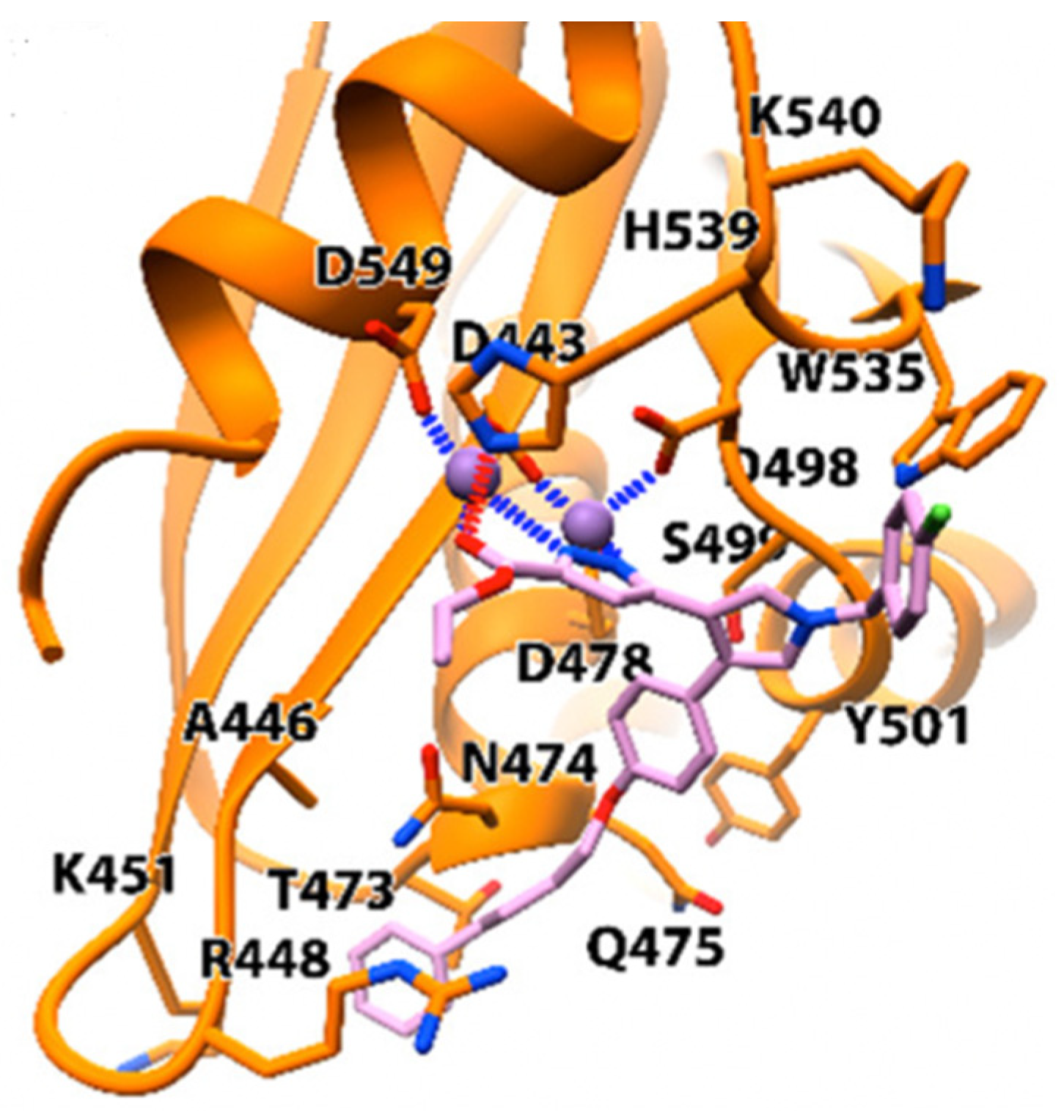
In in vitro screening of RNase H inhibitory activity, compound 50 showed better inhibitory activity of 0.27 ± 0.05 μM compared to its ester version 50 (17.8 ± 1.2). However, compound 50 had a better effective concentration (EC50), higher cytotoxicity, and selectivity index; hence, compound 50 was chosen for analysis in the docking study, as shown in Figure 12. The nitrogen of pyrazole ring and ester moiety formed ligands with two magnesium ions in the core of RNase H ligands. The ester moiety also had hydrogen interaction with H539, while the substituted benzyl group was possibly linked with W535 and K540 by π–cation interaction. The phenyl moiety was linked with Y501, S499, Q475, E478, N474, and Q475. The scaffold exhibited high potency with the HIV-1 RT pocket and could be optimized for drug development.
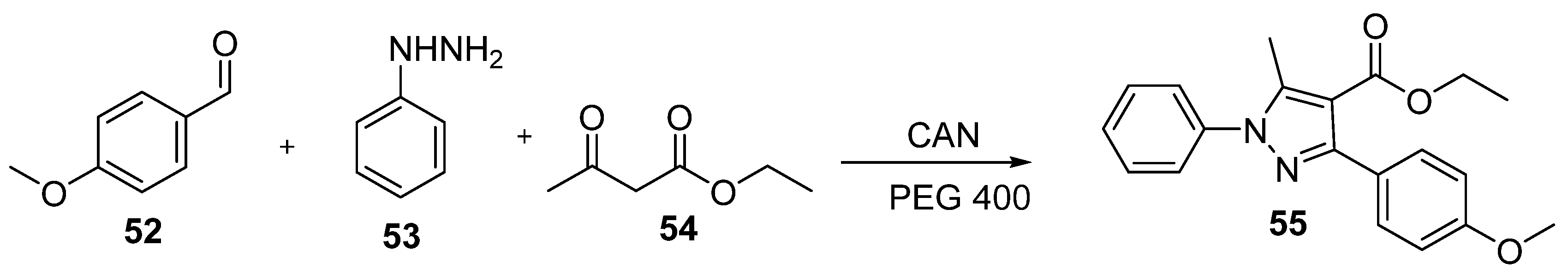
Annually, the influenza virus causes respiratory infection, known as flu, in millions of people in the United States and worldwide. A vaccine is available to prevent the massive outbreak of the disease, but it does not efficiently protect children and elders from the infection. Hence, inhibitors were continuously developed for a long-term battle against influenza viruses. Among many molecular drug targets of influenza viruses, only neuraminidase (NA) inhibitors were approved for treatment [50,51]. Meng et al. [52] synthesized pyrazole derivatives as neuraminidase inhibitors. As shown in Scheme 12, 4-methoxy substituted benzaldehyde 52, phenylhydrazine 53, and ethyl acetoacetate 54 were allowed to react in the presence of cerium (IV) ammonium nitrate (CAN) and PEG 400 in a one-pot synthesis. The reaction time and yield were optimized with CAN compared to other catalysts to yield pyrazole derivative 55.
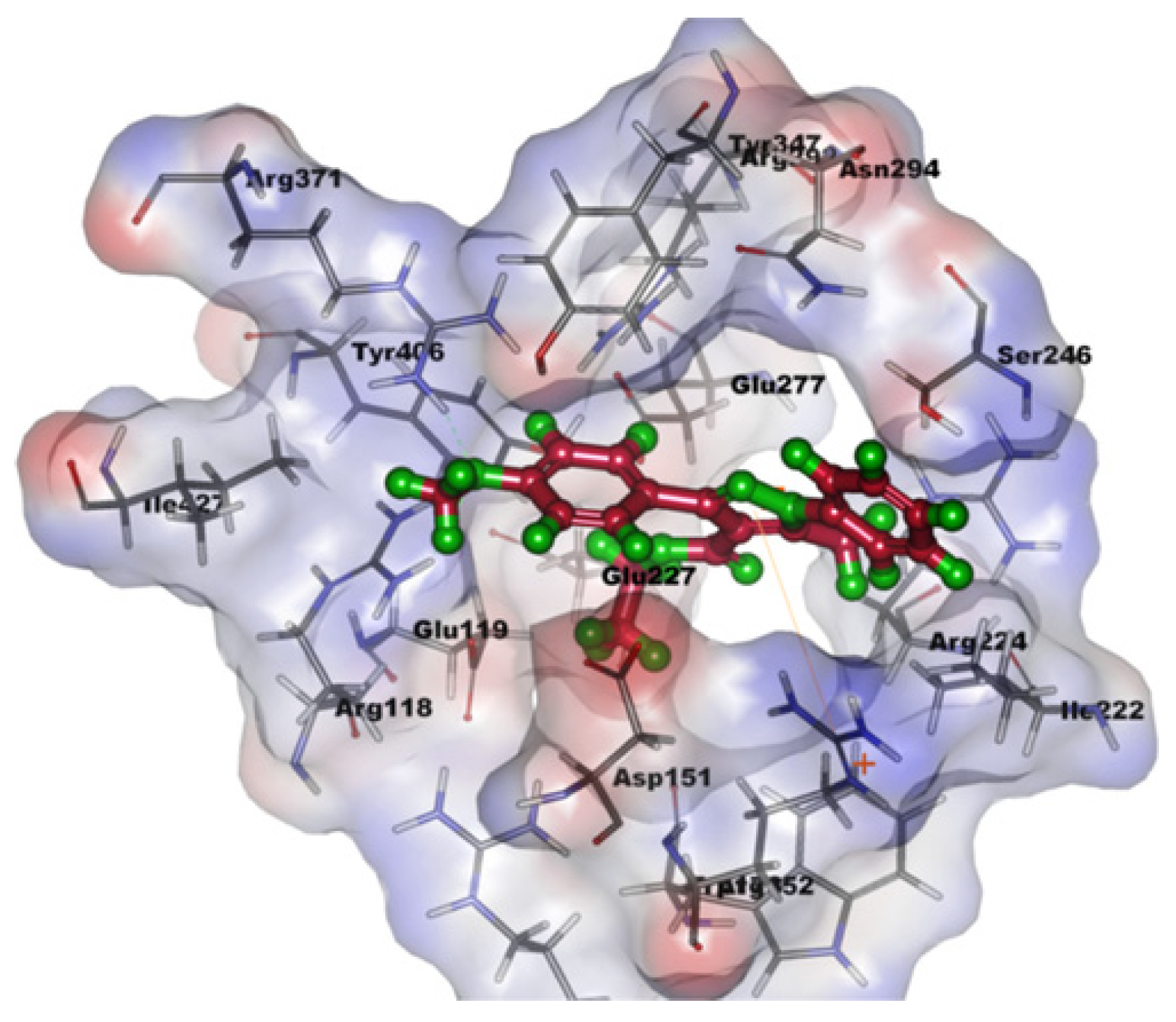
As shown in Table 7, compound 55 has a high potency to inhibit the replication of viruses with an IC50 of 5.4 ± 0.34 μM and a high selectivity index of 102.20 μM in the assay with the influenza A (H1N1) virus. Additionally, the analog also showed great potency (1.32 ± 0.06 μM) against neuraminidase. As shown in Figure 13, the molecular docking of compound 55 in the 3D structure of neuraminidase (NA) was observed. Compound 55 can fit into the binding pocket of neuraminidase and form two hydrogen bonds with residues Arg371 and Arg118 from the methoxy of the compound. The aromatic ring also stabilized with Arg152 through π–cation interaction. With all of the interacted factors in the binding site of NA, compound 55 efficiently exerted inhibitory activity against the targeted protein.
Hepatitis B is another viral infection that is caused by a member of the Hepadnaviridae family. Hepatitis B virus (HBV) has infected millions of people worldwide and the deaths of more than 700,000 people per year. Its replication process is similar to that of RNA retrovirus using the reverse transcription of RNA progenome. Jia et al. [53] developed non-nucleoside HBV inhibitors to combat HBV. As shown in Scheme 13, diethyl carbonate 56 coupled with 4-methylpentan-2-one to produce diketone 57. Compound 57 reacted with 1,1-dimethoxy-N, N-dimethylmethanamine for α-substitution to furnish compound 58. The substituted hydrazine condensed with compound 58 for pyrazole ring formation of compound 59. Pyrazole derivative 60 was produced via base hydrolysis of compound 59. The carboxylic acid of 60 continued to react with an aryl-substituted amine under the coupling condition to form amide 61.
As shown in Table 8, compound 61 demonstrated excellent cytotoxicity in the SAR study. Comparing the inhibitory activity, compound 61 seemed to inhibit viral proteins (HBeAg) of HBV more effectively with a lower IC50 (2.22 μM) and corresponding selectivity index (37.69 μM) than the surface antigen (HBsAg). According to the study, compound 61 showed moderate inhibitory activity of approximately 50% against HBV DNA replication. As a result, compound 61 needs further investigation to optimize its inhibitory properties in targeting viral proteins and inhibiting the fusing of viral DNA into host cells.
6. Quinoline
Quinoline is a heterocycle made of benzene and a fused pyridine ring found in shale oil, coal, and petroleum. This heterocycle attracted attention in organic chemistry for its feasible synthesis and availability for substitution modifications. Quinoline derivatives have been employed in various applications, including those in the pharmaceutical, bioorganic, and industrial chemistry fields [54]. Quinoline skeletons exhibited a broad range of antiviral activities and were submitted for potential clinical applications.
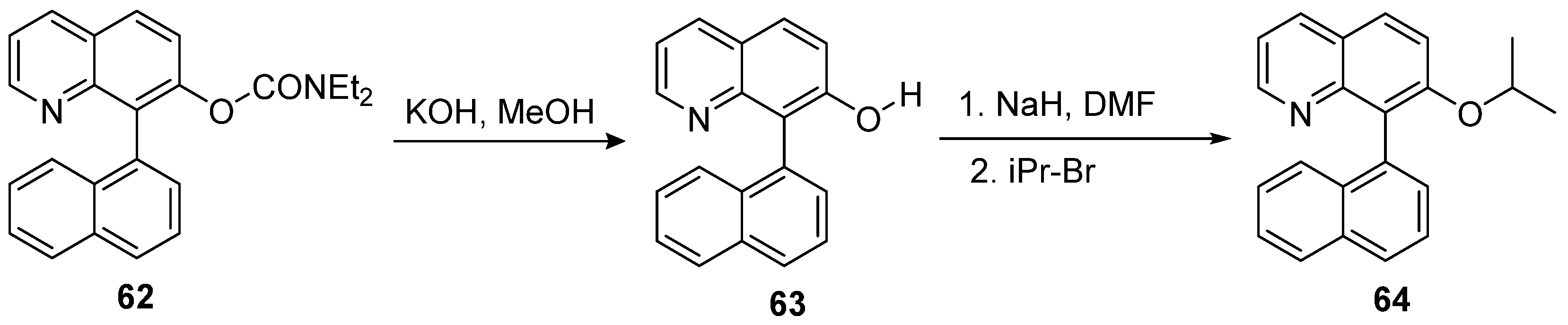
With the progress in developing inhibitors at the RNase H of HIV RT, quinoline scaffolds also joined the race to combat HIV-1. While HIV RT synthesizes DNA from viral RNA, RNase H plays a crucial role in degrading RNA to form double-stranded DNA. Overacker et al. [55] synthesized quinoline derivatives to target HIV RNase H. As shown in Scheme 14, the methanolysis of carbamate moiety in compound 62 took place to afford quinol derivative 63, which then underwent Williamson ether synthesis, converting to isopropyl ether to furnish product 64.
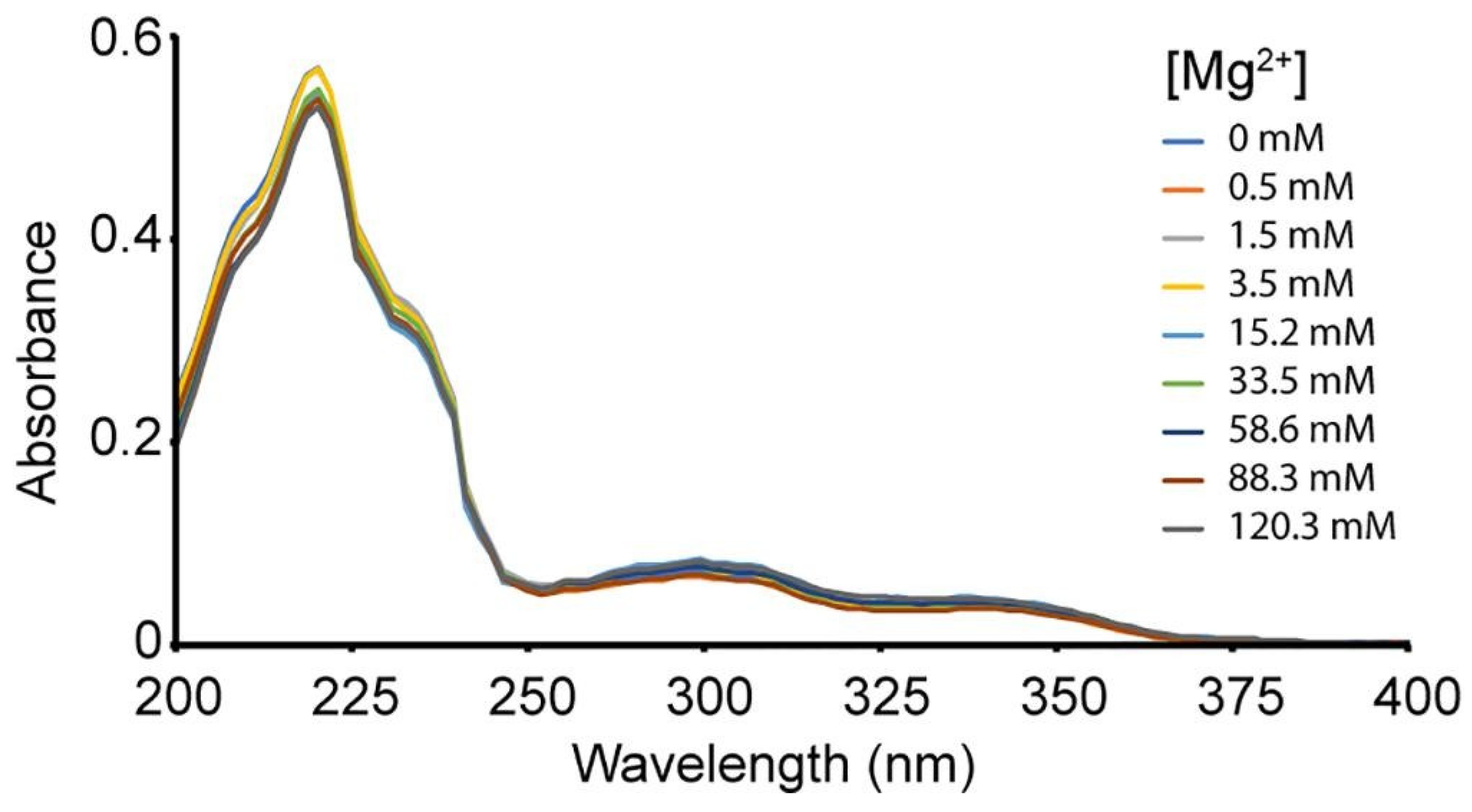
As shown in Table 9, quinoline derivative 64 required a low concentration for antiviral activity and a high concentration for cytotoxicity in the in vitro infectivity assay with pseudoviruses. The compound also had a high selectivity index to inhibit the growth of HIV. For the mode of action, the compound showed better inhibition against the HIV-1 RNase H enzyme compared to weak inhibition (>100 μM) in HIV-1 integrase and HIV-1 RT. As shown in Figure 14, no changes could be detected from the UV spectra, indicating that compound 64 did not chelate with Mg2+ ion in RNase H’s core despite varying concentrations. The structure possibly acted differently in a cell environment or worked as a non-competitive inhibitor of RNase H. This compound was a lead compound for further modification to target RNase H.
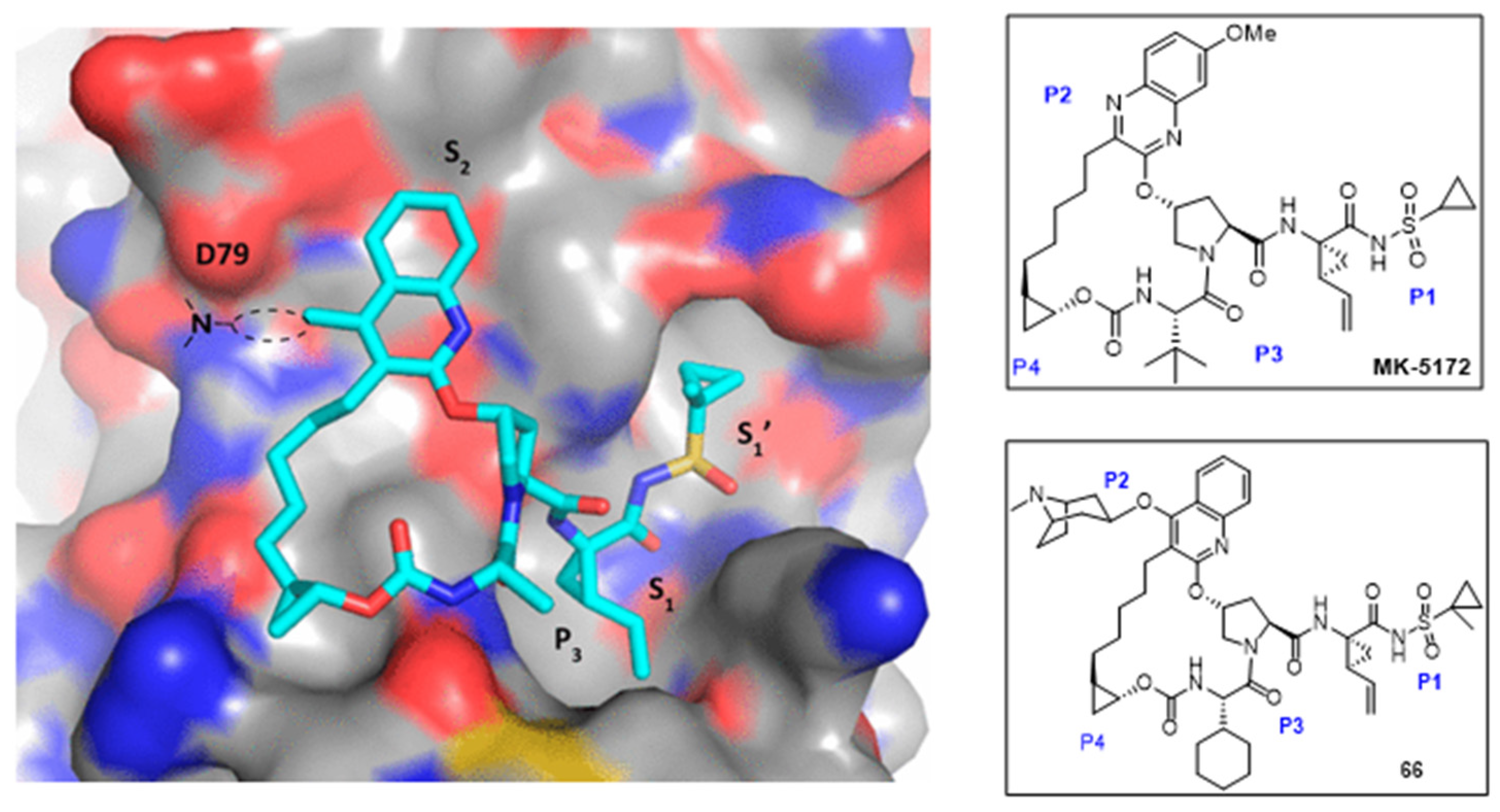
Due to the rapid emergence of hepatitis C virus (HCV) drug resistance, Shah et al. [56] reported the synthesis of quinoline derivatives as HCV drug candidates to target the NS3/4a protease, which plays crucial roles in processing viral protein and replicating viral RNA. It also inhibits the production of interferons that can enhance the immune system against viral infection [57]. The new scaffold was developed based on previous clinical candidate MK-5172 (Figure 15), with a quinoline moiety instead of quinoxaline. As shown in Scheme 15, compound 65 was synthesized by linking the amine derivative with quinoline via five steps. The hydroxyl group was protected using N-Boc-4-hydroxypiperidine before the methyl ester was hydrolyzed and coupled with an amine-acyl sulfonamide chain. The N-Boc-piperidine was then deprotected for the addition of bicyclic piperidine to produce product 66.
Compound 66 exhibited improved potency against HCV genotype 3a and A165 mutants compared to reference MK-5172 and had a moderate pharmacokinetic profile in rats in the SAR study. Compared to MK-5172, the replacement of quinoxaline for quinoline in compound 66 enhanced its interaction with the binding pocket HCV NS3 protease. As shown in Figure 15, the substitution at C-4 of quinoline mainly improved the interaction with D79. The replacement of t-butyl with cyclohexane near P3 or modifying cyclopropyl group seemed to not impact the activity of the scaffold in the HCV NS3 protease binding pocket.
Wang et al. [58] reported the synthesis of indoloquinoline or quindoline derivatives as potential anti-influenza A agents to counter drug resistance of viral strains. As shown in Scheme 16, the 11-chloroquindoline 67 reacted with benzene-1,2-diamine to produce compound 68. In the study, benzene-1,2-diamine could be changed to 1,3 and 1,4-diamine, which then further affected the position of carboxyphenylboronic acid for different analogs of the scaffold. The final product, compound 69, was produced by the coupling reaction of compound 68 and 2-carboxyphenylboronic acid.
Compound 69 exhibited superior properties compared to other derivatives in this scaffold with low cytotoxicity. As shown in Figure 16, the plaque reduction assay showed that plaque development was effectively inhibited, indicating that the compound could inhibit the replication of influenza H1N1 and H3N1. In the confocal imaging, compound 69 was detected in the cytoplasm to target viral neuraminidase and prevent the import of viruses. Compound 69 could interfere with cellular signaling pathways that are essential for viral replication and improve the survival rate of the mouse model in a histopathology study. The quindoline with boronic acid possessed broad anti-influenza A activity for future references and applications.
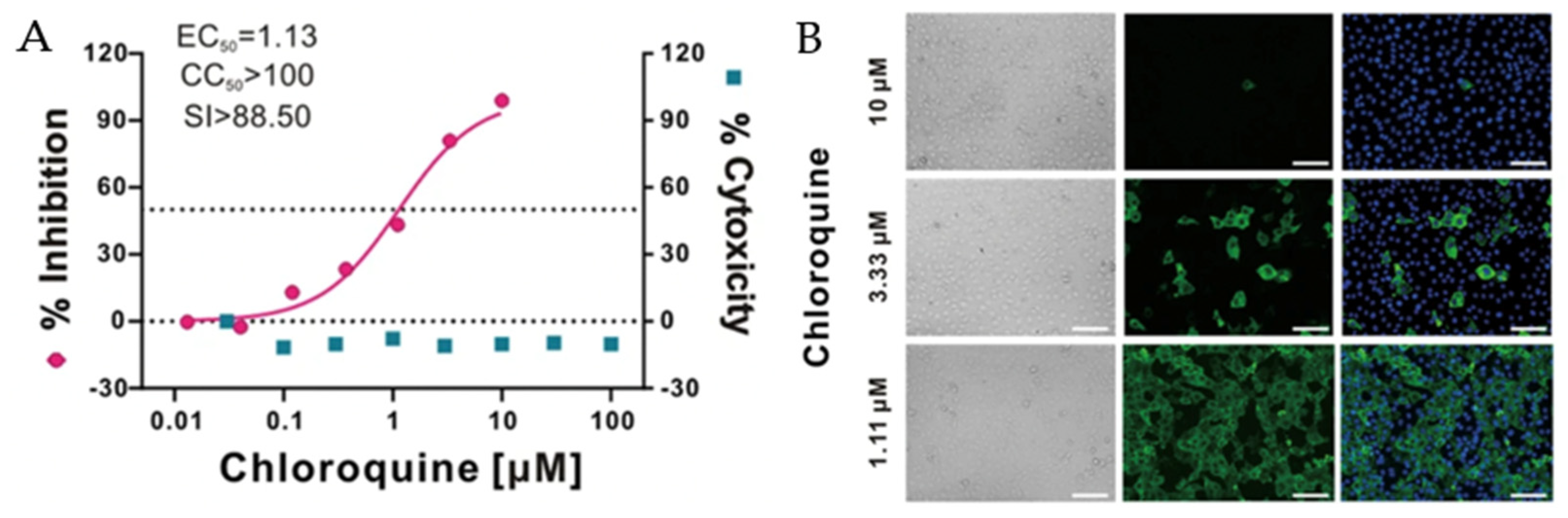
In the early study of COVID-19, besides Remdesivir®, Chloroquine®, an anti-malarial drug (Figure 17), exhibited antiviral activity against 2019-n-CoVs infection in vitro. As shown in Figure 18, Chloroquine® could effectively inhibit viral infection at a low concentration and with good cytotoxicity compared to other FDA-approved antiviral agents. Additionally, Chloroquine® also effectively reduced viral copies with 10 μM after 48 h post-infection in the nucleoprotein through immunofluorescence assay [43].
As shown in Figure 18, Chloroquine showed its effectiveness in blocking virus infection at EC50 = 1.13 µM concentration with a high selectivity index (SI). The immunofluorescence assay also confirmed the efficiency of Chloroquine in inhibiting viruses, as the viral nucleoproteins were eliminated completely with 10 µM. Together with Chloroquine®, Hydroxychloroquine® was also tested for its anti-2019-nCoV activity. Hydroxychloroquine possessed less toxicity in cells compared to Chloroquine® with anti-inflammatory properties [59]. However, both Chloroquine® and Hydroxychloroquine® have side effects, interfering with lysosomal activity and causing cardiac and skeletal muscle problems [60].
7. Conclusions
Nitrogen-containing heterocycle derivatives have a long history against a broad spectrum of viral agents. Their structures are similar to the biological components that are vital for the infection mechanism and replication of viruses. Incorporating nitrogen-containing heterocycles enhances the binding affinity of the scaffolds by increasing interaction with residues and matching with the pocket shape to deactivate the function of targeted enzymes. This article summarized the history, synthesis, and antiviral applications of various nitrogen-containing heterocycle scaffolds, such as indoles, pyrroles, pyrimidines, pyrazoles, and quinolines. The scaffolds were applied to a variety of viruses, ranging from HIV and HCV/HBV to VZV/HSV, SARS-CoV, and influenza viruses. Some approved scaffolds exhibit multiple functions in targeting different viruses, similar to applying Remdesivir®, Chloroquine®, and Hydrochloroquine® for the recent SARS-Co-V 2 viruses. Most scaffolds exhibited potential antiviral activity that could lead to further optimization and in vivo studies for drug development. For example, for HCV treatment, FDA-approved drugs such as Grazoprevir, Voxilaprevir, and Glecaprevir were modified from scaffold 66. Viruses have high rates of emergence that demand continual drug discovery of new antiviral agents. Research on nitrogen heterocycles will progressively expand for future drug development.
Funding
This work was funded in part by a Georgia State University training grant (NIH T34GM131939) to support the undergraduate student TNT.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
M.H. expresses gratitude for the support received through the Georgia State University Brains, Behavior Seed Grant, Atlanta Clinical and Translational Science Institute Healthcare Innovation Seed Grant, and Georgia Research Alliance Ventures Phase 1 Grant for the support. M.H. and T.N.T. are also thankful for GSU Chemistry Department support.
Conflicts of Interest
The authors have no conflict of interest.
References
- Ghosh, A.K.; Osswald, H.L.; Prato, G. Recent Progress in the Development of HIV-1 Protease Inhibitors for the Treatment of HIV/AIDS. J. Med. Chem. 2016, 59, 5172–5208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krammer, F.; Palese, P. Advances in the development of influenza virus vaccines. Nat. Rev. Drug Discov. 2015, 14, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Bolles, M.; Donaldson, E.; Baric, R. SARS-CoV and emergent coronaviruses: Viral determinants of interspecies transmission. Curr. Opin. Virol. 2011, 1, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zou, P.; Wu, F.; Lu, L.; Jiang, S. Development of small-molecule viral inhibitors targeting various stages of the life cycle of emerging and re-emerging viruses. Front. Med. 2017, 11, 449–461. [Google Scholar] [CrossRef] [Green Version]
- Korkmaz, A.; Bursal, E. An in vitro and in silico study on the synthesis and characterization of novel bis(sulfonate) derivatives as tyrosinase and pancreatic lipase inhibitors. J. Mol. Struct. 2022, 1259, 132734. [Google Scholar] [CrossRef]
- Cetin, A.; Bursal, E.; Türkan, F. 2-methylindole analogs as cholinesterases and glutathione S-transferase inhibitors: Synthesis, biological evaluation, molecular docking, and pharmacokinetic studies. Arab. J. Chem. 2021, 14, 103449. [Google Scholar] [CrossRef]
- Zhang, M.Z.; Chen, Q.; Yang, G.F. A review on recent developments of indole-containing antiviral agents. Eur. J. Med. Chem. 2015, 89, 421–441. [Google Scholar] [CrossRef]
- Mohana Roopan, S.; Sompalle, R. Synthetic chemistry of pyrimidines and fused pyrimidines: A review. Synth. Commun. 2016, 46, 645–672. [Google Scholar] [CrossRef]
- Fascio, M.L.; Errea, M.I.; D’Accorso, N.B. Imidazothiazole and related heterocyclic systems. Synthesis, chemical and biological properties. Eur. J. Med. Chem. 2015, 90, 666–683. [Google Scholar] [CrossRef]
- Shalini, K.; Sharma, P.K.; Kumar, N. Imidazole and its biological activities: A review. Der Chem. Sin. 2010, 1, 36–47. [Google Scholar]
- Singh, N.; Pandurangan, A.; Rana, K.; Anand, P.; Ahamad, A.; Tiwari, A.K. Benzimidazole: A short review of their antimicrobial activities. Int. Curr. Pharm. J. 2012, 1, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.; Kant, V. A review on biological activity of imidazole and thiazole moieties and their derivatives. Sci. Int. 2013, 1, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, V.; Gumber, D.; Abbot, V.; Dhiman, S.; Sharma, P. Pyrrole: A resourceful small molecule in key medicinal hetero-aromatics. RSC Adv. 2015, 5, 15233–15266. [Google Scholar] [CrossRef]
- Sharma, V.; Chitranshi, N.; Agarwal, A.K. Significance and biological importance of pyrimidine in the microbial world. Int. J. Med. Chem. 2014, 2014, 1–31. [Google Scholar] [CrossRef] [Green Version]
- Raffa, D.; Maggio, B.; Raimondi, M.V.; Cascioferro, S.; Plescia, F.; Cancemi, G.; Daidone, G. Recent advanced in bioactive systems containing pyrazole fused with a five membered heterocycle. Eur. J. Med. Chem. 2015, 97, 732–746. [Google Scholar] [CrossRef]
- Marella, A.; Tanwar, O.P.; Saha, R.; Ali, M.R.; Srivastava, S.; Akhter, M.; Shaquiquzzaman, M.; Alam, M.M. Quinoline: A versatile heterocyclic. Saudi Pharm. J. 2013, 21, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Sundberg, R.J. Indoles; Academic Press: London, UK; San Diego, CA, USA, 1996. [Google Scholar]
- Hassam, M.; Basson, A.E.; Liotta, D.C.; Morris, L.; van Otterlo, W.A.L.; Pelly, S.C. Novel Cyclopropyl-Indole Derivatives as HIV Non-Nucleoside Reverse Transcriptase Inhibitors. ACS Med. Chem. Lett. 2012, 3, 470–475. [Google Scholar] [CrossRef] [Green Version]
- Lavanchy, D. Evolving epidemiology of hepatitis C virus. Clin. Microbiol. Infect. 2011, 17, 107–115. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Liang, X.; Wang, Y.; Qing, J.; Cao, L.; Shang, L.; Yin, Z. The discovery of indole derivatives as novel hepatitis C virus inhibitors. Eur. J. Med. Chem. 2016, 116, 147–155. [Google Scholar] [CrossRef]
- Chen, J.J.; Gershon, A.A.; Li, Z.; Cowles, R.A.; Gershon, M.D. Varicella zoster virus (VZV) infects and establishes latency in enteric neurons. J. Neurovirol. 2011, 17, 578–589. [Google Scholar] [CrossRef] [Green Version]
- Musella, S.; di Sarno, V.; Ciaglia, T.; Sala, M.; Spensiero, A.; Scala, M.C.; Ostacolo, C.; Andrei, G.; Balzarini, J.; Snoeck, R.; et al. Identification of an indol-based derivative as potent and selective varicella zoster virus (VZV) inhibitor. Eur. J. Med. Chem. 2016, 124, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Topalis, D.; Gillemot, S.; Snoeck, R.; Andrei, G. Thymidine kinase and protein kinase in drug-resistant herpesviruses: Heads of a Lernaean Hydra. Drug Resist. Updat. 2018, 37, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Thanigaimalai, P.; Konno, S.; Yamamoto, T.; Koiwai, Y.; Taguchi, A.; Takayama, K.; Yakushiji, F.; Akaji, K.; Chen, S.E.; Naser-Tavakolian, A.; et al. Development of potent dipeptide-type SARS-CoV 3CL protease inhibitors with novel P3 scaffolds: Design, synthesis, biological evaluation, and docking studies. Eur. J. Med. Chem. 2013, 68, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.J.; Jha, R.K.; Amera, G.M.; Jain, M.; Singh, E.; Pathak, A.; Singh, R.P.; Muthukumaran, J.; Singh, A.K. Targeting SARS-CoV-2: A systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2′-O-ribose methyltransferase. J. Biomol. Struct. Dyn. 2021, 39, 2679–2692. [Google Scholar] [CrossRef] [Green Version]
- Thanigaimalai, P.; Konno, S.; Yamamoto, T.; Koiwai, Y.; Taguchi, A.; Takayama, K.; Yakushiji, F.; Akaji, K.; Kiso, Y.; Kawasaki, Y.; et al. Design, synthesis, and biological evaluation of novel dipeptide-type SARS-CoV 3CL protease inhibitors: Structure-activity relationship study. Eur. J. Med. Chem. 2013, 65, 436–447. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.-M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.; Bean, G. The Chemistry of Pyrroles; Academic Press: London, UK, 1997. [Google Scholar]
- Curreli, F.; Kwon, Y.D.; Belov, D.S.; Ramesh, R.R.; Kurkin, A.V.; Altieri, A.; Kwong, P.D.; Debnath, A.K. Synthesis, Antiviral Potency, in Vitro ADMET, and X-ray Structure of Potent CD4 Mimics as Entry Inhibitors That Target the Phe43 Cavity of HIV-1 gp120. J. Med. Chem. 2017, 60, 3124–3153. [Google Scholar] [CrossRef]
- Checkley, M.A.; Luttge, B.G.; Freed, E.O. HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J. Mol. Biol. 2011, 410, 582–608. [Google Scholar] [CrossRef] [Green Version]
- Ezquerra, J.; Pedregal, C.; Rubio, A.; Valenciano, J.; Navio, J.L.G.; Alvarez-Builla, J.; Vaquero, J.J. General method for the synthesis of 5-arylpyrrole-2-carboxylic acids. Tetrahedron Lett. 1993, 34, 6317–6320. [Google Scholar] [CrossRef] [Green Version]
- Kinchington, P.R.; Leger, A.J.; Guedon, J.M.; Hendricks, R.L. Herpes simplex virus and varicella zoster virus, the house guests who never leave. Herpesviridae 2012, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Hilmy, K.M.; Soliman, D.H.; Shahin, E.B.; Abd Alhameed, R. Synthesis and molecular modeling study of novel pyrrole Schiff Bases as anti-HSV-1 agents. Life Sci. J. 2012, 9, 736–745. [Google Scholar]
- Lin, M.I.; Su, B.H.; Lee, C.H.; Wang, S.T.; Wu, W.C.; Dangate, P.; Wang, S.Y.; Huang, W.I.; Cheng, T.J.; Lin, O.A.; et al. Synthesis and inhibitory effects of novel pyrimido-pyrrolo-quinoxalinedione analogues targeting nucleoproteins of influenza A virus H1N1. Eur. J. Med. Chem. 2015, 102, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Turrell, L.; Lyall, J.W.; Tiley, L.S.; Fodor, E.; Vreede, F.T. The role and assembly mechanism of nucleoprotein in influenza A virus ribonucleoprotein complexes. Nat. Commun. 2013, 4, 1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.J. The Pyrimidines; John Wiley & Sons: New York, NY, USA, 1994; pp. 1–8. [Google Scholar]
- Dinakaran, V.S.; Bomma, B.; Srinivasan, K.K. Fused pyrimidines: The heterocycle of diverse biological and pharmacological significance. Der Pharma Chem. 2012, 4, 255–265. [Google Scholar]
- Malancona, S.; Mori, M.; Fezzardi, P.; Santoriello, M.; Basta, A.; Nibbio, M.; Kovalenko, L.; Speziale, R.; Battista, M.R.; Cellucci, A.; et al. 5,6-Dihydroxypyrimidine Scaffold to Target HIV-1 Nucleocapsid Protein. ACS Med. Chem. Lett. 2020, 11, 766–772. [Google Scholar] [CrossRef]
- Levin, J.G.; Mitra, M.; Mascarenhas, A.; Musier-Forsyth, K. Role of HIV-1 nucleocapsid protein in HIV-1 reverse transcription. RNA Biol. 2010, 7, 754–774. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, M.S.; Sayed, A.I.; Khedr, M.A.; Soror, S.H. Design, synthesis, assessment, and molecular docking of novel pyrrolopyrimidine (7-deazapurine) derivatives as non-nucleoside hepatitis C virus NS5B polymerase inhibitors. Bioorg. Med. Chem. 2016, 24, 2146–2157. [Google Scholar] [CrossRef]
- Zhao, C.; Wang, Y.; Ma, S. Recent advances on the synthesis of hepatitis C virus NS5B RNA-dependent RNA-polymerase inhibitors. Eur. J. Med. Chem. 2015, 102, 188–214. [Google Scholar] [CrossRef]
- Mohamed, S.F.; Flefel, E.M.; Amr Ael, G.; Abd El-Shafy, D.N. Anti-HSV-1 activity and mechanism of action of some new synthesized substituted pyrimidine, thiopyrimidine and thiazolopyrimidine derivatives. Eur. J. Med. Chem. 2010, 45, 1494–1501. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, R. Structural Basis of the Potential Binding Mechanism of Remdesivir to SARS-CoV-2 RNA-Dependent RNA Polymerase. J. Phys. Chem. B 2020, 124, 6955–6962. [Google Scholar] [CrossRef] [PubMed]
- Grein, J.; Ohmagari, N.; Shin, D.; Diaz, G.; Asperges, E.; Castagna, A.; Feldt, T.; Green, G.; Green, M.L.; Lescure, F.X.; et al. Compassionate Use of Remdesivir for Patients with Severe COVID-19. N. Engl. J. Med. 2020, 382, 2327–2336. [Google Scholar] [CrossRef] [PubMed]
- Ansari, A.; Ali, A.; Asif, M.; Shamsuzzaman. Review: Biologically active pyrazole derivatives. New J. Chem. 2017, 41, 16–41. [Google Scholar] [CrossRef]
- Eicher, T.; Hauptmann, S.; Speicher, A. The Chemistry of Heterocycles: Structure, Reactions, Syntheses, and Applications; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Christen, M.T.; Menon, L.; Myshakina, N.S.; Ahn, J.; Parniak, M.A.; Ishima, R. Structural basis of the allosteric inhibitor interaction on the HIV-1 reverse transcriptase RNase H domain. Chem. Biol. Drug Des. 2012, 80, 706–716. [Google Scholar] [CrossRef] [Green Version]
- Messore, A.; Corona, A.; Madia, V.N.; Saccoliti, F.; Tudino, V.; De Leo, A.; Scipione, L.; De Vita, D.; Amendola, G.; Di Maro, S.; et al. Pyrrolyl Pyrazoles as Non-Diketo Acid Inhibitors of the HIV-1 Ribonuclease H Function of Reverse Transcriptase. ACS Med. Chem. Lett. 2020, 11, 798–805. [Google Scholar] [CrossRef]
- Blyth, C.C.; Jacoby, P.; Effler, P.V.; Kelly, H.; Smith, D.W.; Robins, C.; Willis, G.A.; Levy, A.; Keil, A.D.; Richmond, P.C. Effectiveness of trivalent flu vaccine in healthy young children. Pediatrics 2014, 133, e1218–e1225. [Google Scholar] [CrossRef] [Green Version]
- David, J.K.; Amber, F. Extremely low vaccine effectiveness against influenza H3N2 in the elderly during the 2012/2013 flu season. J. Infect. Dev. Ctries 2013, 7, 299–301. [Google Scholar] [CrossRef]
- Meng, F.J.; Sun, T.; Dong, W.Z.; Li, M.H.; Tuo, Z.Z. Discovery of Novel Pyrazole Derivatives as Potent Neuraminidase Inhibitors against Influenza H1N1 Virus. Arch. Pharm. 2016, 349, 168–174. [Google Scholar] [CrossRef]
- Jia, H.; Bai, F.; Liu, N.; Liang, X.; Zhan, P.; Ma, C.; Jiang, X.; Liu, X. Design, synthesis and evaluation of pyrazole derivatives as non-nucleoside hepatitis B virus inhibitors. Eur. J. Med. Chem. 2016, 123, 202–210. [Google Scholar] [CrossRef]
- Prajapati, S.M.; Patel, K.D.; Vekariya, R.H.; Panchal, S.N.; Patel, H.D. Recent advances in the synthesis of quinolines: A review. RSC Adv. 2014, 4, 24463–24476. [Google Scholar] [CrossRef]
- Overacker, R.D.; Banerjee, S.; Neuhaus, G.F.; Milicevic Sephton, S.; Herrmann, A.; Strother, J.A.; Brack-Werner, R.; Blakemore, P.R.; Loesgen, S. Biological evaluation of molecules of the azaBINOL class as antiviral agents: Inhibition of HIV-1 RNase H activity by 7-isopropoxy-8-(naphth-1-yl)quinoline. Bioorg. Med. Chem. 2019, 27, 3595–3604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, U.; Jayne, C.; Chackalamannil, S.; Velázquez, F.; Guo, Z.; Buevich, A.; Howe, J.A.; Chase, R.; Soriano, A.; Agrawal, S.; et al. Novel Quinoline-Based P2–P4 Macrocyclic Derivatives As Pan-Genotypic HCV NS3/4a Protease Inhibitors. ACS Med. Chem. Lett. 2014, 5, 264–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiering, N.; D’Arcy, A.; Villard, F.; Simic, O.; Kamke, M.; Monnet, G.; Hassiepen, U.; Svergun, D.I.; Pulfer, R.; Eder, J.; et al. A macrocyclic HCV NS3/4A protease inhibitor interacts with protease and helicase residues in the complex with its full-length target. Proc. Natl. Acad. Sci. USA 2011, 108, 21052–21056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Yin, R.; Zhang, M.; Yu, R.; Hao, C.; Zhang, L.; Jiang, T. Boronic Acid Modifications Enhance the Anti-Influenza A Virus Activities of Novel Quindoline Derivatives. J. Med. Chem. 2017, 60, 2840–2852. [Google Scholar] [CrossRef]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Pereira, B.B. Challenges and cares to promote rational use of chloroquine and hydroxychloroquine in the management of coronavirus disease 2019 (COVID-19) pandemic: A timely review. J. Toxicol. Environ. Health B Crit. Rev. 2020, 23, 177–181. [Google Scholar] [CrossRef] [Green Version]
Scheme 1.
The synthesis of non-nucleoside scaffolds as HIV reverse transcriptase inhibitors.

Figure 1.
In vitro phenotypic assay values and corresponding cytotoxicity values against HIV. The molecular docking study of 5a in the binding site of HIV reverse transcriptase [18].
Figure 1.
In vitro phenotypic assay values and corresponding cytotoxicity values against HIV. The molecular docking study of 5a in the binding site of HIV reverse transcriptase [18].

Scheme 2.
The synthesis of NINS for anti-HVC activity.

Scheme 3.
Synthesis of tryptamine derivative for targeting TK against VZV.

Scheme 4.
The synthesis of dipeptide inhibitor against SAR-CoV 3CLpro [24].
Scheme 4.
The synthesis of dipeptide inhibitor against SAR-CoV 3CLpro [24].

Figure 2.
Molecular docking poses and binding interaction of 17 bound to SARS-CoV 3CLpro [24].
Figure 2.
Molecular docking poses and binding interaction of 17 bound to SARS-CoV 3CLpro [24].

Scheme 5.
The synthesis of pyrrole derivatives as glycoprotein inhibitors of HIV-1.

Figure 3.
(A) Inhibition assay of tested compounds (R) 21a and (S) 21b with the controls NBD-556 and NBD-11021. (B) Infectivity assay of the compounds and controls with CD4-dependent HIV-1ADA [29].
Figure 3.
(A) Inhibition assay of tested compounds (R) 21a and (S) 21b with the controls NBD-556 and NBD-11021. (B) Infectivity assay of the compounds and controls with CD4-dependent HIV-1ADA [29].

Scheme 6.
Synthesis of thymidine kinase inhibitors to target HSV.

Figure 4.
Docking study of 23a and 23d in the binding site of HSV-1 thymidine kinase [33].
Figure 4.
Docking study of 23a and 23d in the binding site of HSV-1 thymidine kinase [33].

Scheme 7.
The synthesis of pyrimido-pyrrolo-quinoxalinedione for inhibiting nucleoprotein of influenza A H1N1 virus.
Scheme 7.
The synthesis of pyrimido-pyrrolo-quinoxalinedione for inhibiting nucleoprotein of influenza A H1N1 virus.

Figure 5.
(a) Chemical structure of nucleozin 3061. (b) The molecular docking study of compound 29 in the influenza A NP [34].
Figure 5.
(a) Chemical structure of nucleozin 3061. (b) The molecular docking study of compound 29 in the influenza A NP [34].

Scheme 8.
Synthesis of nucleocapsid inhibitors as anti-HIV/AIDS activity.

Figure 6.
The molecular interaction of 33c in the HIV nucleocapsid [38].
Figure 6.
The molecular interaction of 33c in the HIV nucleocapsid [38].

Scheme 9.
Synthesis of anti-HCV agents targeting HCV polymerase (NS5B).

Figure 7.
Molecular interaction of compound 39 in the binding site of HCV polymerase [40].
Figure 7.
Molecular interaction of compound 39 in the binding site of HCV polymerase [40].

Figure 8.
Comparison of effective percentages of inhibition against HSV infection among compounds 44, 45, and Acyclovir [42].
Figure 8.
Comparison of effective percentages of inhibition against HSV infection among compounds 44, 45, and Acyclovir [42].

Scheme 10.
Synthesis of pyrimidine derivatives as anti-HSV agents.

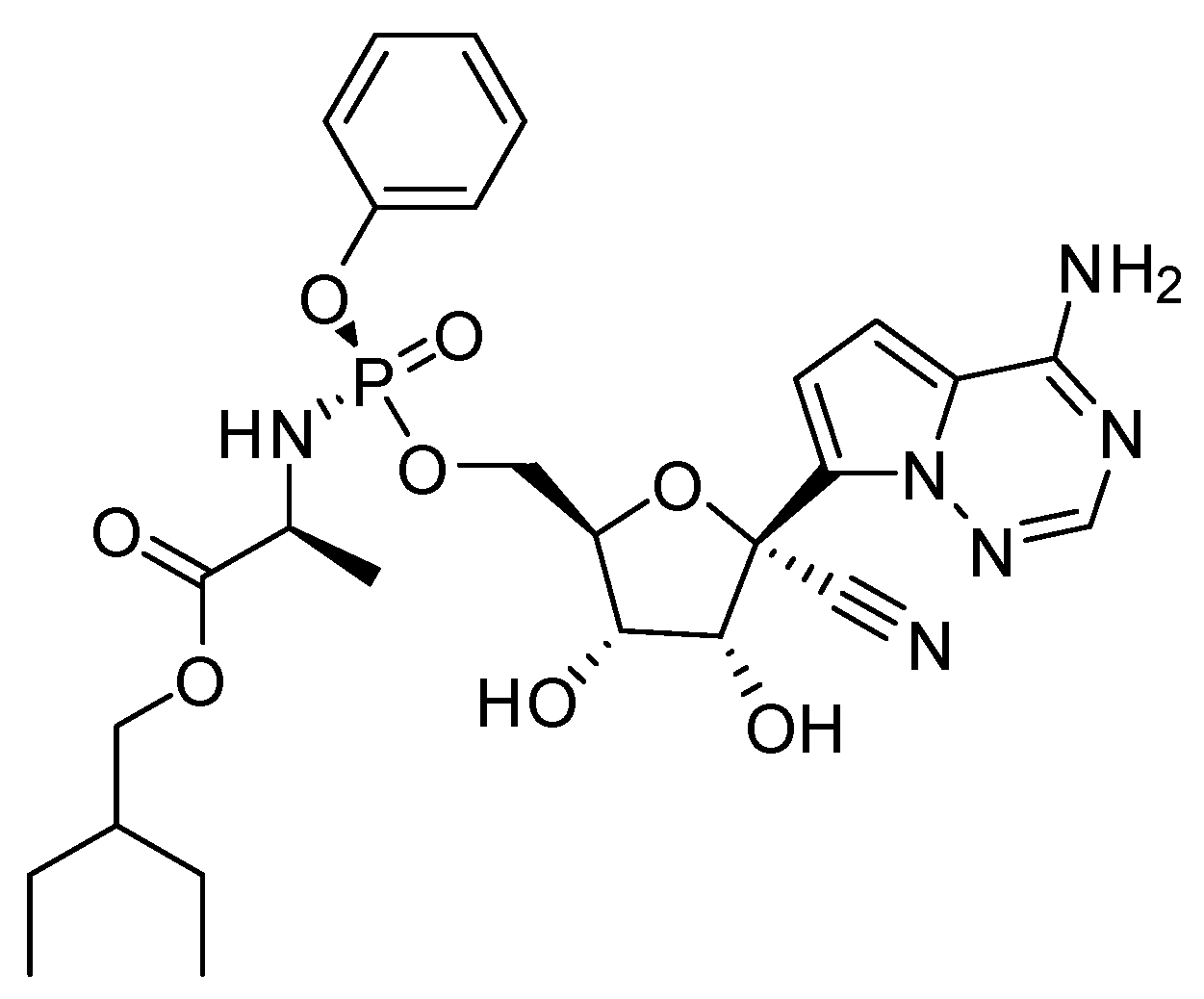
Figure 9.
The molecular structure of Remdesivir®.

Figure 10.
(A) Percentage of inhibition measured by qRT-PCR and cytotoxicity of Remdesivir® when used to treat SARS-CoV-2-infected Vero E6 cells. (B) Immunofluorescence assay of viral infection with Remdesivir® treatment [43].
Figure 10.
(A) Percentage of inhibition measured by qRT-PCR and cytotoxicity of Remdesivir® when used to treat SARS-CoV-2-infected Vero E6 cells. (B) Immunofluorescence assay of viral infection with Remdesivir® treatment [43].

Figure 11.
(A) The binding interaction of ATP with COVID-19 NSP12. (B) The binding interaction of RemTP with COVID-19 NSP12. The green sphere represents the Mg2+ ion [44].
Figure 11.
(A) The binding interaction of ATP with COVID-19 NSP12. (B) The binding interaction of RemTP with COVID-19 NSP12. The green sphere represents the Mg2+ ion [44].

Scheme 11.
Synthesis of inhibitors against HIV-1 RNase H.

Figure 12.
The molecular docking study of compound 50 in the HIV-1 RNase H [49].
Figure 12.
The molecular docking study of compound 50 in the HIV-1 RNase H [49].

Scheme 12.
One-pot synthesis of influenza neuraminidase inhibitor.

Figure 13.
Molecular interaction of compound 55 in the binding site of H1N1 neuraminidase [52].
Figure 13.
Molecular interaction of compound 55 in the binding site of H1N1 neuraminidase [52].

Scheme 13.
Synthesis of pyrazole derivatives as anti-HBV agents.

Scheme 14.
Synthesis of quinoline derivative 64 as HIV RNase H inhibitor.

Figure 14.
UV absorbance spectra of compound 64 with different concentrations of Mg2+ ion [55].
Figure 14.
UV absorbance spectra of compound 64 with different concentrations of Mg2+ ion [55].

Scheme 15.
Synthesis of the inhibitor to target HCV NS3/4a protease.

Figure 15.
Visualization of 66 in the HCV NS3 gt-1b protease active site for interaction with subsites [56].
Figure 15.
Visualization of 66 in the HCV NS3 gt-1b protease active site for interaction with subsites [56].

Scheme 16.
Synthesis of quindoline derivative 69 as anti-influenza A agent.

Figure 16.
(A) Plaque reduction assay at different concentrations of 69 with PR8, Cal09, Minnesota influenza strains in MDCK cells. (B) Corresponding plaque number from the assay with three viruses at different concentrations [58].
Figure 16.
(A) Plaque reduction assay at different concentrations of 69 with PR8, Cal09, Minnesota influenza strains in MDCK cells. (B) Corresponding plaque number from the assay with three viruses at different concentrations [58].

Figure 17.
The molecular structure of Chloroquine® [43].
Figure 17.
The molecular structure of Chloroquine® [43].

Figure 18.
(A) Percentage of inhibition measured by qRT-PCR and cytotoxicity of Chloroquine® when treating 2019 n-CoV infected Vero E6 cells. (B) Immunofluorescence assay of viral infection with Chloroquine® treatment [43].
Figure 18.
(A) Percentage of inhibition measured by qRT-PCR and cytotoxicity of Chloroquine® when treating 2019 n-CoV infected Vero E6 cells. (B) Immunofluorescence assay of viral infection with Chloroquine® treatment [43].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The in vitro SAR study of compounds against HCV [20].
Table 1.
The in vitro SAR study of compounds against HCV [20].
| R1 | EC50 (μM) | CC50 (μM) | SI |
|---|---|---|---|
| Ph | 2.04 ± 0.11 | 35.83 ± 0.25 | 17.56 |
| p-F-Ph | 1.02 ± 0.10 | 46.47 ± 0.24 | 45.56 |
| (R) p-F-Ph | 0.72 ± 0.09 | >50 | >69.44 |
| (S) p-F-Ph | 7.12 ± 0.21 | 34.57 ± 0.53 | 4.86 |
| m-F-Ph | 0.92 ± 0.06 | 21.46 ± 0.34 | 23.33 |
| (R) m-F-Ph | 0.74 ± 0.11 | 31.06 ± 0.37 | 41.97 |
| (S) p-F-Ph | 5.87 ± 0.18 | 23.31 ± 0.30 | 3.97 |
| o-F-Ph | 2.79 ± 0.17 | 34.21 ± 0.43 | 12.26 |
Table 2.
Activity of compound 12 against various viral strains [22].
Table 2.
Activity of compound 12 against various viral strains [22].
| Virus Strain | OKA (TK+) | 07-1 (TK−) | YS-R (TK−) | AD 169 (HMCV) | Davis (HMCV) | KOS (HSV-1) | TK- KOS ACVr 1 (HSV-1) | HSV-2 |
|---|---|---|---|---|---|---|---|---|
| EC50 (μM) | 2.5 | 1.7 | 2.1 | >4 | >4 | >100 | >100 | >1002 |
1 TK− KOS ACVr: HSV-1; KOS and thymidine kinase-deficient acyclovir-resistant.
Table 3.
The SAR study of the compounds against HIV-1 in TZM-bl and MT-2 cells [29].
Table 3.
The SAR study of the compounds against HIV-1 in TZM-bl and MT-2 cells [29].
| Compound | TZM-bl Cells | MT-2-Cells | ||
|---|---|---|---|---|
| IC50 (μM) | CC50 (μM) | IC50 (μM) | CC50 (μM) | |
| (S) 21a | ||||
| (R) 21a | ||||
| (S) 21b | ||||
| (R) 21b | ||||
Table 4.
Anti-HSV-1 activity and docking score of analogs 27a–d and Acyclovir [33].
Table 4.
Anti-HSV-1 activity and docking score of analogs 27a–d and Acyclovir [33].
| Compound | Cytotoxicity (μM) | % Reduction in Cytopathic Effect of HSV-1 | Docking Score |
|---|---|---|---|
| ACV | 10−2 | 96 | −12.74 |
| 23a | 10−2 | 99 | −11.30 |
| 23b | 10−2 | 96 | −11.44 |
| 23c | 10−2 | 94 | -8.98 |
| 23d | 10−2 | 97 | −7.32 |
Table 5.
The inhibiting activity of analogs 33 against HIV nucleocapsid and cells [38].
Table 5.
The inhibiting activity of analogs 33 against HIV nucleocapsid and cells [38].
| ID | NC Inhibition IC50 (μM) | BiCycle Wild Type Strain NL4-3 | HeLa | PMBC | ||
|---|---|---|---|---|---|---|
| EC50 (μM) | CC50 (μM) | SI | CC50 (μM) | SI | ||
| 33a | 167 ± 7 | 1.2± 0.7 | 3.6 ± 0.7 | 3 | 2.6 ± 1.7 | 2 |
| 33b | 22 ± 7 | 0.04 ± 0.03 | 3.0 ± 0.1 | 75 | 2.7 ± 0.1 | 75 |
| 33c | 2 ± 1 | 0.1 ± 0.03 | 3.0 ± 0.5 | 30 | 2.5 ± 0.2 | 25 |
| 33d | 4 ± 1 | 0.1 ± 0.05 | 2.1 ± 0.3 | 21 | 5 ± 2 | 50 |
Table 6.
The calculated percentage of inhibition in the plaque reduction assay of compounds 44 and 45 [42].
Table 6.
The calculated percentage of inhibition in the plaque reduction assay of compounds 44 and 45 [42].
| ID | Concentration (μg/mL) | Viral Count (Control) | Viral Count (Extract) | % Inhibition |
|---|---|---|---|---|
| 44 | 2 | 0.84 × 107 | 0.08 × 107 | 90 |
| 5 | 0.84 × 107 | 0 | 100 | |
| 45 | 2 | 0.84 × 107 | 0.08 × 107 | 90 |
| 5 | 0.84 × 107 | 0.04 × 107 | 95 |
Table 7.
The inhibitory activity of compound 55 against influenza H1N1 virus and neuraminidase.
| Compound | CC50 (μM) | Influenza H1N1 Virus | NA Inhibitor IC50 (μM) | |
|---|---|---|---|---|
| IC50 (μM) | SI (CC50/ IC50) (μM) | |||
| 55 | 552.14 ± 187.34 | 5.4 ± 0.34 | 102.20 | 1.32 ± 0.06 |
Table 8.
The SAR study of compound 61 against HBV.
| Compound | CC50 (μM) | HBsAg | HBeAg | ||
|---|---|---|---|---|---|
| IC50 (μM) | SI | IC50 (μM) | SI | ||
| 61 | 83.67 | 24.33 | 3.44 | 2.22 | 37.69 |
Table 9.
The inhibitory activity of compound 64 against different HIV strains and HIV-1 RNase H.
| Compound | HIV-IC50 (μM) | CC50 (μM) TZM-bl | SI (CC50/ IC50) | IC50 (μM) HIV-1 RNase H | ||
|---|---|---|---|---|---|---|
| HXB2 | YU2 | 89.6 | ||||
| 64 | 6.7 ± 0.9 | 8.9 ± 0.6 | 4.7 ± 1.6 | 68.5 ± 17.1 | 14.6 | 46.8 ± 7.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tran, T.N.; Henary, M. Synthesis and Applications of Nitrogen-Containing Heterocycles as Antiviral Agents. Molecules 2022, 27, 2700. https://doi.org/10.3390/molecules27092700
AMA Style
Tran TN, Henary M. Synthesis and Applications of Nitrogen-Containing Heterocycles as Antiviral Agents. Molecules. 2022; 27(9):2700. https://doi.org/10.3390/molecules27092700
Chicago/Turabian StyleTran, Tuyen N., and Maged Henary. 2022. "Synthesis and Applications of Nitrogen-Containing Heterocycles as Antiviral Agents" Molecules 27, no. 9: 2700. https://doi.org/10.3390/molecules27092700