
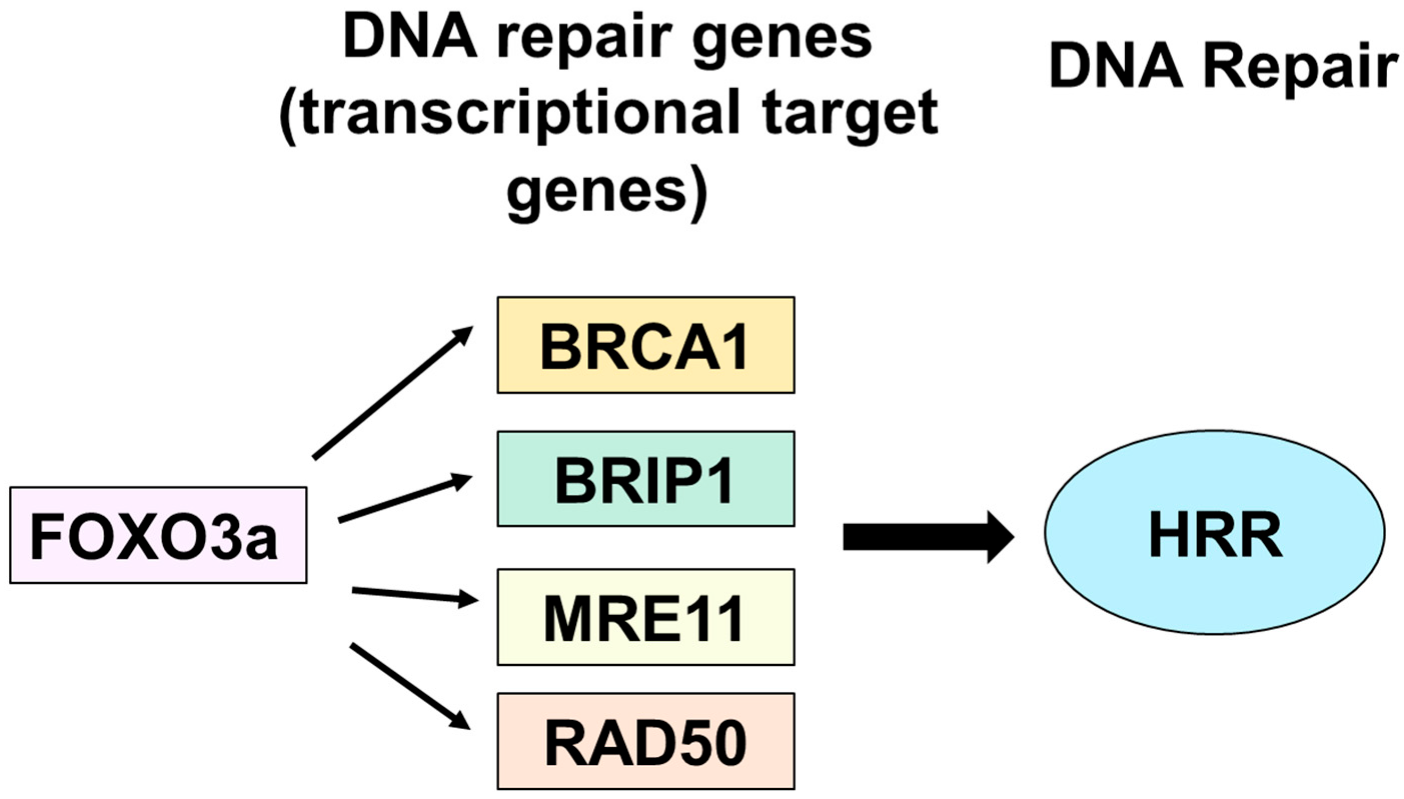
FOXO3a Mediates Homologous Recombination Repair (HRR) via Transcriptional Activation of MRE11, BRCA1, BRIP1, and RAD50

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
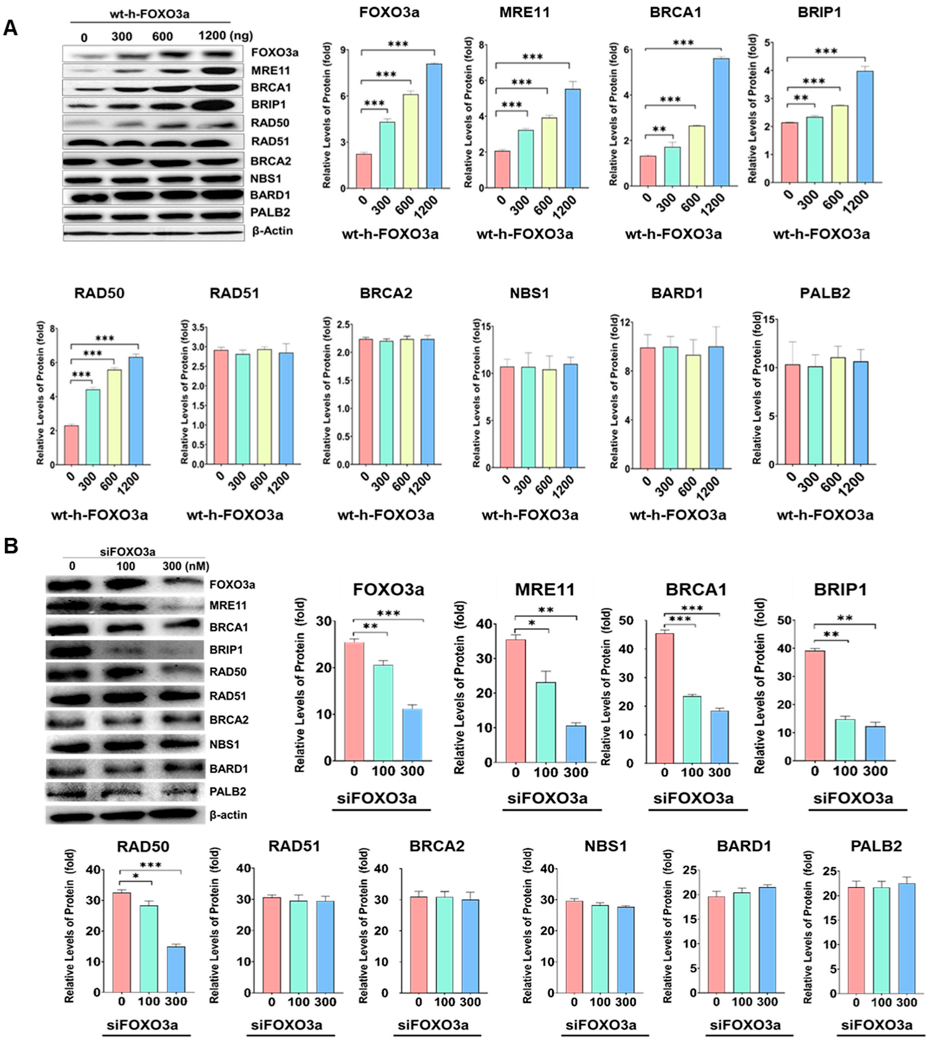
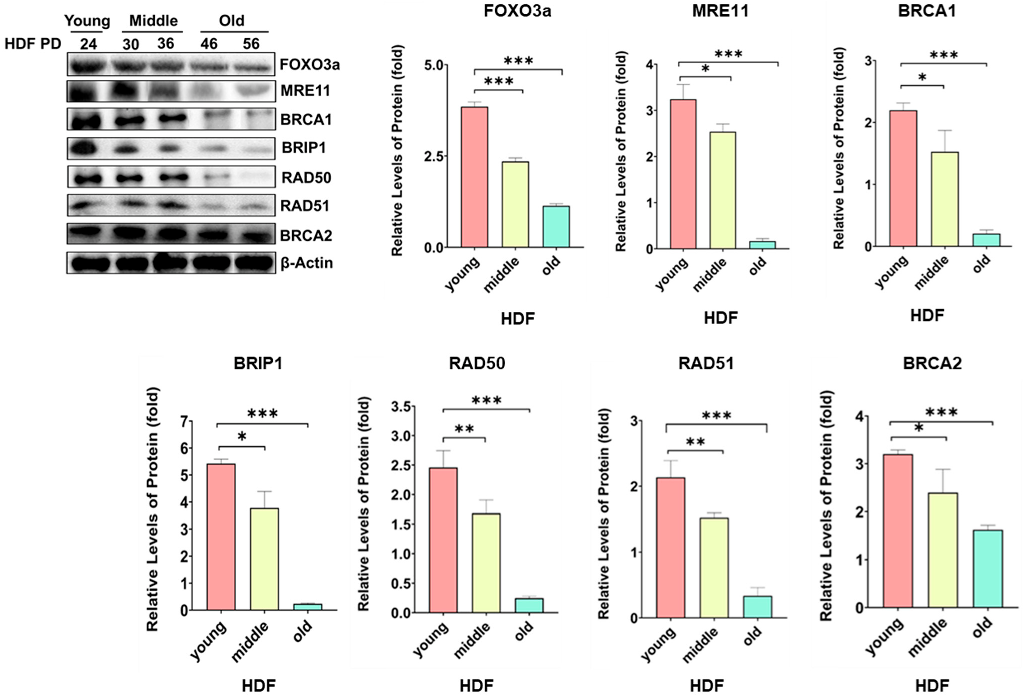
2.1. Protein Levels of MRE11, BRCA1, BRIBP1, and RAD50 Were Regulated by FOXO3a in Cellular-Aging-Dependent Manner in HDF
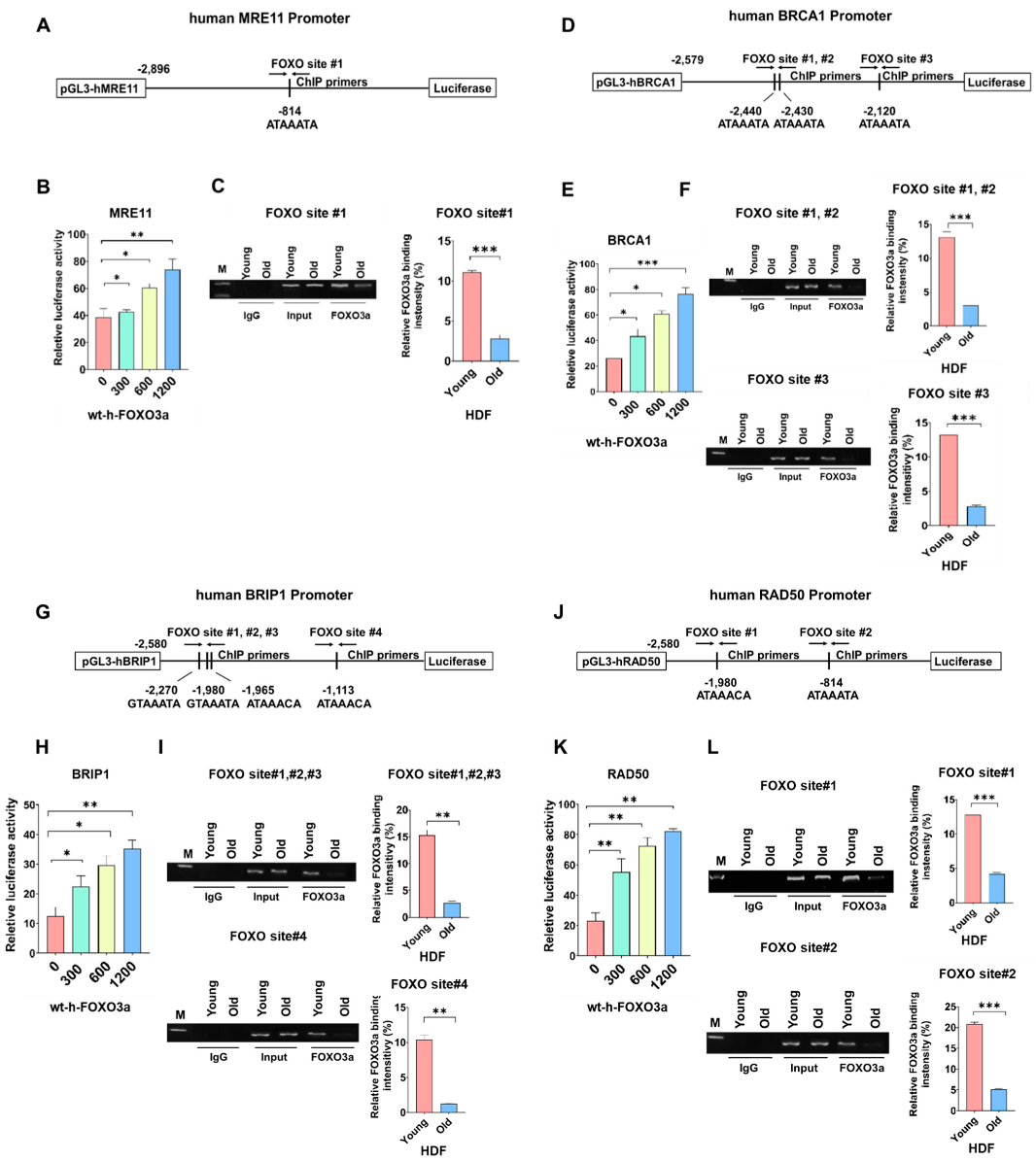
2.2. Promoter Activities of Human MRE11, BRCA1, BRIP1, and RAD50 were FOXO3a-Dependent and ChIP Assay on FOXO Consensus Sites of Human MRE11, BRCA1, BRIP1, and RAD50 Promoters Showed Cellular-Aging-Dependent Decrease in FOXO3a Binding
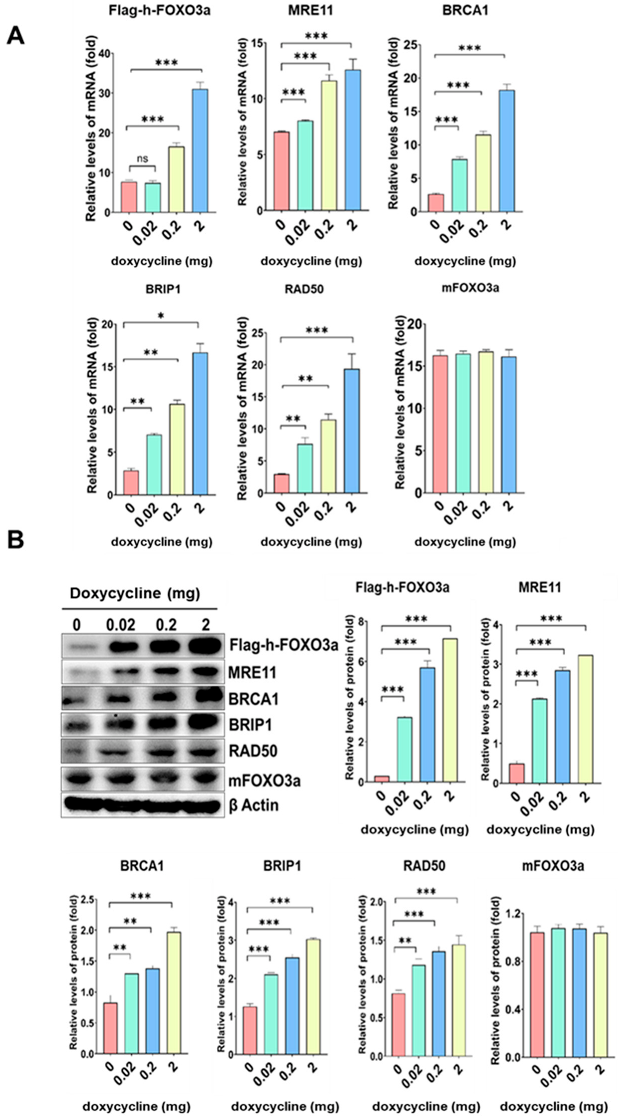
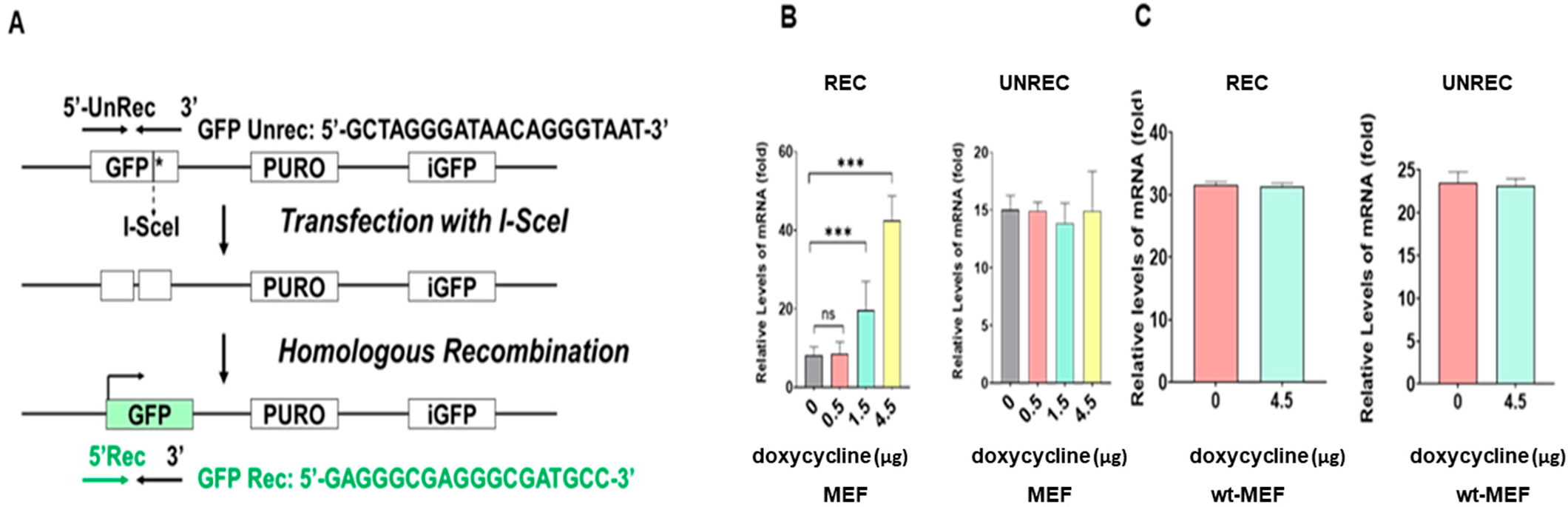
2.3. The mRNA and Protein Levels of MRE11, BRCA1, BRIP1, and RAD50 Increased in a Doxycycline-Dose-Dependent Manner in Tet-On h-FOXO3a Transgenic Mice
2.4. In Vitro HRR Activity Increased in a Doxycycline-Dose-Dependent Manner in MEF Obtained from Embryos of Tet-On h-FOXO3a Transgenic Mice and Decreased during Cellular Aging of HDF
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transfections
4.2. Primary Mouse Embryonic Fibroblasts
4.3. Western Blot Analysis
4.4. Plasmid Constructs
4.5. Promoter Reporter Assay
4.6. ChIP Assay
4.7. RNA Extraction and Quantitative Real-Time RT-PCR (RT-qPCR)
4.8. Development of the Tet-On Flag-h-FOXO3a Transgenic Mice
4.9. In Vitro Homologous Recombination Repair (HRR) Assay
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Lieber, M.R. The Mechanism of Human Nonhomologous DNA End Joining. J. Biol. Chem. 2008, 283, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.D. Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J. 2000, 19, 3398–3407. [Google Scholar] [CrossRef] [PubMed]
- Krejci, L.; Altmannova, V.; Spirek, M.; Zhao, X. Homologous recombination and its regulation. Nucleic Acids Res. 2012, 40, 5795–5818. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyman, C.; Ristic, D.; Kanaar, R. Homologous recombination-mediated double-strand break repair. DNA Repair 2004, 3, 827–833. [Google Scholar] [CrossRef]
- Seluanov, A.; Mittelman, D.; Pereira-Smith, O.M.; Wilson, J.H.; Gorbunova, V. DNA end joining becomes less efficient and more error-prone during cellular senescence. Proc. Natl. Acad. Sci. USA 2004, 101, 7624–7629. [Google Scholar] [CrossRef] [Green Version]
- Vyjayanti, V.; Rao, K.S. DNA double strand break repair in brain: Reduced NHEJ activity in aging rat neurons. Neurosci. Lett. 2006, 393, 18–22. [Google Scholar] [CrossRef]
- Lee, J.-E.; Heo, J.-I.; Park, S.-H.; Kim, J.-H.; Kho, Y.-J.; Kang, H.-J.; Chung, H.Y.; Yoon, J.-L. Calorie restriction (CR) reduces age-dependent decline of non-homologous end joining (NHEJ) activity in rat tissues. Exp. Gerontol. 2011, 46, 891–896. [Google Scholar] [CrossRef]
- Vaidya, A.; Mao, Z.; Tian, X.; Spencer, B.; Seluanov, A.; Gorbunova, V. Knock-In Reporter Mice Demonstrate that DNA Repair by Non-homologous End Joining Declines with Age. PLoS Genet. 2014, 10, e1004511. [Google Scholar] [CrossRef] [Green Version]
- White, R.R.; Sung, P.; Vestal, C.G.; Benedetto, G.; Cornelio, N.; Richardson, C. Double-Strand Break Repair by Interchromosomal Recombination: An In Vivo Repair Mechanism Utilized by Multiple Somatic Tissues in Mammals. PLoS ONE 2013, 8, e84379. [Google Scholar] [CrossRef]
- Sukup-Jackson, M.R.; Kiraly, O.; Kay, J.E.; Na, L.; Rowland, E.A.; Winther, K.E.; Chow, D.N.; Kimoto, T.; Matsuguchi, T.; Jonnalagadda, V.S.; et al. Rosa26-GFP Direct Repeat (RaDR-GFP) Mice Reveal Tissue- and Age-Dependence of Homologous Recombination in Mammals In Vivo. PLoS Genet. 2014, 10, e1004299. [Google Scholar] [CrossRef] [Green Version]
- Mao, Z.; Hine, C.; Tian, X.; Meter, M.V.; Au, M.; Vaidya, A.; Seluanov, A.; Gorbunova, V. SIRT6 promotes DNA repair under stress by activating PARP1. Science 2011, 332, 1443–1446. [Google Scholar] [CrossRef] [Green Version]
- Mao, Z.; Tian, X.; Meter, M.V.; Ke, Z.; Gorbunova, V.; Seluanov, A. Sirtuin 6 (SIRT6) rescues the decline of homologous recombination repair during replicative senescence. Proc. Natl. Acad. Sci. USA 2012, 109, 11800–11805. [Google Scholar] [CrossRef] [Green Version]
- Furuyama, T.; Nakazawa, T.; Nakano, I.; Mori, N. Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem. J. 2000, 349, 629–634. [Google Scholar] [CrossRef]
- Greer, E.L.; Brunet, A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar] [CrossRef] [Green Version]
- Monsalve, M. The Complex Biology of FOXO. Curr. Drug Targets 2011, 12, 1322–1350. [Google Scholar] [CrossRef] [Green Version]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt Promotes Cell Survival by Phosphorylating and Inhibiting a Forkhead Transcription Factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Biggs, W.H.; Meisenhelder, J.; Hunter, T.; Cavenee, W.K.; Arden, K.C. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl. Acad. Sci. USA 1999, 96, 7421–7426. [Google Scholar] [CrossRef] [Green Version]
- Greer, E.L.; Oskoui, P.R.; Banko, M.R.; Maniar, J.M.; Gygi, M.P.; Gygi, S.P.; Brunet, A. The Energy Sensor AMP-activated Protein Kinase Directly Regulates the Mammalian FOXO3 Transcription Factor. J. Biol. Chem. 2007, 282, 30107–30119. [Google Scholar] [CrossRef] [Green Version]
- Essers, M.; Weijzen, S.; De Vries-Smits, A.M.M.; Saarloos, I.; De Ruiter, N.D.; Bos, J.L.; Burgering, B.M.T. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 2004, 23, 4802–4812. [Google Scholar] [CrossRef]
- Oh, S.W.; Mukhopadhyay, A.; Svrzikapa, N.; Jiang, F.; Davis, R.J.; Tissenbaum, H.A. JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of forkhead transcription factor/DAF-16. Proc. Natl. Acad. Sci. USA 2005, 102, 4494–4499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehtinen, M.K.; Yuan, Z.; Boag, P.R.; Yang, Y.; Villén, J.; Becker, E.B.; DiBacco, S.; de la Iglesia, N.; Gygi, S.; Blackwell, T.K.; et al. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell 2006, 125, 987–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.Y.; Zong, C.S.; Xia, W.; Yamaguchi, H.; Ding, Q.; Xie, X.; Lang, J.Y.; Lai, C.C.; Chang, C.J.; Huang, W.C.; et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated deg-radation. Nat. Cell Biol. 2008, 10, 138–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asada, S.; Daitoku, H.; Matsuzaki, H.; Saito, T.; Sudo, T.; Mukai, H.; Iwashita, S.; Kako, K.; Kishi, T.; Kasuya, Y.; et al. Mitogen-activated protein kinases, Erk and p38, phosphorylate and regulate Foxo1. Cell Signal 2007, 19, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef] [Green Version]
- Van der Horst, A.; Tertoolen, L.G.J.; de Vries-Smits, L.M.M.; Frye, R.A.; Medema, R.H.; Burgering, B.M.T. FOXO4 Is Acetylated upon Peroxide Stress and Deacetylated by the Longevity Protein hSir2. J. Biol. Chem. 2004, 279, 28873–28879. [Google Scholar] [CrossRef] [Green Version]
- Calnan, D.R.; Webb, A.E.; White, J.L.; Stowe, T.R.; Goswami, T.; Shi, X.; Espejo, A.; Bedford, M.T.; Gozani, O.; Gygi, S.P.; et al. Methylation by Set9 modulates FoxO3 stability and transcriptional activity. Aging 2012, 4, 462–479. [Google Scholar] [CrossRef] [Green Version]
- Yamagata, K.; Daitoku, H.; Takahashi, Y.; Namiki, K.; Hisatake, K.; Kako, K.; Mukai, H.; Kasuya, Y.; Fukamizu, A. Arginine Methylation of FOXO Transcription Factors Inhibits Their Phosphorylation by Akt. Mol. Cell 2008, 32, 221–231. [Google Scholar] [CrossRef]
- Eijkelenboom, A.; Burgering, B.M.T. FOXOs: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef]
- Tran, H.; Brunet, A.; Grenier, J.M.; Datta, S.R.; Fornace, A.J.; DiStefano, P.S.; Chiang, L.W.; Greenberg, M.E. DNA Repair Pathway Stimulated by the Forkhead Transcription Factor FOXO3a Through the Gadd45 Protein. Science 2002, 296, 530–534. [Google Scholar] [CrossRef]
- White, R.R.; Maslov, A.Y.; Lee, M.; Wilner, S.E.; Levy, M.; Vijg, J. FOXO3a acts to suppress DNA double-strand break-induced mutations. Aging Cell 2020, 19, e13184. [Google Scholar] [CrossRef]
- Shao, L.; Feng, W.; Lee, K.-J.; Chen, B.P.C.; Zhou, D. A Sensitive and Quantitative Polymerase Chain Reaction-Based Cell Free In Vitro Non-Homologous End Joining Assay for Hematopoietic Stem Cells. PLoS ONE 2012, 7, e33499. [Google Scholar] [CrossRef] [Green Version]
- Cuozzo, C.; Porcellini, A.; Angrisano, T.; Morano, A.; Lee, B.; Di Pardo, A.; Messina, S.; Iuliano, R.; Fusco, A.; Santillo, M.R.; et al. DNA Damage, Homology-Directed Repair, and DNA Methylation. PLoS Genet. 2007, 3, e110. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Nguyen, L.W.M.; Martens, J.; Van Hoeck, A.; Cuppen, E. Pan-cancer landscape of homologous recombination deficiency. Nat. Commun. 2020, 11, 5584. [Google Scholar] [CrossRef]
- Royfman, R.; Whiteley, E.; Noe, O.; Morand, S.; Creeden, J.; Stanbery, L.; Hamouda, D.; Nemunaitis, J. BRCA1/2 signaling and homologous recombination deficiency in breast and ovarian cancer. Futur. Oncol. 2021, 17, 2817–2830. [Google Scholar] [CrossRef]
- Meindl, A.; Hellebrand, H.; Wiek, C.; Erven, V.; Wappenschmidt, B.; Niederacher, D.; Freund, M.; Lichtner, P.; Hartmann, L.; Schaal, H.; et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat. Genet. 2010, 42, 410–414. [Google Scholar] [CrossRef]
- Monteiro, L.J.; Khongkow, P.; Kongsema, M.; Morris, J.R.; Man, C.; Weekes, D.; Koo, C.-Y.; Gomes, A.R.; Pinto, P.H.; Varghese, V.; et al. The Forkhead Box M1 protein regulates BRIP1 expression and DNA damage repair in epirubicin treatment. Oncogene 2012, 32, 4634–4645. [Google Scholar] [CrossRef]
- Khongkow, M.; Karunarathna, U.; Gong, C.; Gomes, A.R.; Yagüe, E.; Monteiro, L.J.; Kongsema, M.; Zona, S.; Man, E.P.S.; Tsang, J.W.-H.; et al. FOXM1 targets NBS1 to regulate DNA damage-induced senescence and epirubicin resistance. Oncogene 2013, 33, 4144–4155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Im, J.; Lawrence, J.; Seelig, D.; Nho, R.S. FoxM1-dependent RAD51 and BRCA2 signaling protects idiopathic pulmonary fibrosis fibroblasts from radiation-induced cell death. Cell Death Dis. 2018, 9, 584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halasi, M.; Gartel, A.L. FOX(M1) News—It Is Cancer. Mol. Cancer Ther. 2013, 12, 245–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wierstra, I. FOXM1 (Forkhead box M1) in tumorigenesis: Overexpression in human cancer, implication in tumorigenesis, oncogenic functions, tumor-suppressive properties, and target of anticancer therapy. Adv. Cancer Res. 2013, 119, 191–419. [Google Scholar] [CrossRef]
- Liu, H.; Song, Y.; Qiu, H.; Liu, Y.; Luo, K.; Yi, Y.; Jiang, G.; Lu, M.; Zhang, Z.; Yin, J.; et al. Downregulation of FOXO3a by DNMT1 promotes breast cancer stem cell properties and tumorigenesis. Cell Death Differ. 2019, 27, 966–983. [Google Scholar] [CrossRef] [Green Version]
- Tsai, W.-B.; Chung, Y.M.; Takahashi, Y.; Xu, Z.; Hu, M.C.-T. Functional interaction between FOXO3a and ATM regulates DNA damage response. Nature 2008, 10, 460–467. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.K.; Kim, Y.K.; Song, I.-H.; Baek, S.-H.; Lee, S.-R.; Kim, J.H. Down-Regulation of a Forkhead Transcription Factor, FOXO3a, Accelerates Cellular Senescence in Human Dermal Fibroblasts. J. Gerontol. Ser. A 2005, 60, 4–9. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inci, G.; Warkad, M.S.; Kang, B.-G.; Lee, N.-K.; Suh, H.-W.; Lim, S.S.; Kim, J.; Kim, S.-C.; Lee, J.-Y. FOXO3a Mediates Homologous Recombination Repair (HRR) via Transcriptional Activation of MRE11, BRCA1, BRIP1, and RAD50. Molecules 2022, 27, 8623. https://doi.org/10.3390/molecules27238623
Inci G, Warkad MS, Kang B-G, Lee N-K, Suh H-W, Lim SS, Kim J, Kim S-C, Lee J-Y. FOXO3a Mediates Homologous Recombination Repair (HRR) via Transcriptional Activation of MRE11, BRCA1, BRIP1, and RAD50. Molecules. 2022; 27(23):8623. https://doi.org/10.3390/molecules27238623
Chicago/Turabian StyleInci, Gozde, Madhuri Shende Warkad, Beom-Goo Kang, Na-Kyung Lee, Hong-Won Suh, Soon Sung Lim, Jaebong Kim, Sung-Chan Kim, and Jae-Yong Lee. 2022. "FOXO3a Mediates Homologous Recombination Repair (HRR) via Transcriptional Activation of MRE11, BRCA1, BRIP1, and RAD50" Molecules 27, no. 23: 8623. https://doi.org/10.3390/molecules27238623