
2-(2-(Dimethylamino)vinyl)-4H-pyran-4-ones as Novel and Convenient Building-Blocks for the Synthesis of Conjugated 4-Pyrone Derivatives

 , , , ,
, , , ,
Abstract
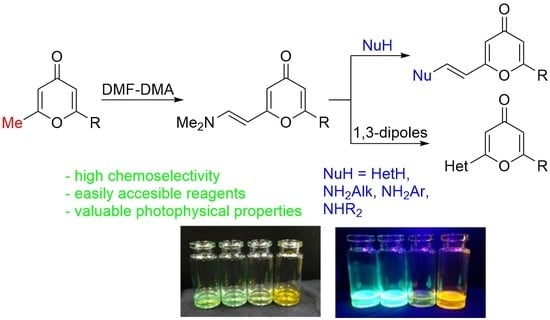
:
1. Introduction
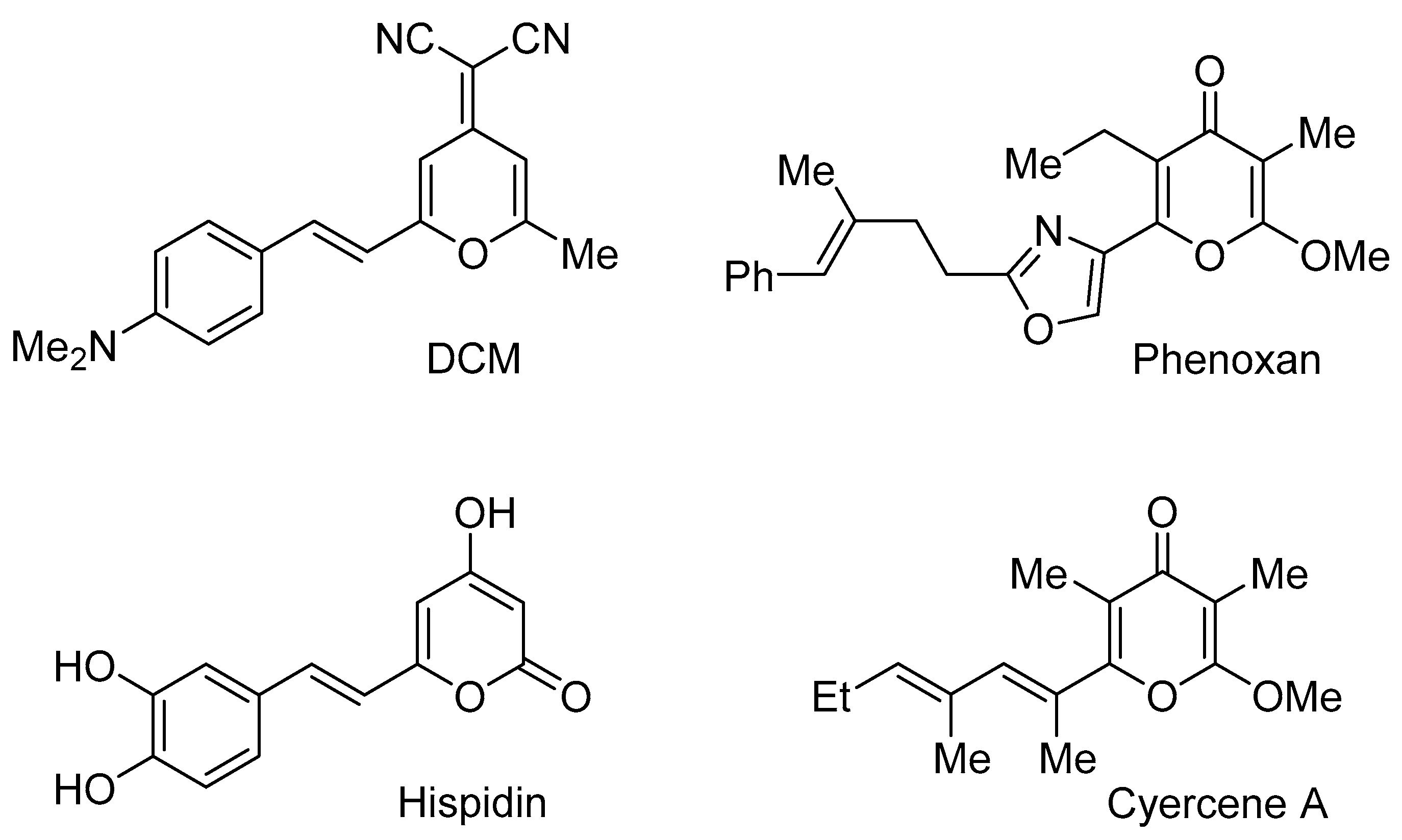
2. Results and Discussion
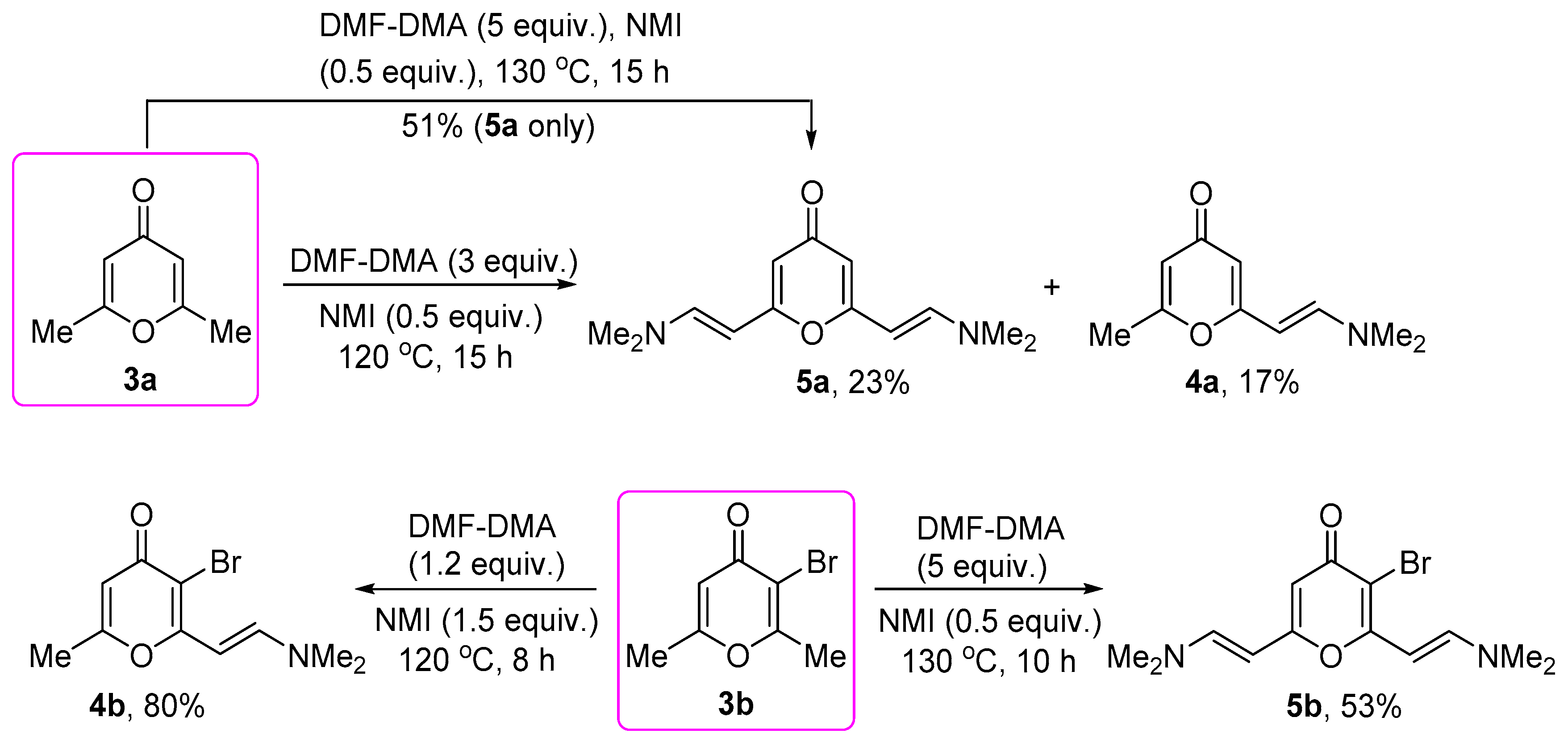
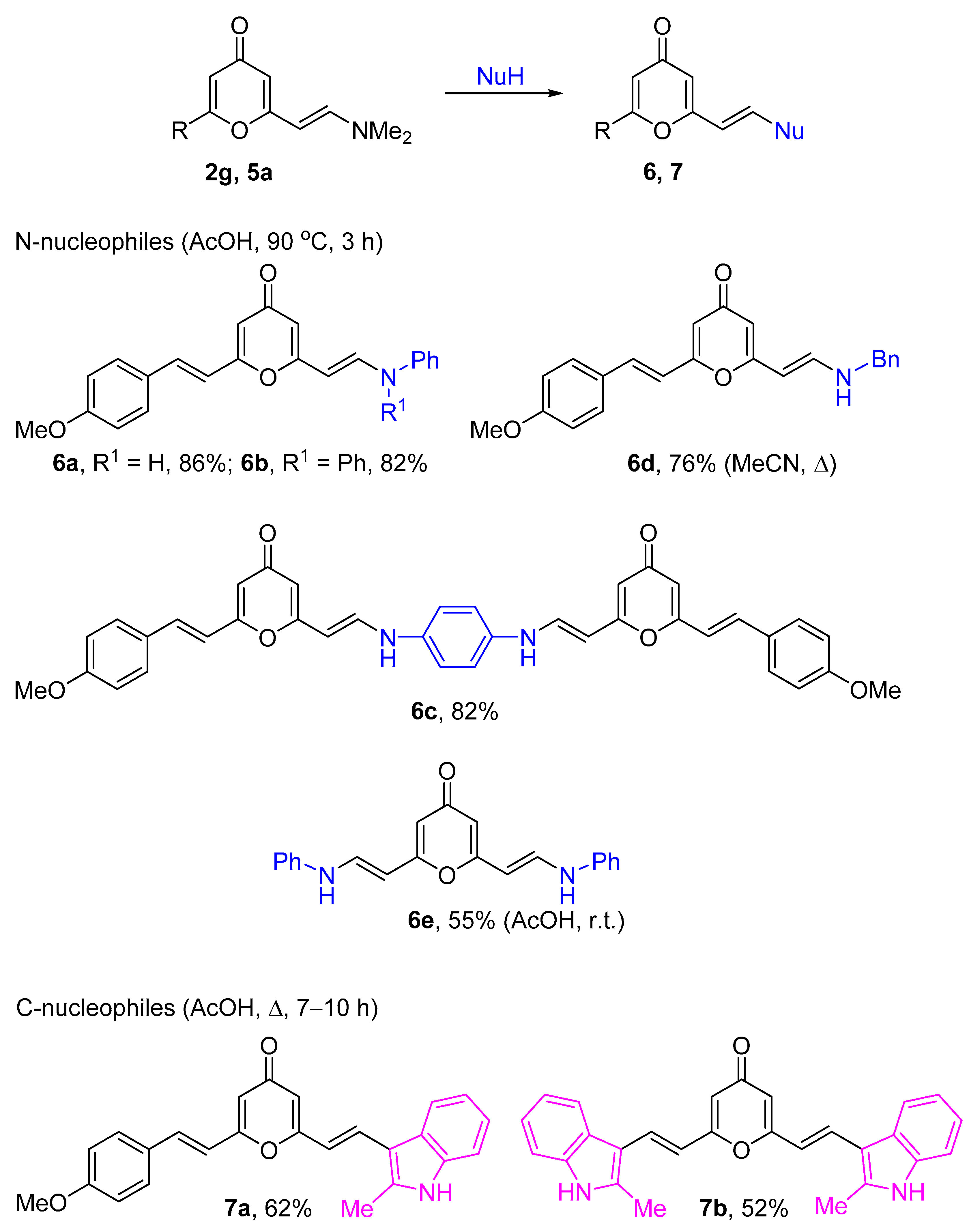
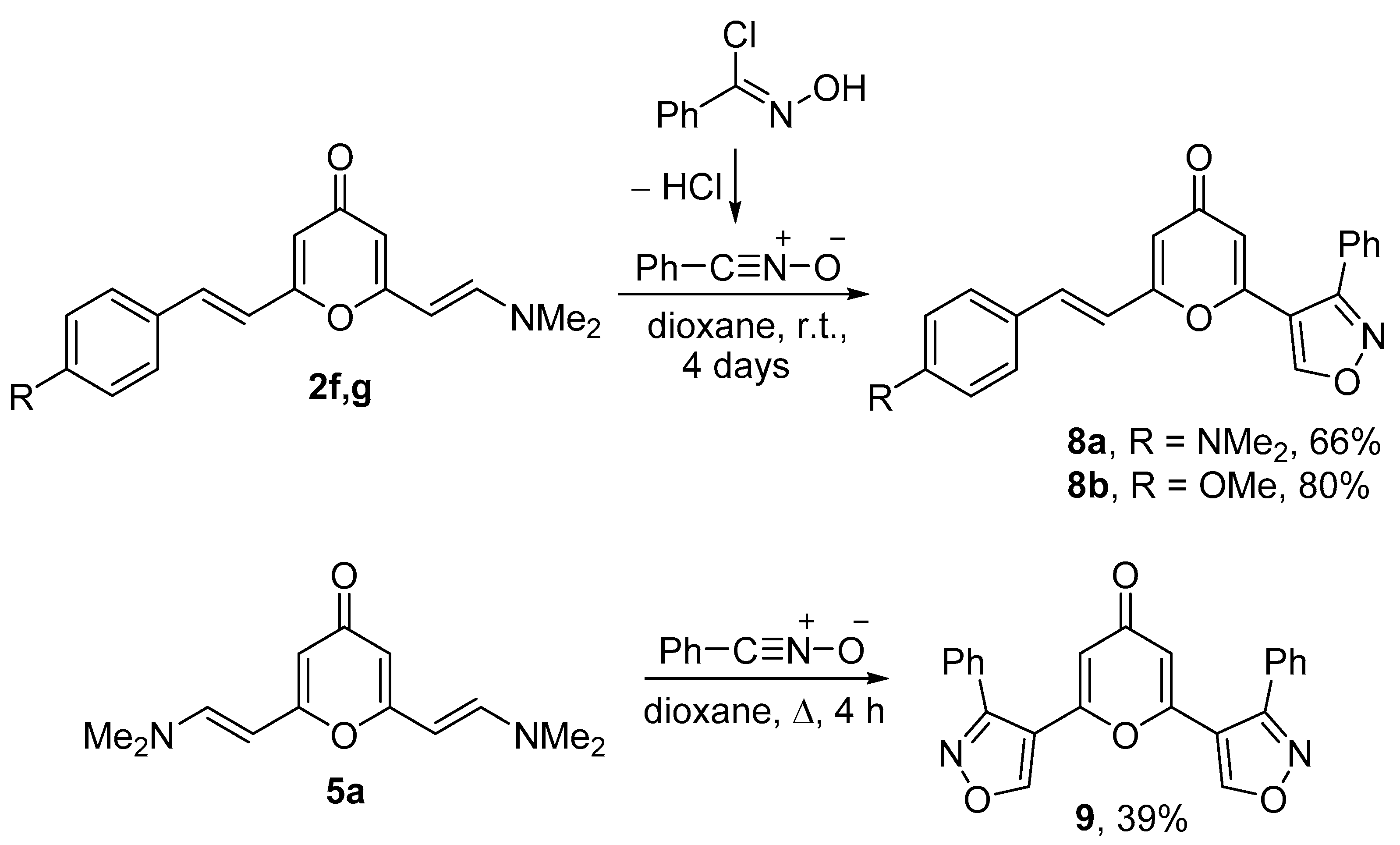
2.1. Synthesis of 2-Enamino-substituted 4-Pyrones and Their Chemical Properties
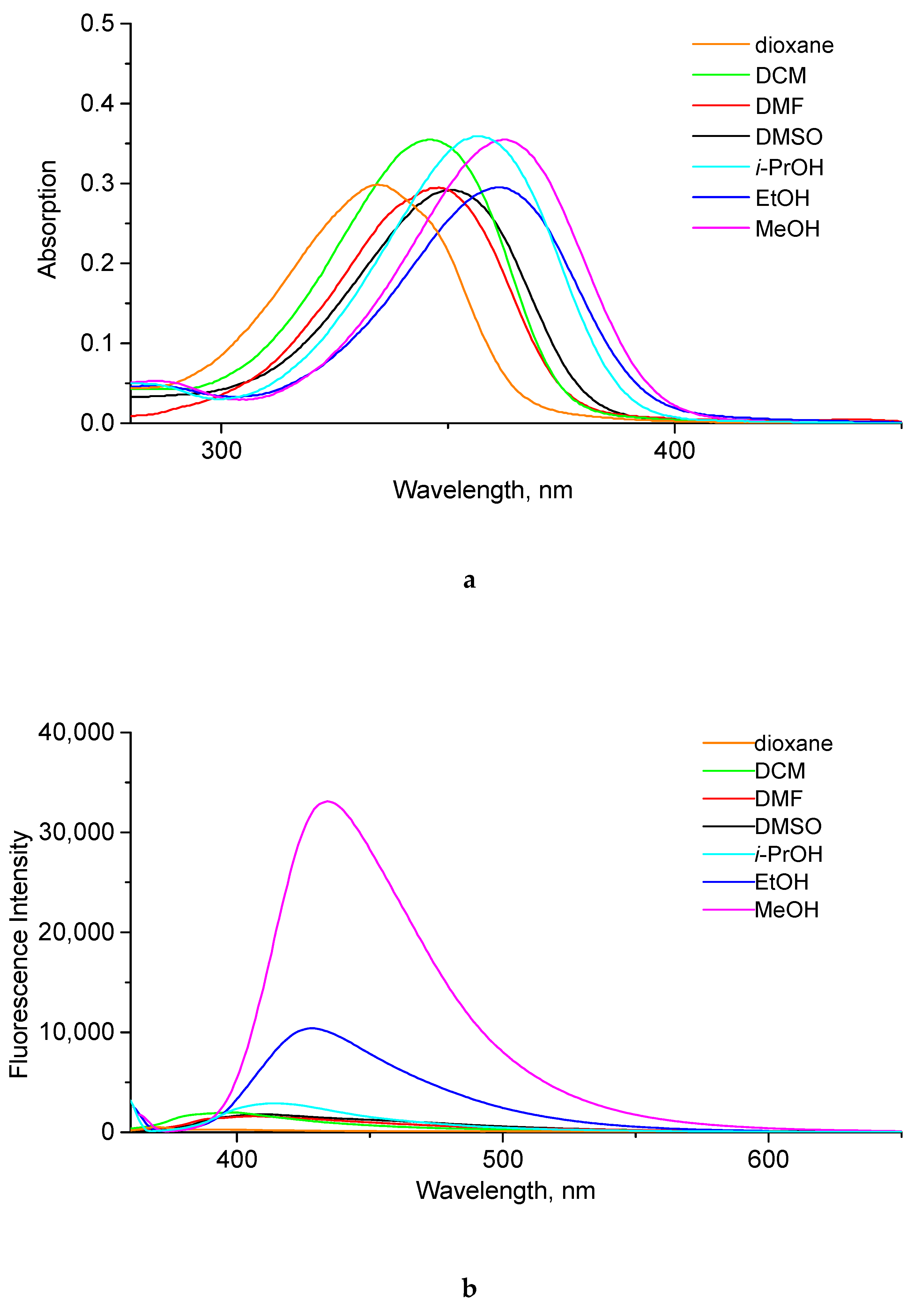
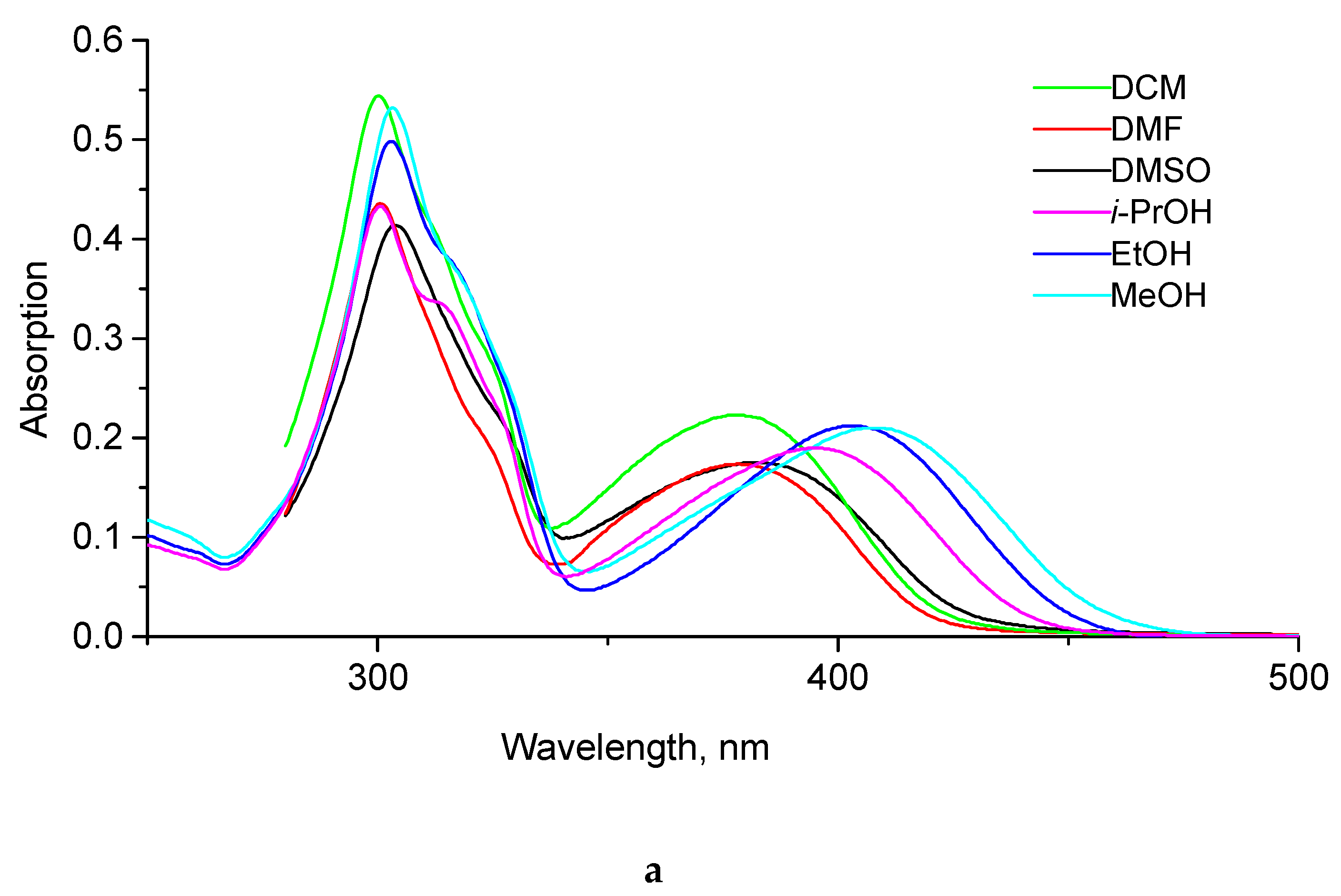
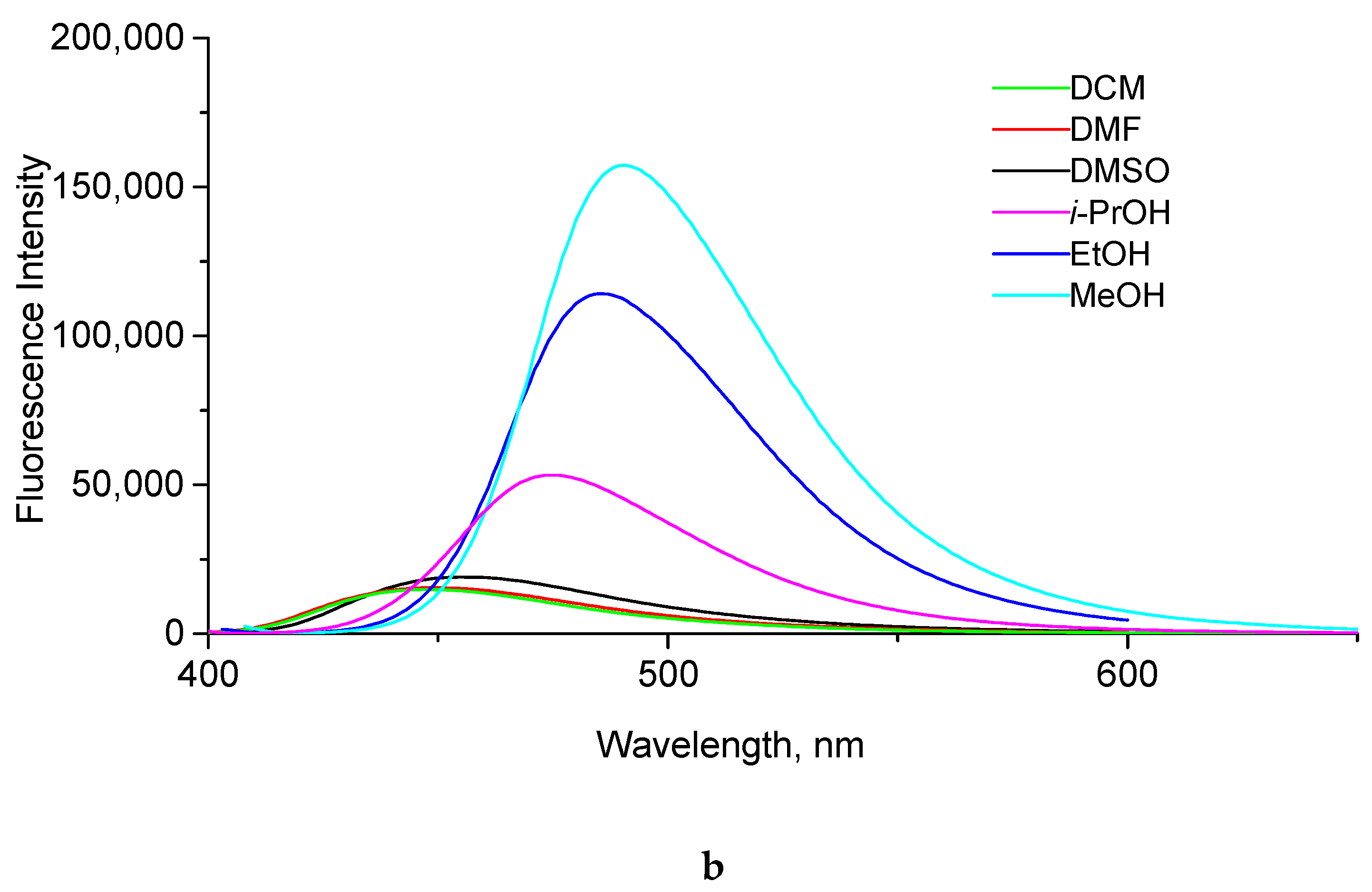
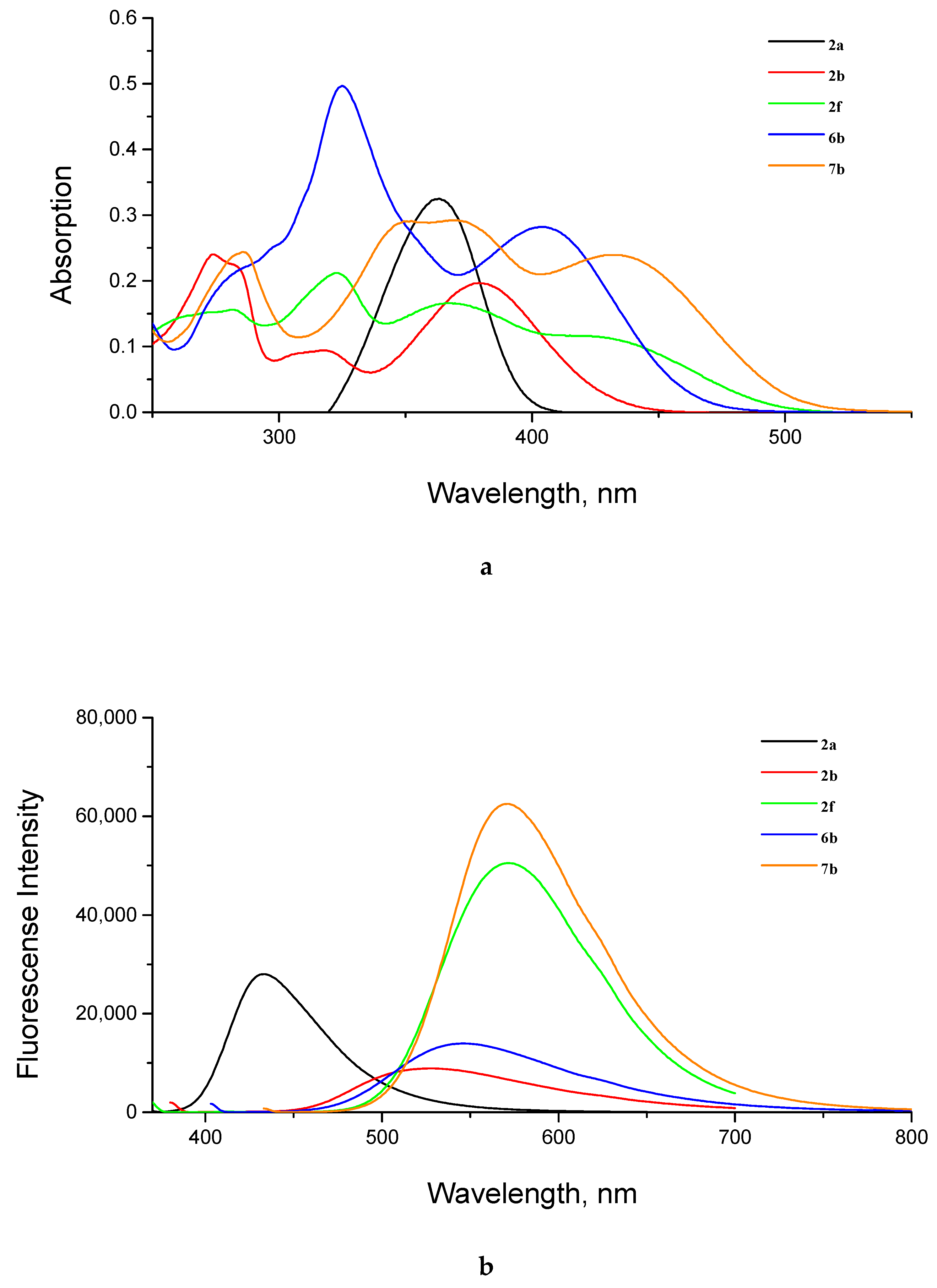
2.2. Photophysical Properties of Products
2.3. Theoretical Calculations of the Absorption and Emission
3. Materials and Methods
3.1. Quantum Mechanical Calculations
3.2. Synthesis of Compounds 2
3.3. Synthesis of Compounds 4
3.4. General Method for the Synthesis of Bis(enamino)-substituted 4-Pyrones 5a,b
3.5. General Method for the Preparation of 2-((E)-4-Methoxystyryl)-6-((E)-2-aminovinyl)-4H-pyran-4-one 6a–c
3.6. Synthesis of Compounds 7a,b
3.7. Synthesis of 6-(3-Phenylisoxazol-4-yl)-4H-pyran-4-ones 8a,b
3.8. Synthesis of compound 9
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Singh, K.S. Pyrone-derived marine natural products: A review on isolation, bio-activities and synthesis. Curr. Org. Chem. 2020, 24, 354–401. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Ohba, S.; Nishiyama, S.; Yamamura, S. Total synthesis of phenoxan and a related pyrone derivative. Tetrahedron Lett. 1996, 37, 2997–3000. [Google Scholar] [CrossRef]
- Schiavone, D.V.; Kapkayeva, D.M.; Murelli, R.P. Investigations into a stoichiometrically equivalent intermolecular oxidopyrylium [5 + 2] cycloaddition reaction leveraging 3-hydroxy-4-pyrone-based oxidopyrylium dimers. J. Org. Chem. 2021, 86, 3826–3835. [Google Scholar] [CrossRef]
- Usachev, S.A.; Nigamatova, D.I.; Mysik, D.K.; Naumov, N.A.; Obydennov, D.L.; Sosnovskikh, V.Y. 2-Aryl-6-polyfluoroalkyl-4-pyrones as promising RF-building-blocks: Synthesis and application for construction of fluorinated azaheterocycles. Molecules 2021, 26, 4415. [Google Scholar] [CrossRef] [PubMed]
- Milyutin, C.V.; Komogortsev, A.N.; Lichitsky, B.V.; Melekhina, V.G.; Minyaev, M.E. Construction of spiro-γ-butyrolactone core via cascade photochemical reaction of 3-hydroxypyran-4-one derivatives. Org. Lett. 2021, 23, 5266–5270. [Google Scholar] [CrossRef]
- Chen, J.; Wu, L.; Wu, J. Kojic acid and maltol: The “transformers” in organic synthesis. Chin. Chem. Lett. 2020, 31, 2993–2995. [Google Scholar] [CrossRef]
- Yasukata, T.; Masui, M.; Ikarashi, F.; Okamoto, K.; Kurita, T.; Nagai, M.; Sugata, Y.; Miyake, N.; Hara, S.; Adachi, Y.; et al. Practical synthetic method for the preparation of pyrone diesters: An efficient synthetic route for the synthesis of dolutegravir sodium. Org. Process Res. Dev. 2019, 23, 565–570. [Google Scholar] [CrossRef]
- Obydennov, D.L.; Khammatova, L.R.; Eltsov, O.S.; Sosnovskikh, V.Y. A chemo- and regiocontrolled approach to bipyrazoles and pyridones via the reaction of ethyl 5-acyl-4-pyrone-2-carboxylates with hydrazines. Org. Biomol. Chem. 2018, 16, 1692–1707. [Google Scholar] [CrossRef]
- Palkina, K.A.; Ipatova, D.A.; Shakhova, E.S.; Balakireva, A.V.; Markina, N.M. Therapeutic potential of hispidin—Fungal and plant polyketide. J. Fungi 2021, 7, 323. [Google Scholar] [CrossRef]
- Purtov, K.V.; Petushkov, V.N.; Baranov, M.S.; Mineev, K.S.; Rodionova, N.S.; Kaskova, Z.M.; Tsarkova, A.S.; Petunin, A.I.; Bondar, V.S.; Rodicheva, E.K.; et al. The chemical basis of fungal bioluminescence. Angew. Chem. Int. Ed. 2015, 54, 8124–8128. [Google Scholar] [CrossRef]
- Zuidema, D.R.; Jones, P.B. Triplet photosensitization in cyercene A and related pyrones. J. Photochem. Photobiol. B Biol. 2006, 83, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Moses, J.E.; Baldwin, J.E.; Adlington, R.M. An efficient synthesis of cyercene A. Tetrahedron Lett. 2004, 45, 6447–6448. [Google Scholar] [CrossRef]
- Krasnaya, Z.A.; Smirnova, Y.V.; Tatikolov, A.S.; Kuz’min, V.A. Synthesis and photonics of ketocyanine dyes, 2,6-bis(4-dimethylaminoalka-1,3-dienyl)-4H-pyran-4-ones and ethoxytridecamethine salts based on them. Russ. Chem. Bull. 1999, 48, 1329–1334. [Google Scholar] [CrossRef]
- Obydennov, D.L.; Simbirtseva, A.E.; Sosnovskikh, V.Y. Synthesis of 4-oxo-6-styryl-4H-pyran-2-carbonitriles and their application for the construction of new 4-pyrone derivatives. Res. Chem. Intermed. 2022, 48, 2155–2179. [Google Scholar] [CrossRef]
- Rahimpour, K.; Zarenezhad, H.; Teimuri-Mofrad, R. Transition metal-catalyzed C–N cross-coupling reaction of bromine-substituted pyranylidene derivatives: Synthesis, characterization, and optical properties study of pyran-based chromophores. J. Iran. Chem. Soc. 2020, 17, 2627–2636. [Google Scholar] [CrossRef]
- Cao, Y.; Xi, Y.; Teng, X.; Li, Y.; Yan, X.; Chen, L. Alkoxy substituted D-p-A dimethyl-4-pyrone derivatives: Aggregation induced emission enhancement, mechanochromic and solvatochromic properties. Dyes Pigm. 2017, 137, 75–83. [Google Scholar] [CrossRef]
- Cao, Y.; Chen, L.; Xi, Y.; Li, Y.; Yan, X. Stimuli-responsive 2,6-diarylethene-4H-pyran-4-one derivatives: Aggregation induced emission enhancement, mechanochromism and solvatochromism. Mater. Lett. 2018, 212, 225–230. [Google Scholar] [CrossRef]
- Liu, D.; Cao, Y.; Yan, X.; Wang, B. Two stimulus responsive carbazole substituted D–π–A pyrone compounds exhibiting mechanochromism and solvatochromism. Res. Chem. Intermed. 2019, 45, 2429–2439. [Google Scholar] [CrossRef]
- Pecourneau, J.; Losantos, R.; Monari, A.; Parant, S.; Pasc, A.; Mourer, M. Synthesis and photoswitching properties of bioinspired dissymmetric γ-pyrone, an analogue of cyclocurcumin. J. Org. Chem. 2021, 86, 8112–8126. [Google Scholar] [CrossRef]
- Guo, Z.; Zhu, W.; Tian, H. Dicyanomethylene-4H-pyranchromophores for OLED emitters, logic gates and optical chemosensors. Chem. Commun. 2012, 48, 6073–6084. [Google Scholar] [CrossRef]
- Casimiro, L.; Maisonneuve, S.; Retailleau, P.; Silvi, S.; Xie, J.; Métivier, R. Photophysical properties of 4-dicyanomethylene-2-methyl-6-(p-dimethylamino-styryl)-4H-pyran revisited: Fluorescence versus photoisomerization. Chem. Eur. J. 2020, 26, 14341–14350. [Google Scholar] [CrossRef] [PubMed]
- Hoche, J.; Schulz, A.; Dietrich, L.M.; Humeniuk, A.; Stolte, M.; Schmidt, D.; Brixner, T.; Würthner, F.; Mitric, R. The origin of the solvent dependence of fluorescence quantum yields in dipolar merocyanine dyes. Chem. Sci. 2019, 10, 11013–11022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.-J.; Hua, Y.; Xu, J.-Q.; Zhang, L.-X.; Wang, S.; Kang, Y.-F. Near-infrared fluorescence probe with a large Stokes shift for selectively imaging of hydrogen peroxide in living cells and in vivo. Dyes Pigm. 2022, 197, 109930. [Google Scholar] [CrossRef]
- Krasnaya, Z.A.; Smirnova, Y.V.; Shvedova, L.A.; Tatikolov, A.S.; Kuz’min, V.A. Synthesis and spectroscopic properties of cross-conjugated ketones and meso-substituted tridecamethine salts containing the pyran or pyridone fragment. Russ. Chem. Bull. 2003, 52, 2029–2037. [Google Scholar] [CrossRef]
- Obydennov, D.L.; Simbirtseva, A.E.; Piksin, S.E.; Sosnovskikh, V.Y. 2,6-Dicyano-4-pyrone as a novel and multifarious building block for the synthesis of 2,6-bis(hetaryl)-4-pyrones and 2,6-bis(hetaryl)-4-pyridinols. ACS Omega 2020, 5, 33406–33420. [Google Scholar] [CrossRef]
- Obydennov, D.L.; Usachev, B.I.; Sosnovskikh, V.Y. Reactions of 2-mono- and 2,6-disubstituted 4-pyrones with phenylhydrazine as general method for the synthesis of 3-(N-phenylpyrazolyl)indoles. Chem. Heterocycl. Compd. 2015, 50, 1388–1403. [Google Scholar] [CrossRef]
- Usachev, B. 2-(Trifluoromethyl)-4H-pyran-4-ones: Convenient, available and versatile building-blocks for regioselective syntheses of trifluoromethylated organic compounds. J. Fluor. Chem. 2015, 172, 80–91. [Google Scholar] [CrossRef]
- Zatsikha, Y.V.; Yakubovskyi, V.P.; Shandura, M.P.; Kovtun, Y.P. Functionalized bispyridoneannelated BODIPY—Bright long-wavelength fluorophores. Dyes Pigm. 2015, 114, 215–221. [Google Scholar] [CrossRef]
- Bosson, J.; Labrador, G.M.; Besnard, C.; Jacquemin, D.; Lacour, J. Chiral near-infrared fluorophores by self-promoted oxidative coupling of cationic helicenes with amines/enamines. Angew. Chem. 2021, 133, 8815–8820. [Google Scholar] [CrossRef]
- Wang, D.; Guo, X.; Wu, H.; Wu, Q.; Wang, H.; Zhang, X.; Hao, E.; Jiao, L. Visible light excitation of BODIPYs enables dehydrogenative enamination at their α-positions with aliphatic amines. J. Org. Chem. 2020, 85, 8360–8370. [Google Scholar] [CrossRef]
- Baleeva, N.S.; Zaitseva, S.O.; Mineev, K.S.; Khavroshechkina, A.V.; Zagudaylova, M.B.; Baranov, M.S. Enamine–azide [2+3]-cycloaddition as a method to introduce functional groups into fluorescent dyes. Tetrahedron Lett. 2019, 60, 456–459. [Google Scholar] [CrossRef]
- Lugovik, K.I.; Popova, A.V.; Eltyshev, A.K.; Benassi, E.; Belskaya, N.P. Synthesis of thiazoles bearing aryl enamine/aza-enamine side chains: Effect of the π-conjugated spacer structure and hydrogen bonding on photophysical properties. Eur. J. Org. Chem. 2017, 2017, 4175–4187. [Google Scholar] [CrossRef]
- Stanovnik, B.; Svete, J. Synthesis of heterocycles from alkyl 3-(dimethylamino)propenoates and related enaminones. Chem. Rev. 2004, 104, 2433–2480. [Google Scholar] [CrossRef]
- Ghosh, C.K.; Bhattacharyya, S.; Ghosh, C.; Patra, A. Benzopyrans. Part 41. Reactions of 2-(2-dimethylaminovinyl)-1-benzopyran-4-ones with various dienophiles. J. Chem. Soc. Perkin Trans. 1999, 1, 3005–3013. [Google Scholar] [CrossRef]
- Gümüş, M.; Koca, İ. Enamines and dimethylamino imines as building blocks in heterocyclic synthesis: Reactions of DMF-DMA reagent with different functional groups. Chemistryselect 2020, 5, 12377–12397. [Google Scholar] [CrossRef]
- Beliaev, N.A.; Shafikov, M.Z.; Efimov, I.V.; Beryozkina, T.V.; Lubec, G.; Dehaen, W.; Bakulev, V.A. Design and synthesis of imidazoles linearly connected to carbocyclic and heterocyclic rings via a 1,2,3-triazole linker. Reactivity of β-azolyl enamines towards heteroaromatic azides. New J. Chem. 2018, 42, 7049–7059. [Google Scholar] [CrossRef]
- Brouwer, A.M. Standards for photoluminescence quantum yield measurements in solution (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 2213–2228. [Google Scholar] [CrossRef] [Green Version]
- Fabian, J. TDDFT-calculations of Vis/NIR absorbing compounds. Dyes Pigm. 2010, 84, 36–53. [Google Scholar] [CrossRef]
- Chipem, F.A.S.; Mishra, A.; Krishnamoorthy, G. The role of hydrogen bonding in excited state intramolecular charge transfer. Phys. Chem. Chem. Phys. 2012, 14, 8775–8790. [Google Scholar] [CrossRef]
- Clark, B.P.; Ross, W.J.; Todd, A. 6-Substituted Pyranone Compounds and Their Use as Pharmaceuticals. U.S. Patent 4471129, 30 July 1984. [Google Scholar]
- Coffin, A.; Ready, J.M. Selective synthesis of (+)-dysoline. Org. Lett. 2019, 21, 648–651. [Google Scholar] [CrossRef]
- Chen, C.H.; Klubek, K.P.; Shi, J. Red Organic Electroluminescent Materials. U.S. Patent 5908581, 1 June 1999. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Parthasarathy, V.; Castet, F.; Pandey, R.; Mongin, O.; Das, P.K.; Blanchard-Desce, M. Unprecedented intramolecular cyclization in strongly dipolar extended merocyanine dyes: A route to novel dyes with improved transparency, nonlinear optical properties and thermal stability. Dyes Pigm. 2016, 130, 70–78. [Google Scholar] [CrossRef]
- Barca, G.M.J.; Bertoni, C.; Carrington, L.; Datta, D.; De Silva, N.; Deustua, J.E.; Fedorov, D.G.; Gour, J.R.; Gunina, A.O.; Guidez, E.; et al. Recent developments in the general atomic and molecular electronic structure system. J. Chem. Phys. 2020, 152, 154102. [Google Scholar] [CrossRef] [Green Version]
- Bode, B.M.; Gordon, M.S. Macmolplt: A graphical user interface for GAMESS. J. Mol. Graph. Model. 1998, 16, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | NMI, equiv. | Time, h | Temp., °C | Yield of 2a, % |
|---|---|---|---|---|
| 1 | 3 | 8 | 100 | 15 |
| 2 | 1 | 15 | 120 | 40 |
| 3 | 0.5 | 20 | 120 | 67 |
| 4 | 0.25 | 25 | 120 | 72 |
| 5 | – | 25 | 120 | 57 |
| 6 | 0.25 | 10 | 130 | 54 |
| Entry | R | Product | Temp., °C | Equiv. of NMI | Time, h | Yield, % |
|---|---|---|---|---|---|---|
| 1 | t-Bu | 2a | 120 | 0.25 | 15 | 72 |
| 2 | Ph | 2b | 120 | 0.25 | 12 | 53 |
| 3 | CF3 | 2c | 100 | 0.25 | 5 | 43, 12 a |
| 4 | CO2Me | 2d | 120 | 4 | 6 | 27 b, 8 c |
| 5 | PhCH = CH | 2e | 100 | 0.25 | 4 | 22 |
| 6 | 4-Me2NC6H4CH = CH | 2f | 120 | 3 | 3 | 72 |
| 7 | 4-MeOC6H4CH = CH | 2g | 120 | 3 | 3 | 51 |
| Compound | Solvent | λabs, nm a | λem, nm b | ε, M−1cm−1 | Stokes Shift, nm | QY, % c |
|---|---|---|---|---|---|---|
 | dioxane | 334 | 370 | 29,900 | 26 | <0.1 d |
| CH2Cl2 | 345 | 399 | 35,500 | 54 | <0.1 | |
| DMF | 348 | 407 | 29,500 | 59 | <0.1 | |
| DMSO | 350 | 408 | 29,200 | 58 | 0.2 | |
| i-PrOH | 356 | 415 | 35,900 | 59 | 0.3 | |
| EtOH | 361 | 428 | 29,500 | 67 | 1.4 | |
| MeOH | 363 | 434 | 35,500 | 71 | 3.6 |
| Compound | Solvent | λabs, nm a | λem,nm b | ε, M−1cm−1 | Stokes Shift, nm | QY, % c |
|---|---|---|---|---|---|---|
 | CH2Cl2 | 300 | 54,400 | |||
| 378 | 447 | 22,300 | 69 | 3.0 | ||
| DMF | 300 | 43,600 | ||||
| 378 | 450 | 17,400 | 72 | 4.1 | ||
| DMSO | 304 | 41,400 | ||||
| 381 | 455 | 17,500 | 74 | 2.3 | ||
| i-PrOH | 301 | 43,300 | ||||
| 395 | 475 | 19,000 | 80 | 11 | ||
| EtOH | 303 | 49,800 | ||||
| 403 | 485 | 21,200 | 82 | 21 | ||
| MeOH | 303 | 53,200 | ||||
| 408 | 490 | 21,000 | 83 | 28 |
| Compd. | Structure | λabs, nm a | λem, nm b | ε, M−1 cm−1 | Stokes Shift, nm | QY, % c |
|---|---|---|---|---|---|---|
| 2a |  | 364 | 433 | 32,500 | 70 | 3.0 |
| 2b |  | 274 | 24,000 | |||
| 317 | 9400 | |||||
| 380 | 527 | 19,700 | 147 | 3.4 | ||
| 2f |  | 323 | 21,200 | |||
| 368 | 572 | 16,600 | 204 | 18 | ||
| 6b |  | 325 | 49,700 | |||
| 404 | 546 | 28,200 | 142 | 3.4 | ||
| 7b |  | 286 | 24,400 | |||
| 375–348 (plateau) | 29,000 | |||||
| 433 | 571 | 23,900 | 138 | 15 |
| Entry | Compd. | Solvent | λabs, nm | fGS | λem, nm | fS1 | μGS, D | μS1, D | μ(GS->S1), D |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 4a | Vacuum | 286 | 0.718 | 293 b | 0.744 b | 8.808 | 4.246 | −4.562 |
| 2 | DMSO | 331 | 1.070 | 361 | 1.092 | 0.097 | 3.114 | 3.017 | |
| 3 | EtOH | 340 | 1.072 | 367 | 1.082 | 1.102 | 4.372 | 3.270 | |
| 4 | MeOH | 342 | 1.076 | 369 | 1.086 | 1.246 | 4.407 | 3.161 | |
| 5 | 5a | Vacuum | 252 | 0.631 | 315 | 0.363 | 10.772 | 13.084 | 2.312 |
| 6 | DMSO | 302 | 1.891 | 394 | 0.507 | 0.190 | 2.386 | 2.196 | |
| 7 | EtOH | 307 | 1.903 | 408 | 0.529 | 1.407 | 3.882 | 2.475 | |
| 8 | MeOH | 308 | 1.921 | 411 | 0.537 | 1.556 | 4.033 | 2.477 |
| Entry | Compd. | Charge | Vacuum | DMSO | EtOH | MeOH |
|---|---|---|---|---|---|---|
| 1 | 4a | GS | −0.537 | −0.640 | −0.731 | −0.746 |
| 2 | S1 | −0.303 | −0.711 | −0.823 | −0.839 | |
| 3 | delta(GS-S1) | −0.234 | 0.071 | 0.092 | 0.093 | |
| 4 | 5a | GS | −0.549 | −0.660 | −0.758 | −0.776 |
| 5 | S1 | −0.634 | −0.779 | −0.905 | −0.925 | |
| 6 | delta(GS-S1) | 0.085 | 0.119 | 0.147 | 0.149 |
| Entry | Compound | HOMO | LUMO |
|---|---|---|---|
| 1 | 4a |  −5.36 eV |  −1.36 eV |
| 2 | 5a |  −5.06 eV |  −1.41 eV |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Obydennov, D.L.; Nigamatova, D.I.; Shirinkin, A.S.; Melnikov, O.E.; Fedin, V.V.; Usachev, S.A.; Simbirtseva, A.E.; Kornev, M.Y.; Sosnovskikh, V.Y. 2-(2-(Dimethylamino)vinyl)-4H-pyran-4-ones as Novel and Convenient Building-Blocks for the Synthesis of Conjugated 4-Pyrone Derivatives. Molecules 2022, 27, 8996. https://doi.org/10.3390/molecules27248996
Obydennov DL, Nigamatova DI, Shirinkin AS, Melnikov OE, Fedin VV, Usachev SA, Simbirtseva AE, Kornev MY, Sosnovskikh VY. 2-(2-(Dimethylamino)vinyl)-4H-pyran-4-ones as Novel and Convenient Building-Blocks for the Synthesis of Conjugated 4-Pyrone Derivatives. Molecules. 2022; 27(24):8996. https://doi.org/10.3390/molecules27248996
Chicago/Turabian StyleObydennov, Dmitrii L., Diana I. Nigamatova, Alexander S. Shirinkin, Oleg E. Melnikov, Vladislav V. Fedin, Sergey A. Usachev, Alena E. Simbirtseva, Mikhail Y. Kornev, and Vyacheslav Y. Sosnovskikh. 2022. "2-(2-(Dimethylamino)vinyl)-4H-pyran-4-ones as Novel and Convenient Building-Blocks for the Synthesis of Conjugated 4-Pyrone Derivatives" Molecules 27, no. 24: 8996. https://doi.org/10.3390/molecules27248996