
The Formation of Inherently Chiral Calix[4]quinolines by Doebner–Miller Reaction of Aldehydes and Aminocalixarenes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
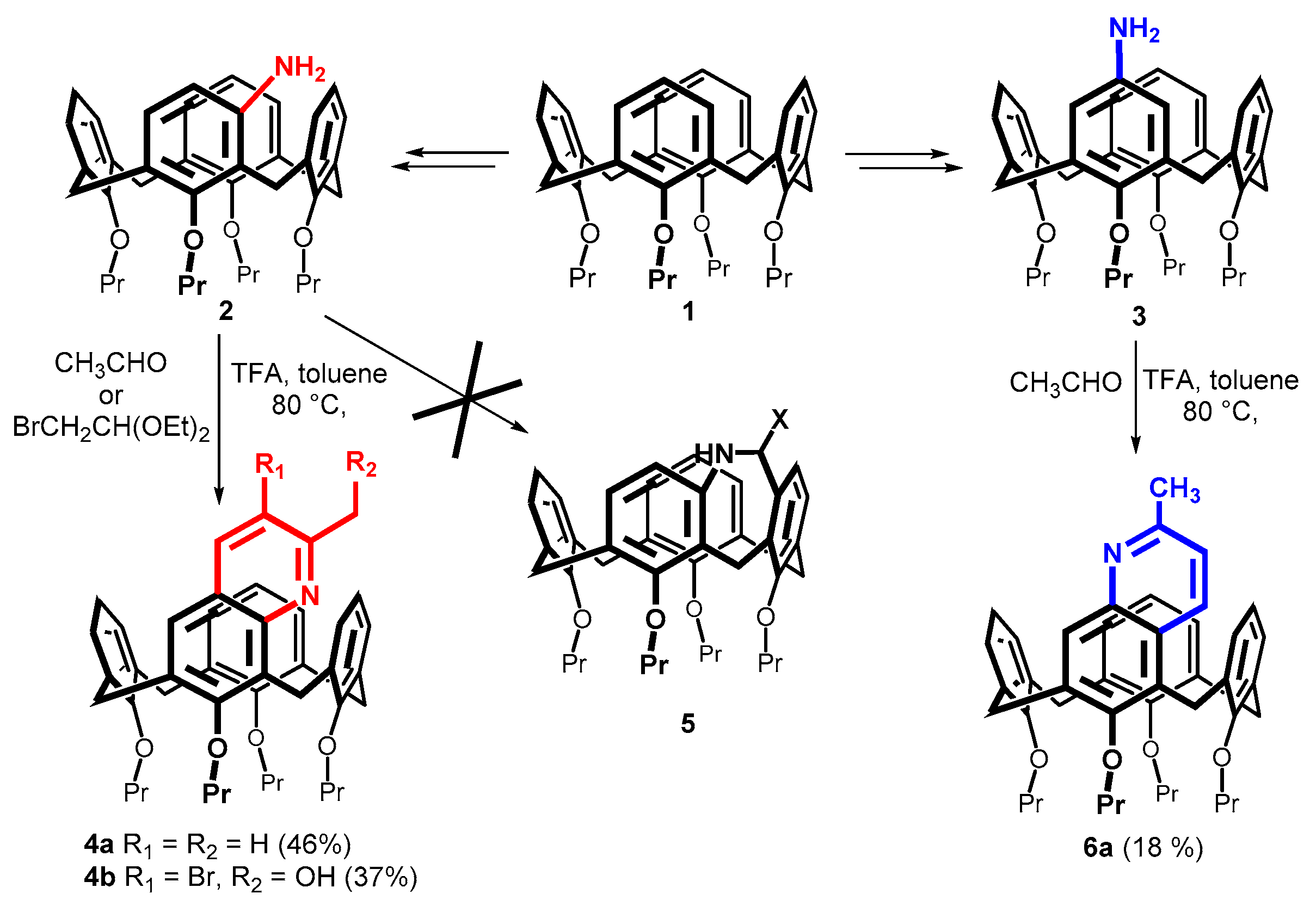
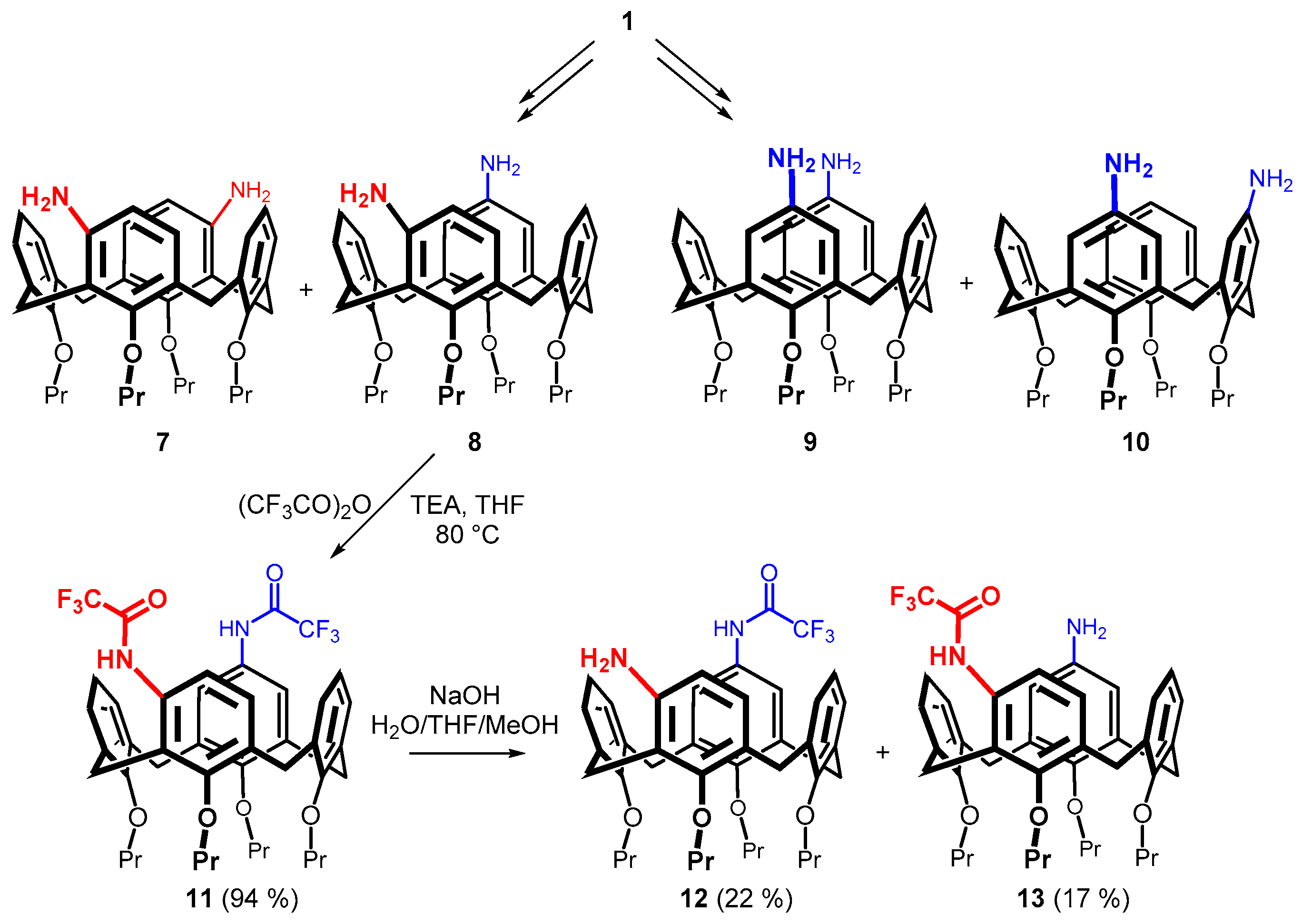
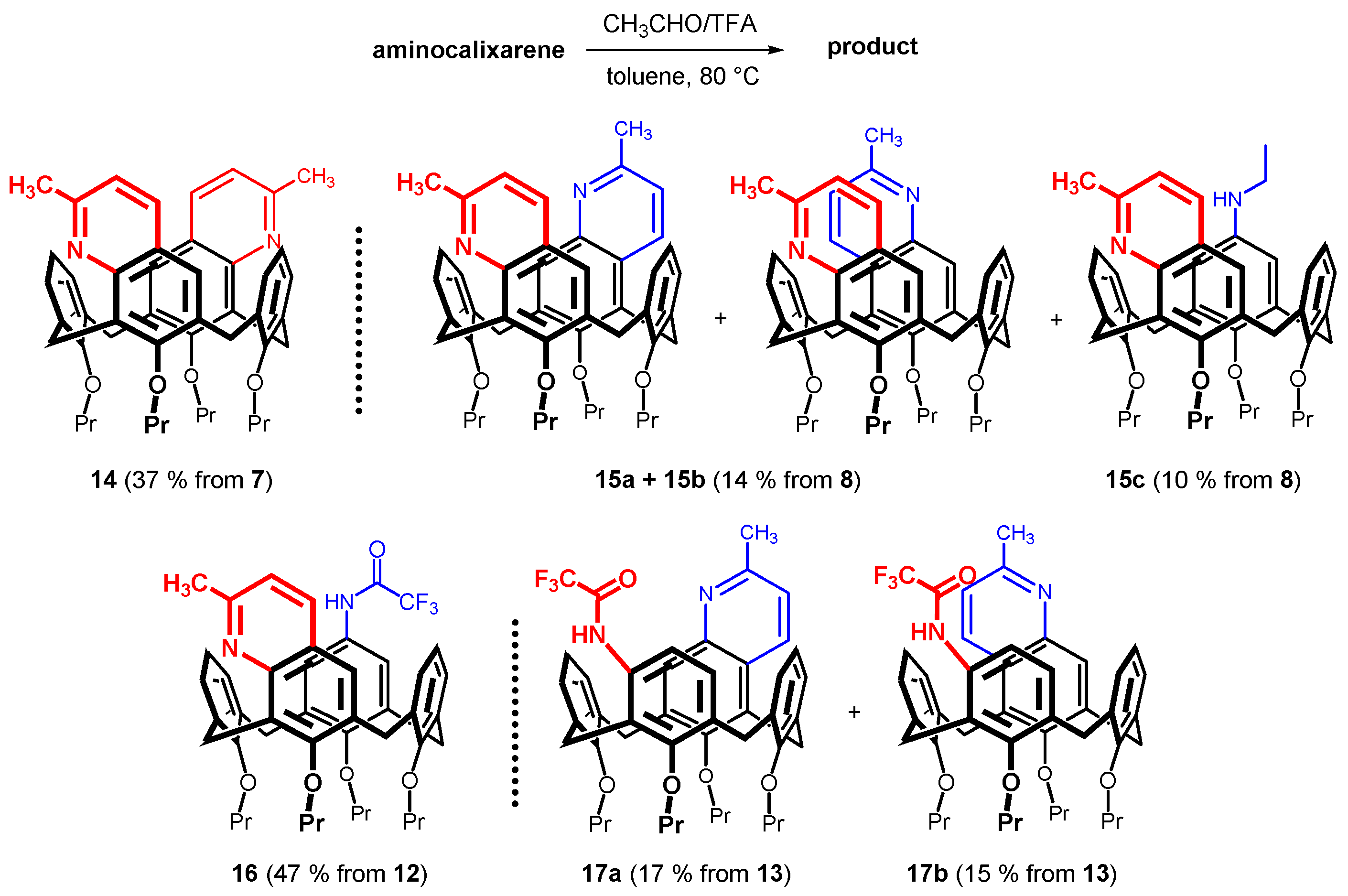
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Synthetic Procedures
3.2.1. Quinoline Derivative 4a
3.2.2. Quinoline Derivative 4b
3.2.3. Quinoline Derivative 6a
3.2.4. Bis-Quinoline Derivative 14
3.2.5. Quinoline Derivatives 15a and 15b
3.2.6. Quinoline Derivative 15c
3.2.7. Quinoline Derivative 16
3.2.8. Quinoline Derivative 17a
3.2.9. Quinoline Derivative 17b
3.3. Chiral Separation
3.4. NMR Titrations
3.5. X-ray Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gutsche, C.D. Calixarenes: An Introduction; RSC Publishing: Cambridge, UK, 2008. [Google Scholar]
- Mandolini, L.; Ungaro, R. Calixarenes in Action; Imperial College Press: London, UK, 2000. [Google Scholar]
- Vicens, J.; Harrowfield, J.; Baklouti, L. Calixarenes in the Nanoworld; Springer: Dordrecht, The Netherlands, 2007. [Google Scholar]
- Neri, P.; Sessler, J.L.; Wang, M.X. Calixarenes and Beyond; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Leray, I.; Valeur, B. Calixarene-Based Fluorescent Molecular Sensors for Toxic Metals. Eur. J. Inorg. Chem. 2009, 24, 3525–3535. [Google Scholar] [CrossRef]
- Siddiqui, S.; Cragg, P.J. Design and Synthesis of Transition Metal and Inner Transition Metal Binding Calixarenes. Mini-Rev. Org. Chem. 2009, 6, 283–299. [Google Scholar] [CrossRef]
- Mutihac, L.; Buschmann, H.-J.; Mutihac, R.-C.; Schollmeyer, E. Complexation and separation of amines, amino acids, and peptides by functionalized calix[n]arenes. J. Incl. Phenom. Macrocyclic Chem. 2005, 51, 1–10. [Google Scholar] [CrossRef]
- Lhotak, P. Anion receptors based on calixarenes. Top. Curr. Chem. 2005, 255, 65–95. [Google Scholar]
- Kongor, A.R.; Mehta, V.A.; Modi, K.M.; Panchal, M.K.; Dey, S.A.; Panchal, U.S.; Jain, V.K. Calix-Based Nanoparticles: A Review. Top. Curr. Chem. 2016, 374, 1–46. [Google Scholar] [CrossRef]
- Szumna, A. Inherently chiral concave molecules-from synthesis to applications. Chem. Soc. Rev. 2010, 39, 4274–4285. [Google Scholar] [CrossRef] [PubMed]
- Arnott, G.E. Inherently Chiral Calixarenes: Synthesis and Applications. Chem.-Eur. J. 2018, 24, 1744–1754. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-Y.; Xu, Y.-W.; Liu, Y.-W.; Su, C.-Y. Inherently chiral calixarenes. Synthesis, optical resolution, chiral recognition and asymmetric catalysis. Int. J. Mol. Sci. 2011, 12, 429–455. [Google Scholar] [CrossRef]
- Lhoták, P. Direct meta substitution of calix[4]arenes. Org. Biomol. Chem. 2022, 20, 7377–7390. [Google Scholar] [CrossRef]
- Slavik, P.; Dudic, M.; Flidrova, K.; Sykora, J.; Cisarova, I.; Bohm, S.; Lhotak, P. Unprecedented Meta-Substitution of Calixarenes: Direct Way to Inherently Chiral Derivatives. Org. Lett. 2012, 14, 3628–3631. [Google Scholar] [CrossRef]
- Ikeda, A.; Yoshimura, M.; Lhotak, P.; Shinkai, S. Synthesis and optical resolution of naphthalene-containing inherently chiral calix[4] arenes derived by intramolecular ring closure or stapling of proximal phenyl units. J. Chem. Soc. Perkin Trans. 1996, 1, 1945–1950. [Google Scholar] [CrossRef]
- Hueggenberg, W.; Seper, A.; Oppel, I.M.; Dyker, G. Multifold photocyclization reactions of styrylcalix[4]arenes. Eur. J. Org. Chem. 2010, 6786–6797. [Google Scholar] [CrossRef]
- Elaieb, F.; Semeril, D.; Matt, D.; Pfeffer, M.; Bouit, P.-A.; Hissler, M.; Gourlaouen, C.; Harrowfield, J. Calix[4]arene-fused phospholes. Dalton Trans. 2017, 46, 9833–9845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tlusty, M.; Dvorakova, H.; Cejka, J.; Kohout, M.; Lhotak, P. Regioselective formation of the quinazoline moiety on the upper rim of calix[4]arene as a route to inherently chiral systems. New J. Chem. 2020, 44, 6490–6500. [Google Scholar] [CrossRef]
- Miao, R.; Zheng, Q.-Y.; Chen, C.-F.; Huang, Z.-T. Efficient Syntheses and Resolutions of Inherently Chiral Calix[4]quinolines in the Cone and Partial-Cone Conformation. J. Org. Chem. 2005, 70, 7662–7671. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Ledeboer, M.W.; Duffy, J.P.; Pierce, A.C.; Zuccola, H.J.; Block, E.; Shlyakter, D.; Hogan, J.K.; Bennani, Y.L. A novel chemotype of kinase inhibitors: Discovery of 3,4-ring fused 7-azaindoles and deazapurines as potent JAK2 inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Xiang, J.; Dang, Q.; Guo, S.; Bai, X. Design and synthesis of a tetracyclic pyrimidine-fused benzodiazepine library. J. Comb. Chem. 2006, 8, 381–387. [Google Scholar] [CrossRef]
- Tlusty, M.; Slavik, P.; Kohout, M.; Eigner, V.; Lhotak, P. Inherently Chiral Upper-Rim-Bridged Calix[4]arenes Possessing a Seven Membered Ring. Org. Lett. 2017, 19, 2933–2936. [Google Scholar] [CrossRef]
- Tlusty, M.; Eigner, V.; Babor, M.; Kohout, M.; Lhotak, P. Synthesis of upper rim-double-bridged calix[4]arenes bearing seven membered rings and related compounds. RSC Adv. 2019, 9, 22017–22030. [Google Scholar] [CrossRef] [Green Version]
- Liska, A.; Flidrova, K.; Lhotak, P.; Ludvik, J. Influence of structure on electrochemical reduction of isomeric mono- and di-, nitro- or nitrosocalix[4]arenes. Monatsh. Chem. 2015, 146, 857–862. [Google Scholar] [CrossRef]
- Wu, Y.-C.; Liu, L.; Li, H.-J.; Wang, D.; Chen, Y.-J. Skraup−Doebner−Von Miller Quinoline Synthesis Revisited: Reversal of the Regiochemistry for γ-Aryl-β,γ-unsaturated α-Ketoesters. J. Org. Chem. 2006, 71, 6592–6595. [Google Scholar] [CrossRef] [PubMed]
- Su, L.-L.; Zheng, Y.-W.; Wang, W.-G.; Chen, B.; Wei, X.-Z.; Wu, L.-Z.; Tung, C.-H. Photocatalytic Synthesis of Quinolines via Povarov Reaction under Oxidant-Free Conditions. Org. Lett. 2022, 24, 1180–1185. [Google Scholar] [CrossRef] [PubMed]
- Kelderman, E.; Verboom, W.; Engbersen, J.F.; Reinhoudt, D.N.; Heesink, G.J.; van Hulst, N.F.; Derhaeg, L.; Persoons, A. Nitrocalix[4] arenes as molecules for second-order nonlinear optics. Angew. Chem. Int. Ed. Engl. 1992, 31, 1075–1077. [Google Scholar] [CrossRef] [Green Version]
- van Wageningen, A.M.; Snip, E.; Verboom, W.; Reinhoudt, D.N.; Boerrigter, H. Synthesis and Application of Iso (thio) cyanate-Functionalized Calix[4] arenes. Liebigs Ann. 1997, 1997, 2235–2245. [Google Scholar] [CrossRef] [Green Version]
- Flidrova, K.; Bohm, S.; Dvorakova, H.; Eigner, V.; Lhotak, P. Dimercuration of Calix[4]arenes: Novel Substitution Pattern in Calixarene Chemistry. Org. Lett. 2014, 16, 138–141. [Google Scholar] [CrossRef] [PubMed]
- The Binding Constants Were Calculated Using the Bindfit Application Freely. Available online: http://supramolecular.org (accessed on 30 October 2022).
- Palatinus, L.; Chapuis, G. SUPERFLIP–a computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Crystallogr. 2007, 40, 786–790. [Google Scholar] [CrossRef] [Green Version]
- Betteridge, P.; Carruthers, J.; Cooper, R.; Prout, K.; Watkin, D. CRYSTALS version 12: Software for guided crystal structure analysis. J. Appl. Crystallogr. 2003, 36, 1487. [Google Scholar] [CrossRef]
- Rohlíček, J.; Hušák, M. MCE2005–a new version of a program for fast interactive visualization of electron and similar density maps optimized for small molecules. J. Appl. Crystallogr. 2007, 40, 600–601. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tlustý, M.; Eigner, V.; Dvořáková, H.; Lhoták, P. The Formation of Inherently Chiral Calix[4]quinolines by Doebner–Miller Reaction of Aldehydes and Aminocalixarenes. Molecules 2022, 27, 8545. https://doi.org/10.3390/molecules27238545
Tlustý M, Eigner V, Dvořáková H, Lhoták P. The Formation of Inherently Chiral Calix[4]quinolines by Doebner–Miller Reaction of Aldehydes and Aminocalixarenes. Molecules. 2022; 27(23):8545. https://doi.org/10.3390/molecules27238545
Chicago/Turabian StyleTlustý, Martin, Václav Eigner, Hana Dvořáková, and Pavel Lhoták. 2022. "The Formation of Inherently Chiral Calix[4]quinolines by Doebner–Miller Reaction of Aldehydes and Aminocalixarenes" Molecules 27, no. 23: 8545. https://doi.org/10.3390/molecules27238545