
Potential Anticancer Activity of the Furanocoumarin Derivative Xanthotoxin Isolated from Ammi majus L. Fruits: In Vitro and In Silico Studies

 ,
,  , ,
, ,
Abstract
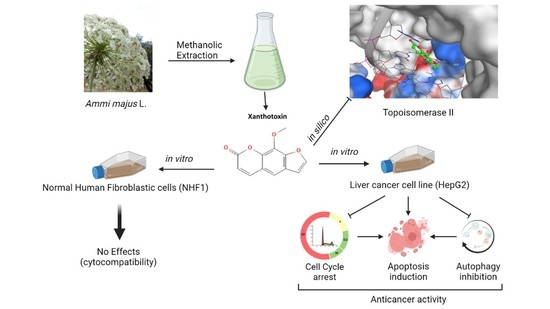
:
1. Introduction
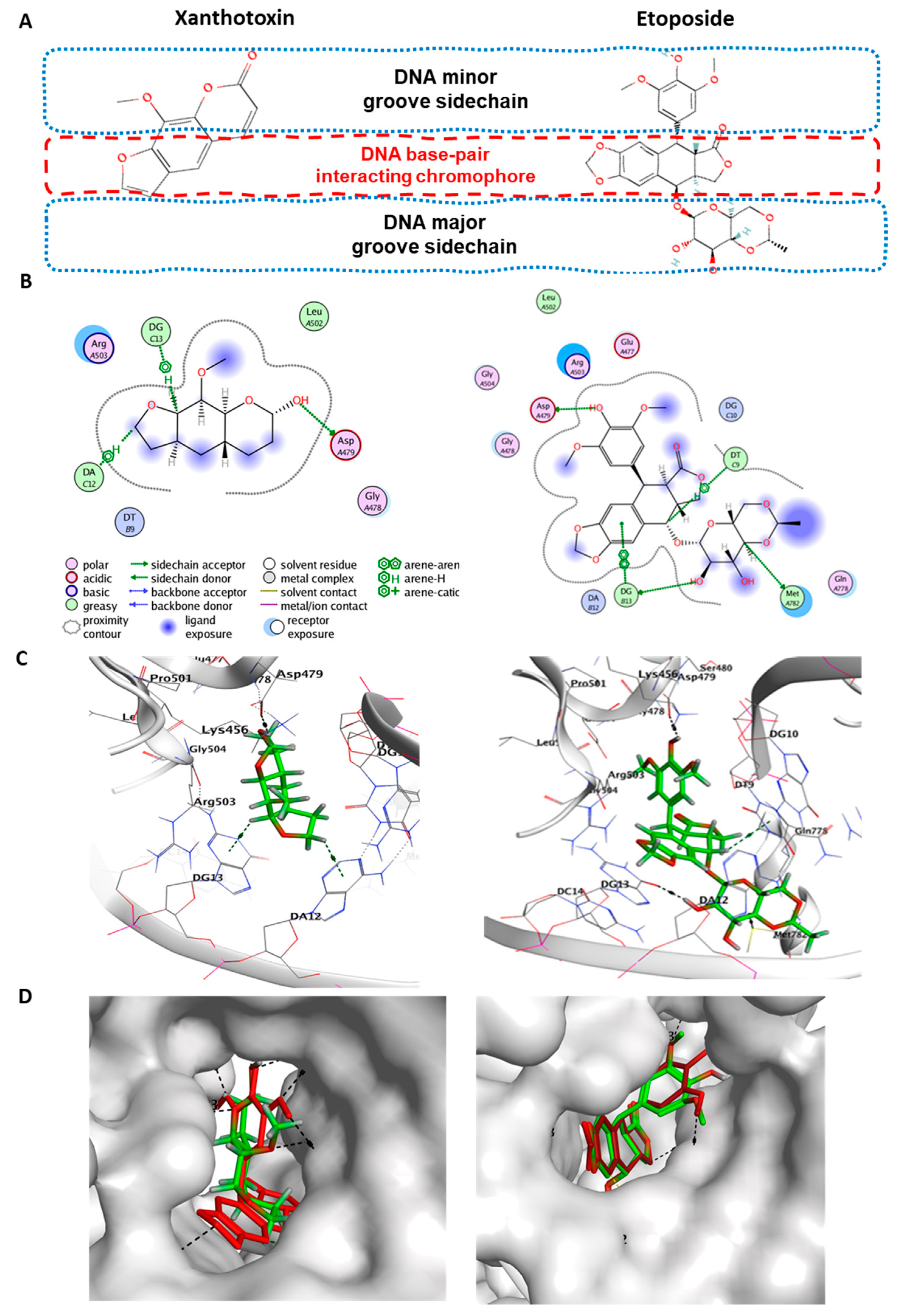
2. Results
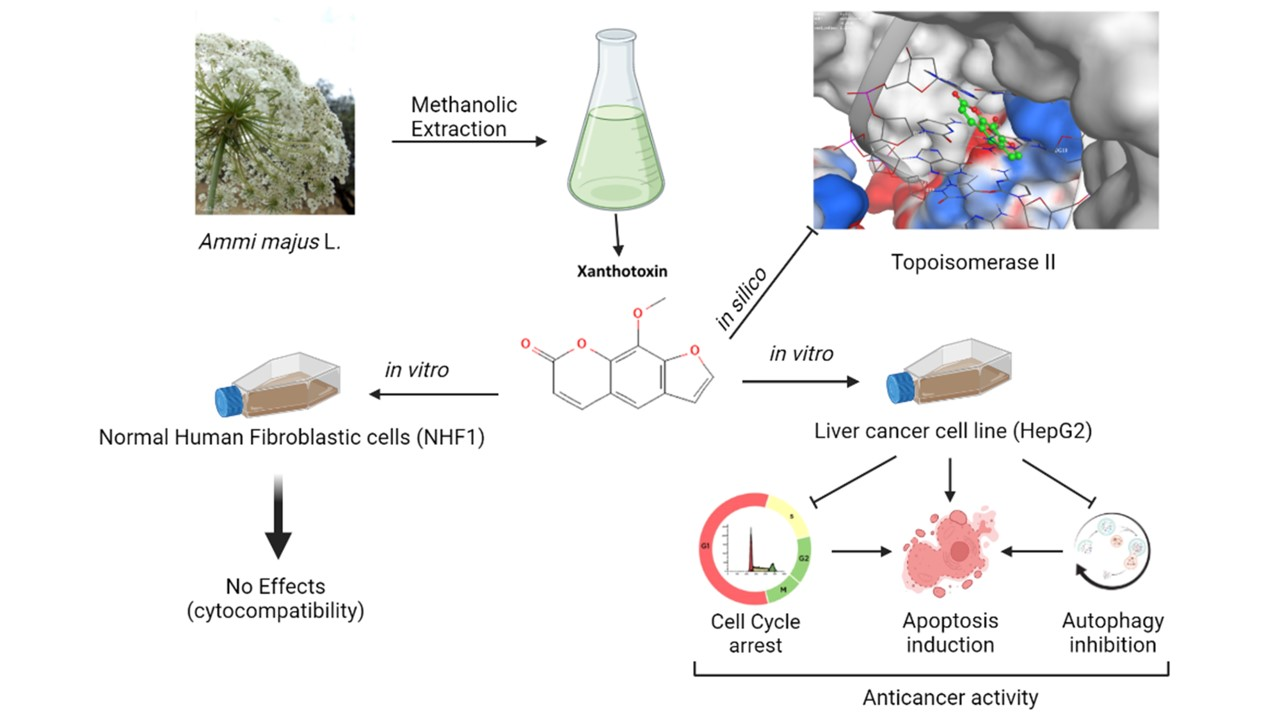
2.1. Cellular Cytocompatibility and Cytotoxicity of AME and Its Fractions
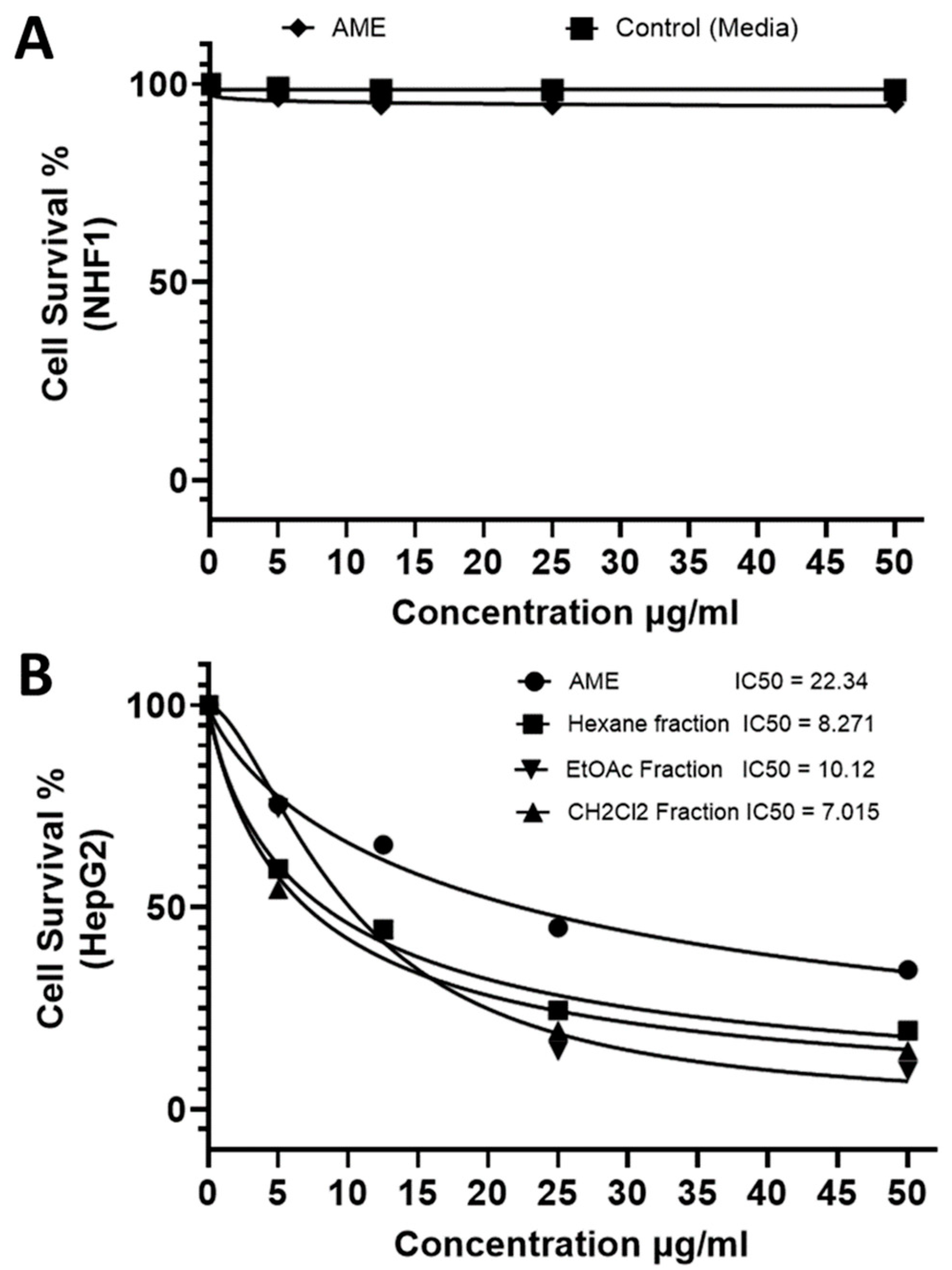
2.2. Identification and Elucidation of the Isolated Compounds
- Xanthotoxin
- β–sitosterol
- Isoarnottinin
- Marmesin
- Imperatorin
- Ammirin
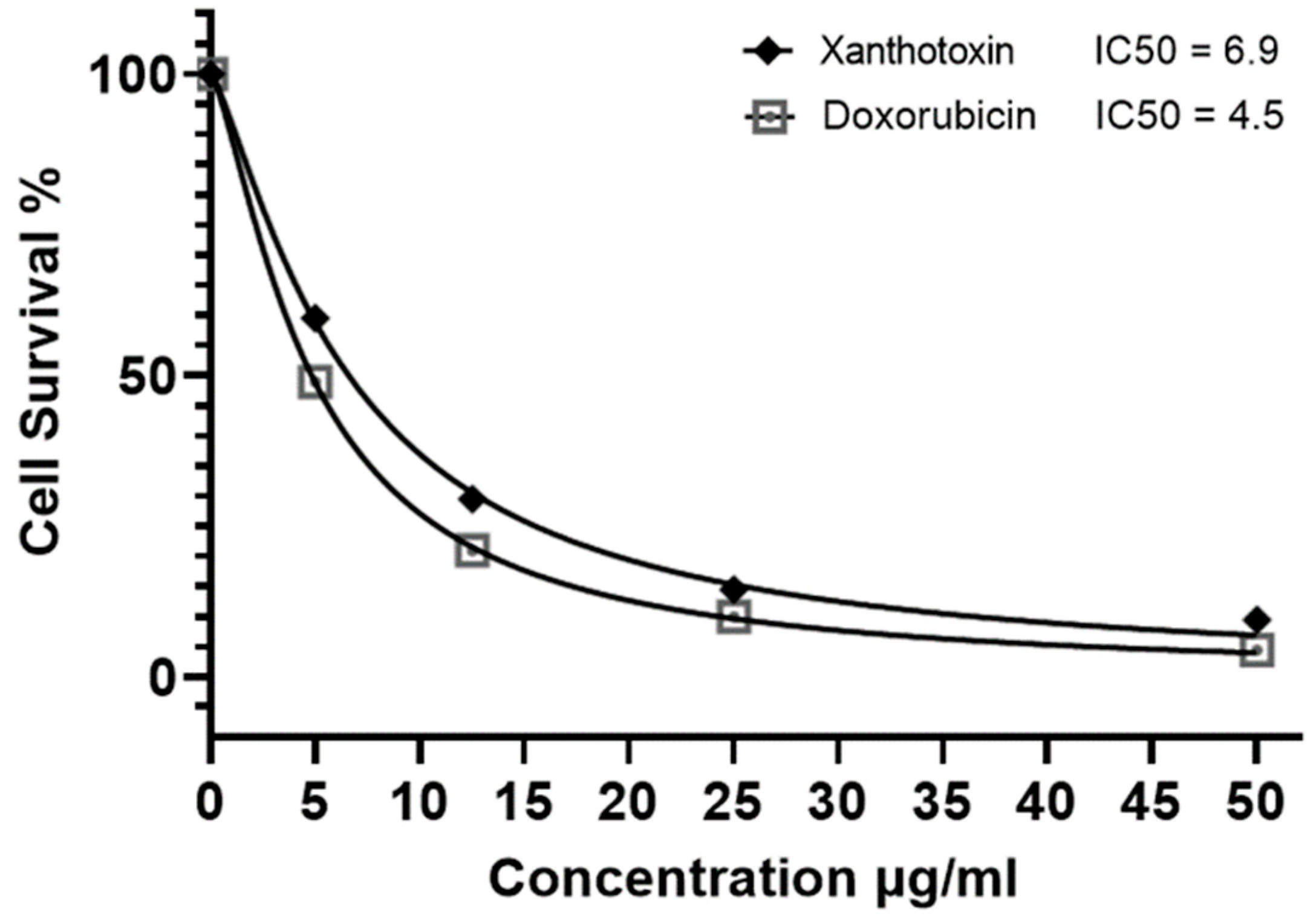
2.3. Cytotoxic Activity of Xanthotoxin against HepG2 Cell Line
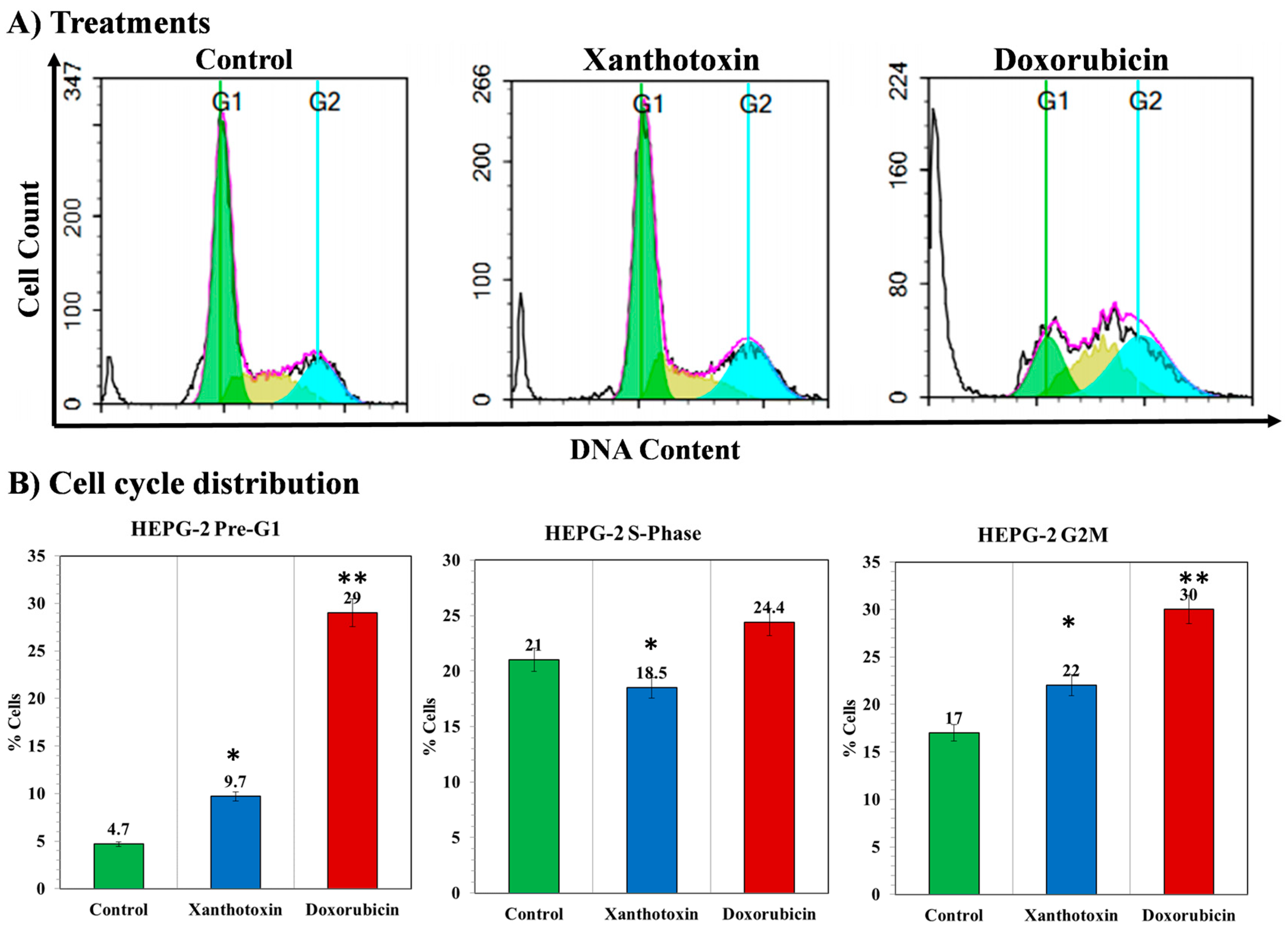
2.4. The Effects of Xanthotoxin on Cell Cycle Kinetics
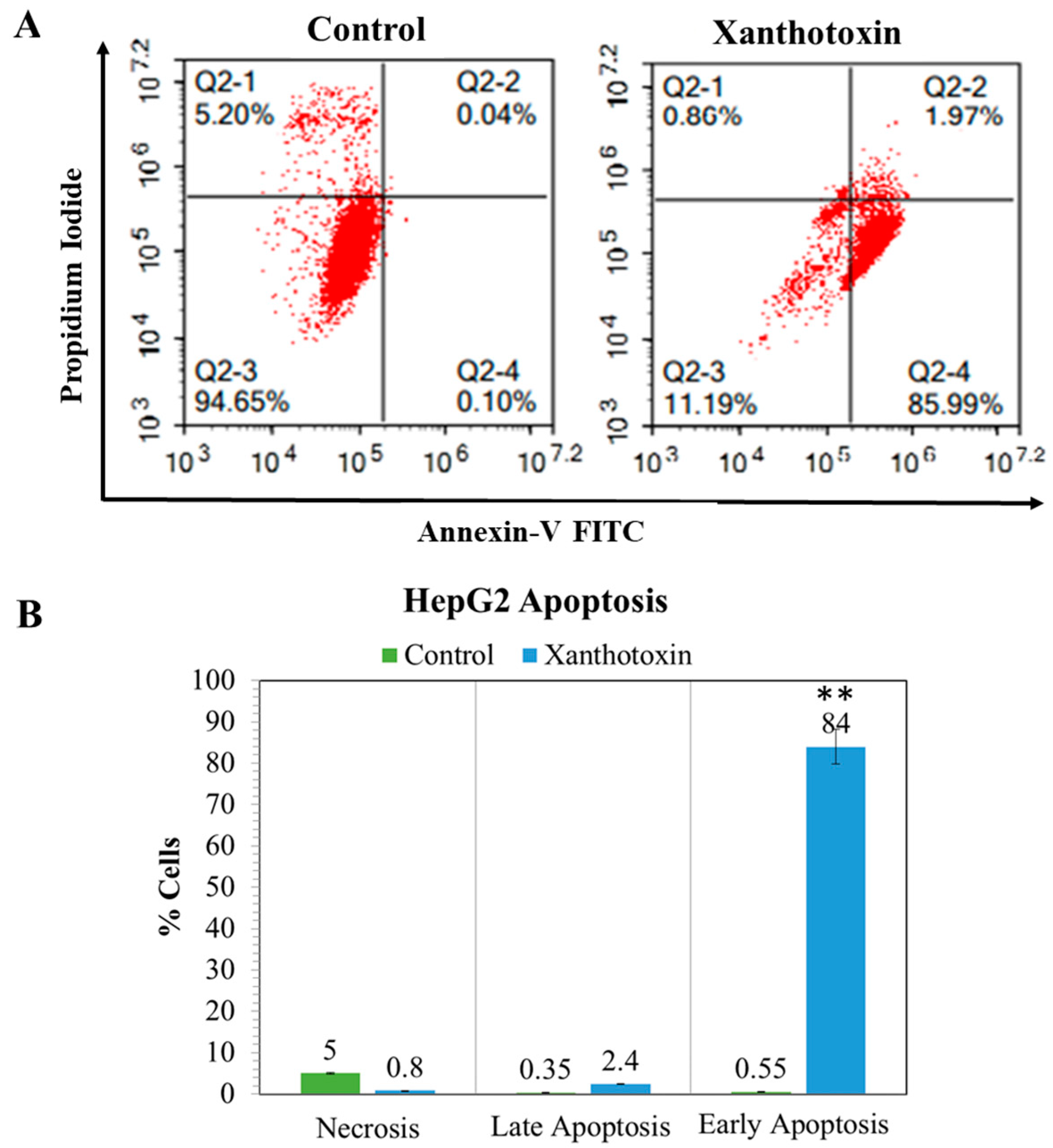
2.5. The Effects of Xanthotoxin on Programmed Cell Death
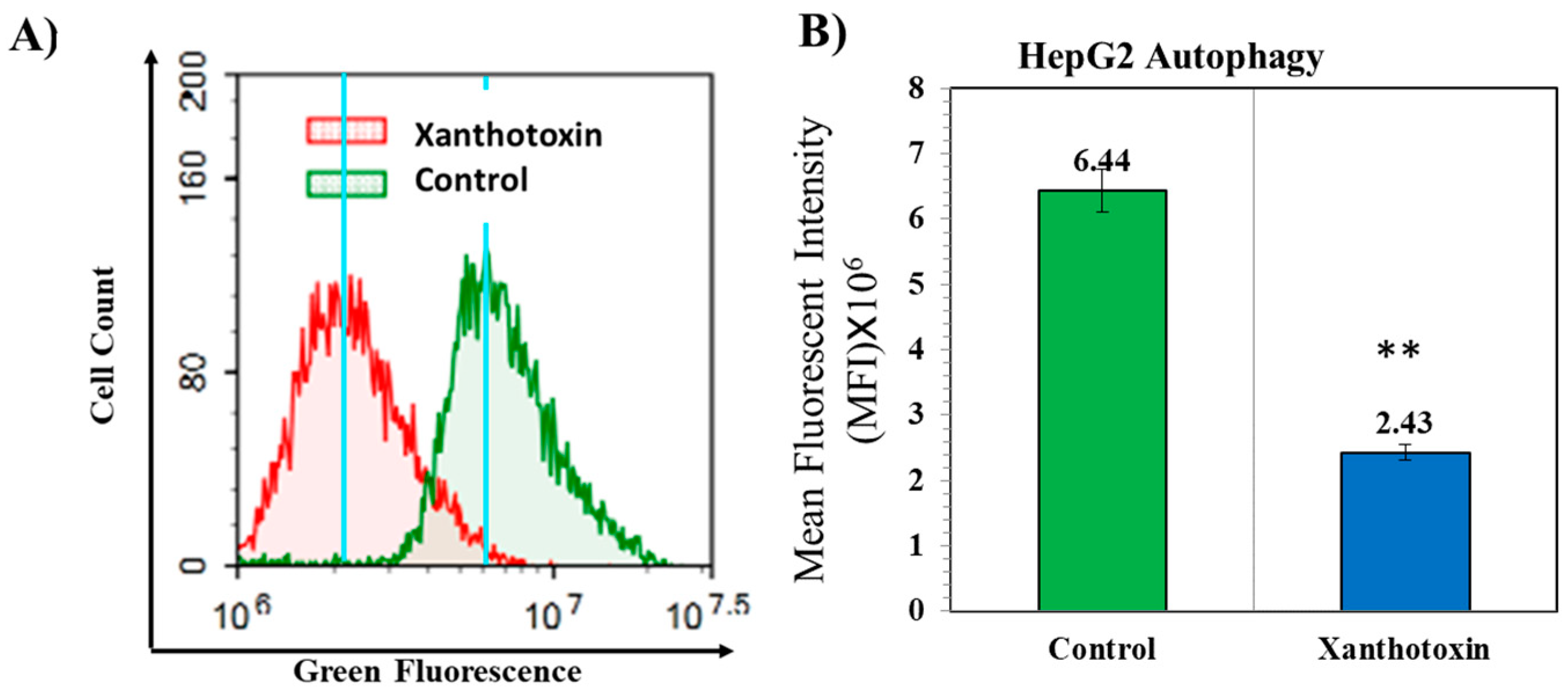
2.6. The Effects of Xanthotoxin on Cellular Autophagy
2.7. Molecular Docking of Xanthotoxin on Topoisomerase IIb Enzyme
3. Discussion
4. Materials and Methods
4.1. Chemical Studies
4.1.1. Plant Material
4.1.2. Extraction and Fractionation of the Plant Material
4.1.3. Purification and Isolation of the Compounds
4.2. Biological Evaluation
4.2.1. Cell Culture
4.2.2. Cytocompatibility and Cytotoxicity Assessments
4.2.3. Cell Cycle Kinetics Analysis
4.2.4. Apoptosis Analysis by Flow Cytometry
4.2.5. Autophagy Assay
4.3. Molecular Docking Analysis
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Yang, J.D.; Mohamed, E.A.; Aziz, A.O.A.; Shousha, H.I.; Hashem, M.B.; Nabeel, M.M.; Abdelmaksoud, A.H.; Elbaz, T.M.; Afihene, M.Y.; Duduyemi, B.M.; et al. Characteristics, management, and outcomes of patients with hepatocellular carcinoma in Africa: A multicountry observational study from the Africa Liver Cancer Consortium. Lancet Gastroenterol. Hepatol. 2017, 2, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Li, S. Clinical characteristics and prognosis of 2887 patients with hepatocellular carcinoma: A single center 14 years experience from China. Medicine 2019, 98, e14070. [Google Scholar] [CrossRef] [PubMed]
- Ingle, P.V.; Samsudin, S.Z.; Chan, P.Q.; Ng, M.K.; Heng, L.X.; Yap, S.C.; Chai, A.S.H.; Wong, A.S.Y. Development and novel therapeutics in hepatocellular carcinoma: A review. Ther. Clin. Risk Manag. 2016, 12, 445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.R.; Qiu, H.C.; Hu, Y.; Wang, Y.; Wang, Y.T. Herbal Medicine Offered as an Initiative Therapeutic Option for the Management of Hepatocellular Carcinoma. Phyther. Res. 2016, 30, 863–877. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Pezzuto, J.M. Natural Products as a Vital Source for the Discovery of Cancer Chemotherapeutic and Chemopreventive Agents. Med. Princ. Pract. 2016, 25, 41–59. [Google Scholar] [CrossRef] [PubMed]
- Moreno, F.S.; Heidor, R.; Pogribny, I.P. Nutritional Epigenetics and the Prevention of Hepatocellular Carcinoma with Bioactive Food Constituents. Nutr. Cancer 2016, 68, 719–733. [Google Scholar] [CrossRef] [PubMed]
- Rawat, D.; Shrivastava, S.; Naik, R.A.; Chhonker, S.K.; Mehrotra, A.; Koiri, R.K. An Overview of Natural Plant Products in the Treatment of Hepatocellular Carcinoma. Anticancer Agents Med. Chem. 2018, 18, 1838–1859. [Google Scholar] [CrossRef] [PubMed]
- Fathallah, N.; Raafat, M.M.; Issa, M.Y.; Abdel-Aziz, M.M.; Bishr, M.; Abdelkawy, M.A.; Salama, O. Bio-guided fractionation of prenylated benzaldehyde derivatives as potent antimicrobial and antibiofilm from Ammi majus L. fruits-associated Aspergillus amstelodami. Molecules 2019, 24, 4118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kausar, H.; Abidin, L.; Mujeeb, M.; Aqil, M.; Alam, O. Factorial design-guided optimization of extraction of therapeutically active furanocoumarin khellin from Ammi majus L. fruits. Pharmacogn. Mag. 2020, 16, 835. [Google Scholar] [CrossRef]
- Hossain, M.A.; Al Touby, S. Ammi majus an Endemic Medicinal Plant: A Review of the Medicinal Uses, Pharmacological and Phytochemicals. Ann. Toxicol. 2020, 2, 9–14. [Google Scholar] [CrossRef]
- Duddeck, H.; Kaiser, M. 13C NMR spectroscopy of coumarin derivatives. Org. Magn. Reson. 1982, 20, 55–72. [Google Scholar] [CrossRef]
- Karawya, M.S.; Khayyal, S.E.; Youssef, G.F. Estimation of xanthotoxin, imperatorin and bergapten in Ammi majus fruits and formulations. Planta Med. 1970, 18, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Selim, Y.; Ouf, N. Anti-inflammatory new coumarin from the Ammi majus L. Org. Med. Chem. Lett. 2012, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Al-Hadhrami, R.M.S.; Hossain, M.A. Evaluation of antioxidant, antimicrobial and cytotoxic activities of seed crude extracts of Ammi majus grown in Oman. Egypt. J. Basic Appl. Sci. 2016, 3, 329–334. [Google Scholar] [CrossRef] [Green Version]
- Grigg, R.; Knight, J.A.; Roffey, P. NMR solvent shifts and structure elucidation in coumarins. Tetrahedron 1966, 22, 3301–3304. [Google Scholar] [CrossRef]
- Razavi, S.M.; Zarrini, G.; Rad, G.F. Isoarnottinin 4′-glucoside, a glycosylated coumarin from Prangos uloptera, with biological activity. Russ. J. Bioorg. Chem. 2011, 37, 240–243. [Google Scholar] [CrossRef]
- Hani, M.; Elgamal, A.; Nagwa, M.; Shalaby, M.; Duddeck, H. Isolation of two Adjuncts from the Fruits of Ammi majus L. Nat. Prod. Lett. 1993, 3, 209–212. [Google Scholar] [CrossRef]
- Abu-Mustafa, E.A.; Fayez, M.B.E. Natural Coumarins. I. Marmesin and Marmesinin, Further Products from the Fruits of Ammi majus L. J. Org. Chem. 1961, 26, 161–166. [Google Scholar] [CrossRef]
- Wang, Q.; Zhong, S.; Wu, H.; Wu, Q. In vitro anti-cancer effect of marmesin by suppression of PI3K/Akt pathway in esophagus cancer cells. Esophagus 2021, 19, 163–174. [Google Scholar] [CrossRef]
- Luszczki, J.J.; Glowniak, K.; Czuczwar, S.J. Imperatorin enhances the protective activity of conventional antiepileptic drugs against maximal electroshock-induced seizures in mice. Eur. J. Pharmacol. 2007, 574, 133–139. [Google Scholar] [CrossRef]
- Abu-Mustafa, E.A.; El-Bay, F.K.A.; Fayez, M.B.E. Ammirin, a new coumarin constituent from Ammi majus L. fruits-Natural coumarins, part XVI. Naturwissenschaften 1975, 62, 39–40. [Google Scholar] [CrossRef] [PubMed]
- El-Fadl, H.M.A.; Hagag, N.M.; El-Shafei, R.A.; Khayri, M.H.; El-Gedawy, G.; El Maksoud, A.I.A.; Mohamed, D.D.; Mohamed, D.D.; El Halfawy, I.; Khoder, A.I.; et al. Effective Targeting of Raf-1 and Its Associated Autophagy by Novel Extracted Peptide for Treating Breast Cancer Cells. Front. Oncol. 2021, 11, 3317. [Google Scholar] [CrossRef] [PubMed]
- Sordet, O.; Goldman, A.; Pommier, Y. Topoisomerase II and tubulin inhibitors both induce the formation of apoptotic topoisomerase I cleavage complexes. Mol. Cancer Ther. 2006, 5, 3139–3144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.C.; Li, T.K.; Farh, L.; Lin, L.Y.; Lin, T.S.; Yu, Y.J.; Yen, T.J.; Chiang, C.W.; Chan, N.L. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 2011, 333, 459–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishina, S.; Yamauchi, A.; Kawaguchi, T.; Kaku, K.; Goto, M.; Sasaki, K.; Hara, Y.; Tomiyama, Y.; Kuribayashi, F.; Torimura, T.; et al. Dipeptidyl Peptidase 4 Inhibitors Reduce Hepatocellular Carcinoma by Activating Lymphocyte Chemotaxis in Mice. Cmgh 2019, 7, 115–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arafah, M.W.; Almutairi, B.; Al-Zharani, M.; Alkahtane, A.A.; Al-Otibi, F.O.; Ali, D.; Alghamdi, W.M.; Alanazi, I.S.; Aljarba, N.H.; Alhoshani, N.M.; et al. The protective effect of Ammi visnaga extract against human hepatic cancer. J. King Saud Univ. Sci. 2021, 33, 101540. [Google Scholar] [CrossRef]
- Voykov, P.; Chaushev, H.; Panova, N. Psoralens: Pharmaceutical applications in treating proliferative skin diseases. Scr. Sci. Pharm. 2015, 2, 55. [Google Scholar]
- Konstantinow, A.; Balda, B.R. Treatment of cutaneous T-cell lymphoma with extracorporeal photochemotherapy. J. Eur. Acad. Dermatol. Venereol. 1997, 9, 111–117. [Google Scholar] [CrossRef]
- Mirzaei, S.A.; Gholamian Dehkordi, N.; Ghamghami, M.; Amiri, A.H.; Dalir Abdolahinia, E.; Elahian, F. ABC-transporter blockage mediated by xanthotoxin and bergapten is the major pathway for chemosensitization of multidrug-resistant cancer cells. Toxicol. Appl. Pharmacol. 2017, 337, 22–29. [Google Scholar] [CrossRef]
- Shokoohinia, Y.; Hosseinzadeh, L.; Alipour, M.; Mostafaie, A.; Mohammadi-Motlagh, H.R. Comparative Evaluation of Cytotoxic and Apoptogenic Effects of Several Coumarins on Human Cancer Cell Lines: Osthole Induces Apoptosis in p53-Deficient H1299 Cells. Adv. Pharmacol. Sci. 2014, 2014, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Yang, B.; Zhou, Q.; Wu, Y.; Shang, D.; Guo, Y.; Song, Z.; Zheng, Q.; Xiong, J. Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis 2013, 34, 1343–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.T.; Zhou, T.T.; Liu, B.; Bao, J.K. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Das, C.K.; Banerjee, I.; Mandal, M. Pro-survival autophagy: An emerging candidate of tumor progression through maintaining hallmarks of cancer. In Seminars in Cancer Biology; Elsevier: Amsterdam, The Netherlands, 2020; Volume 66, pp. 59–74. [Google Scholar]
- Yu, S.; Wang, Y.; Jing, L.; Claret, F.X.; Li, Q.; Tian, T.; Liang, X.; Ruan, Z.; Jiang, L.; Yao, Y.; et al. Autophagy in the “inflammation-carcinogenesis” pathway of liver and HCC immunotherapy. Cancer Lett. 2017, 411, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.S.; Vats, S.; Chia, A.Y.-Q.Q.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef] [PubMed]
- McClendon, A.K.; Osheroff, N. DNA topoisomerase II, genotoxicity, and cancer. Mutat. Res. Mol. Mech. Mutagen. 2007, 623, 83–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghanem, A.; Emara, H.A.; Muawia, S.; Abd El Maksoud, A.I.; Al-Karmalawy, A.A.; Elshal, M.F. Tanshinone IIA synergistically enhances the antitumor activity of doxorubicin by interfering with the PI3K/AKT/mTOR pathway and inhibition of topoisomerase II: In vitro and molecular docking studies. New J. Chem. 2020, 44, 17374–17381. [Google Scholar] [CrossRef]
- Das, S.; Tripathi, N.; Siddharth, S.; Nayak, A.; Nayak, D.; Sethy, C.; Bharatam, P.V.; Kundu, C.N. Etoposide and doxorubicin enhance the sensitivity of triple negative breast cancers through modulation of TRAIL-DR5 axis. Apoptosis 2017, 22, 1205–1224. [Google Scholar] [CrossRef]
- Balbaa, S.I.; Hilal, S.H.; Haggag, M.Y. Separation of ammajin and marmesin from the fruits of Ammi majus and their chemical estimation. Planta Med. 1973, 23, 191–195. [Google Scholar] [CrossRef]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, S.A.; Elshal, M.F.; Kumosani, T.A.; Aldahlawi, A.M.; Basbrain, T.A.; Alshehri, F.A.; Choudhry, H. L-asparaginase isolated from Phaseolus vulgaris seeds exhibited potent anti-acute lymphoblastic leukemia effects in-vitro and low immunogenic properties in-vivo. Int. J. Environ. Res. Public Health 2016, 13, 1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eliaa, S.G.; Al-Karmalawy, A.A.; Saleh, R.M.; Elshal, M.F. Empagliflozin and Doxorubicin Synergistically Inhibit the Survival of Triple-Negative Breast Cancer Cells via Interfering with the mTOR Pathway and Inhibition of Calmodulin: In Vitro and Molecular Docking Studies. ACS Pharmacol. Transl. Sci. 2020, 3, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Thomé, M.P.; Filippi-Chiela, E.C.; Villodre, E.S.; Migliavaca, C.B.; Onzi, G.R.; Felipe, K.B.; Lenz, G. Ratiometric analysis of Acridine Orange staining in the study of acidic organelles and autophagy. J. Cell Sci. 2016, 129, 4622–4632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elia, S.G.; Al-Karmalawy, A.A.; Nasr, M.Y.; Elshal, M.F. Loperamide potentiates doxorubicin sensitivity in triple-negative breast cancer cells by targeting MDR1 and JNK and suppressing mTOR and Bcl-2: In vitro and molecular docking study. J. Biochem. Mol. Toxicol. 2021, 36, e22938. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (a) | ||||||||
| Position | C1 | C3 | C4 | C5 | C6 | |||
| δH | δC | δH | δH | δC | δH | δH | δC | |
| 1 | - | - | - | - | - | - | - | - |
| 2 | - | 161.84 | - | - | 169.90 | - | - | 163. |
| 3 | 6.38 (d, 12) | 113.82 | 6.08 (d, 7.2) | 6.40 (d, 8.5) | 100.12 | 6.40 (d, 9.5) | 7.05 (d, 7.2) | 105.50 |
| 4 | 7.78 (d, 12) | 142.50 | 7.62 (d, 8.5) | 7.80 (d, 7.9) | 144.83 | 7.79 (d, 9.6) | 7.61 (d, 7.2) | 145.21 |
| 5 | 7.37 (s) | 114.82 | 7.05 (d, 8.0) | 6.85 (s) | 130.55 | - | 7.28 (s) | 130.21 |
| 6 | - | 126.26 | 7.27 (d, 9.3) | - | 127.94 | - | - | 124.82 |
| 7 | - | 147.16 | - | - | 164.55 | - | - | 163 |
| 8 | - | 132.80 | - | 7.72 (s) | 111.50 | 7.38 (s) | 7.28 (s) | 117 |
| 1′ | - | - | 3.57 (d, 7.7) | - | - | - | - | - |
| 2′ | 7.70 (d, 4) | 147.80 | 5.37 (m) | 4.21 (m) | 95.90 | 7.72 (d, 2.3) | 4.76 (m) | 95 |
| 3′ | 6.83 (d, 4) | 107.32 | - | 2.42 (d, 0.8) | 21.50 | 6.84 (d, 2.2) | 2.35 (d, 0.7) | 41 |
| 4′ | 4.30 (s) | 50.10 | 1.70 (s) | - | 62.10 | - | - | 130.15 |
| 5′ | - | - | 4.20 (s) | 1.28 (s) | 19.97 | - | 6.07 (s) | 115.47 |
| 6′ | - | - | - | 1.65 (s) | 19.97 | - | 1.27 (s) | 20.10 |
| 1a | - | 145.26 | - | - | 145.51 | - | - | 110.34 |
| 4a | - | 116.18 | - | - | 107.50 | - | - | 142.96 |
| 1″ | - | - | - | - | - | 1.57 (s) | - | - |
| 2″ | - | - | - | - | - | 5.39 (m) | - | - |
| 3″ | - | - | - | - | - | - | - | - |
| 4″ | - | - | - | - | - | 1.28 (s) | - | - |
| 5″ | - | - | - | - | - | 4.33 (d, 2.3) | - | - |
| (b) | ||||||||
| Position | C2 | |||||||
| δH | ||||||||
| 1 | 1.51 (m) | |||||||
| 2 | 1.60 (m) | |||||||
| 3 | 3.55 (m). | |||||||
| 4 | 2.01 (d, 5.1). | |||||||
| 5, 10, 13 | No proton | |||||||
| 6 | 5.37 (m) | |||||||
| 18 | 0.83 (s) | |||||||
| 19 | 1.03 (s) | |||||||
| 21 | 0.94 (d, 6.5) | |||||||
| 26 | 0.88 (d, 8.0) | |||||||
| 27 | 0.85 (d, 1.8) | |||||||
| 28 | 1.43–1.24 (m) | |||||||
| 29 | 1.24 (m) | |||||||
| Compound | a S | b RMSD (Å ) | Amino Acid/Bond | Distance (Å) | E (Kcal/mol) |
|---|---|---|---|---|---|
| Xanthotoxin | −5.72 | 1.51 | Asp479/H–donor | 2.87 | −3.01 |
| DA12/H–pi | 3.98 | −1.20 | |||
| DG13/H–pi | 3.72 | −0.07 | |||
| Etoposide | −7.31 | 1.93 | ASP479/H–donor | 2.56 | −3.0 |
| MET782/H–donor | 3.26 | −0.4 | |||
| DG13/H–donor | 2.96 | −2.8 | |||
| DG13/pi–pi | 2.72 | −3.1 | |||
| DA12/H–pi | 3.79 | −1.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Issa, M.Y.; Elshal, M.F.; Fathallah, N.; Abdelkawy, M.A.; Bishr, M.; Salama, O.; Abulfadl, Y.S. Potential Anticancer Activity of the Furanocoumarin Derivative Xanthotoxin Isolated from Ammi majus L. Fruits: In Vitro and In Silico Studies. Molecules 2022, 27, 943. https://doi.org/10.3390/molecules27030943
Issa MY, Elshal MF, Fathallah N, Abdelkawy MA, Bishr M, Salama O, Abulfadl YS. Potential Anticancer Activity of the Furanocoumarin Derivative Xanthotoxin Isolated from Ammi majus L. Fruits: In Vitro and In Silico Studies. Molecules. 2022; 27(3):943. https://doi.org/10.3390/molecules27030943
Chicago/Turabian StyleIssa, Marwa Y., Mohamed F. Elshal, Noha Fathallah, Mostafa A. Abdelkawy, Mokhtar Bishr, Osama Salama, and Yasmin S. Abulfadl. 2022. "Potential Anticancer Activity of the Furanocoumarin Derivative Xanthotoxin Isolated from Ammi majus L. Fruits: In Vitro and In Silico Studies" Molecules 27, no. 3: 943. https://doi.org/10.3390/molecules27030943