
Theoretical Studies of Leu-Pro-Arg-Asp-Ala Pentapeptide (LPRDA) Binding to Sortase A of Staphylococcus aureus


Abstract
:1. Introduction
2. Results
2.1. PDB Structure Validation
2.1.1. Evidence from the Literature
The Intrinsic Motions and Partial Disorder
The Use in Molecular Modeling
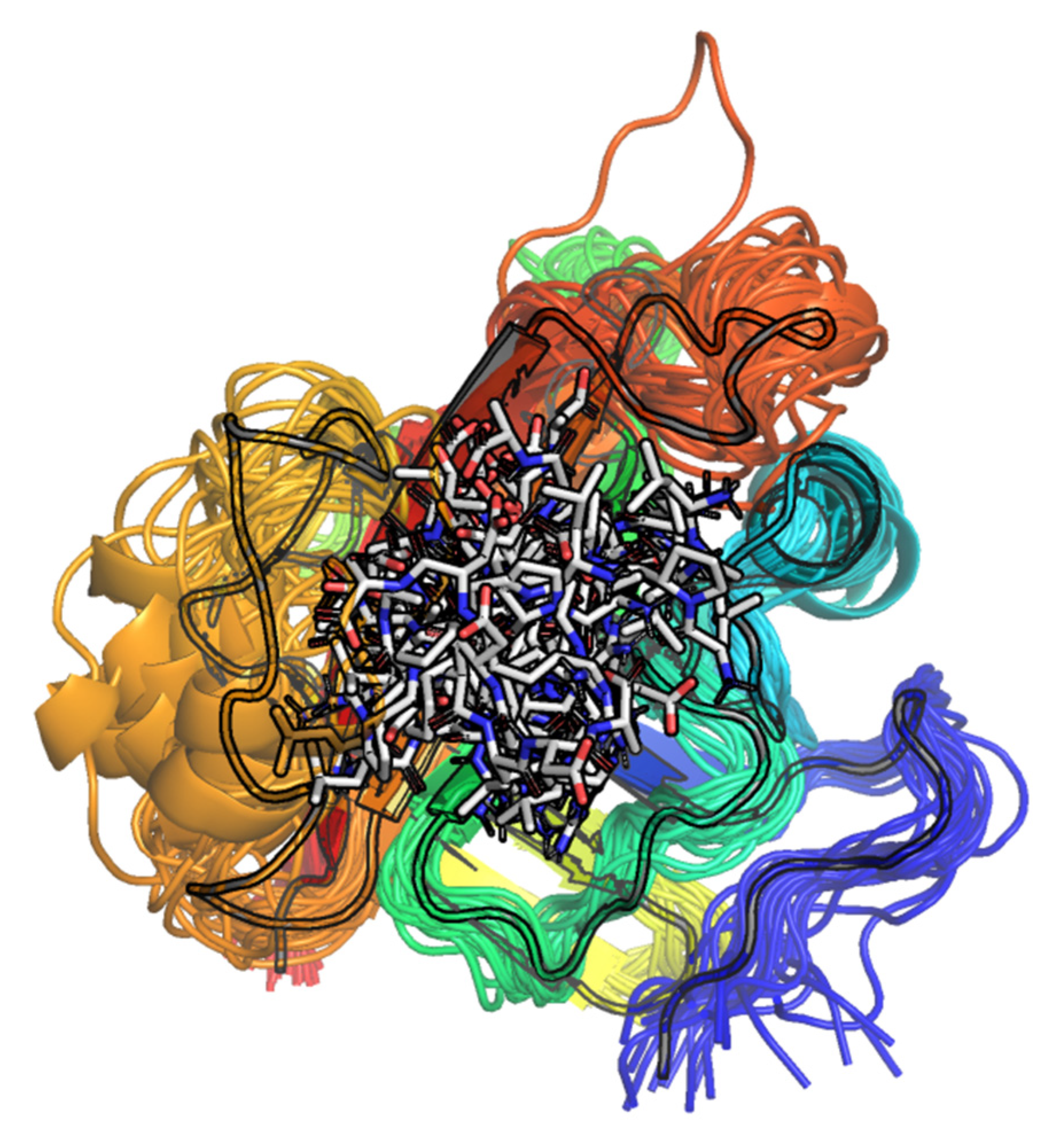
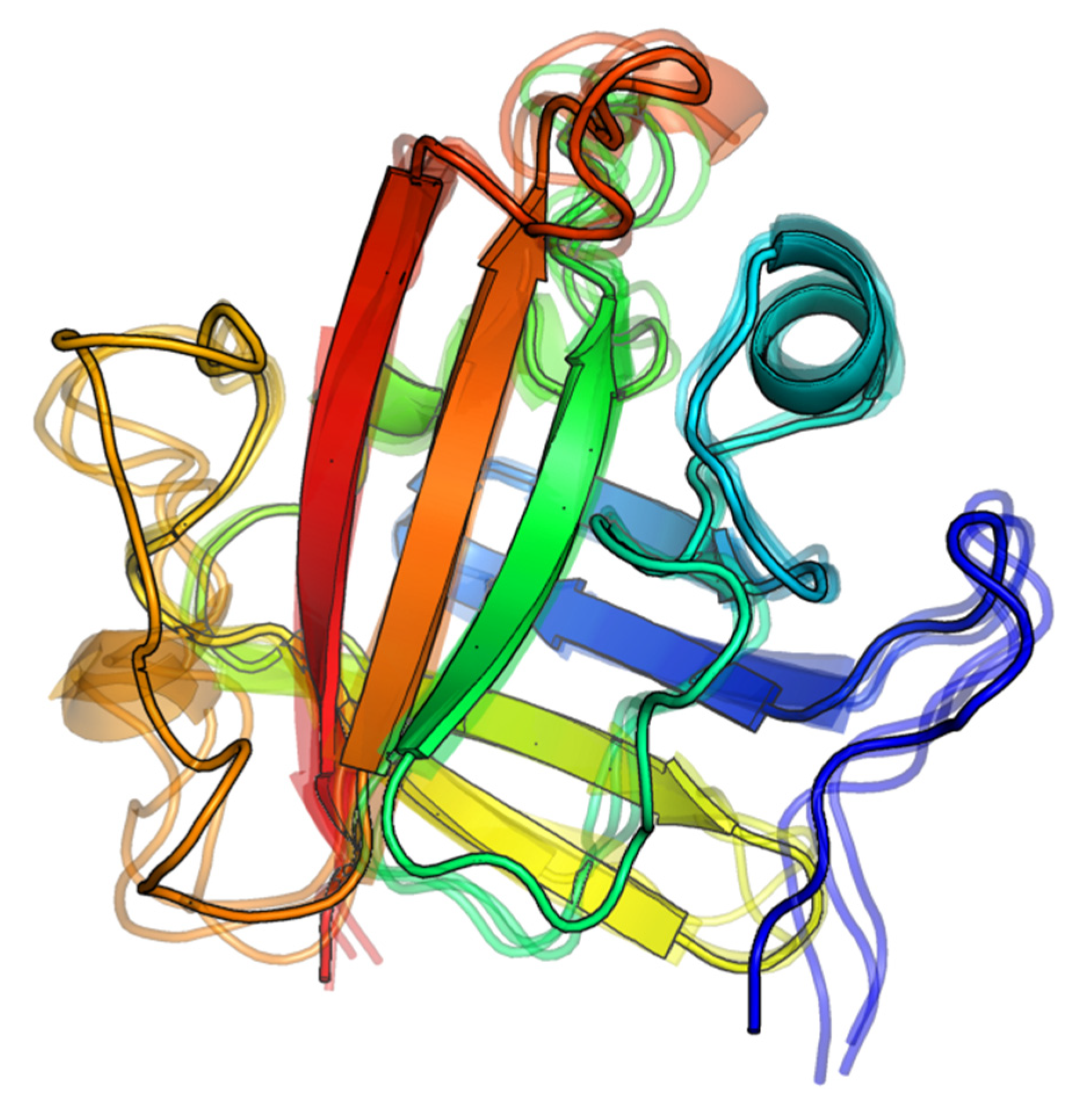
2.1.2. Visual Analysis
The Influence of the β6/β7 Loop Conformation
Polypeptide Containing Structures
Other Complexes
2.1.3. Ligand–SrtA Complex Structure Validation Criteria
- The same and opposite charged groups proximity;
- Location of hydrophobic parts/residues—in pockets or in the solute;
- Reasonable placement of N- and C-termini, assuming continuation of the polypeptide chain;
- Reasonable proximity of the catalytically broken bond of an oligopeptide to Cys184 (or its place in case of C184A mutant)—the bonds between T and G in LPxTG and between D and A in LPRDA;
- If the position of binding close to the catalytically relevant one is sought, the side chain of x in LPxTG should not be restricted by the protein surface;
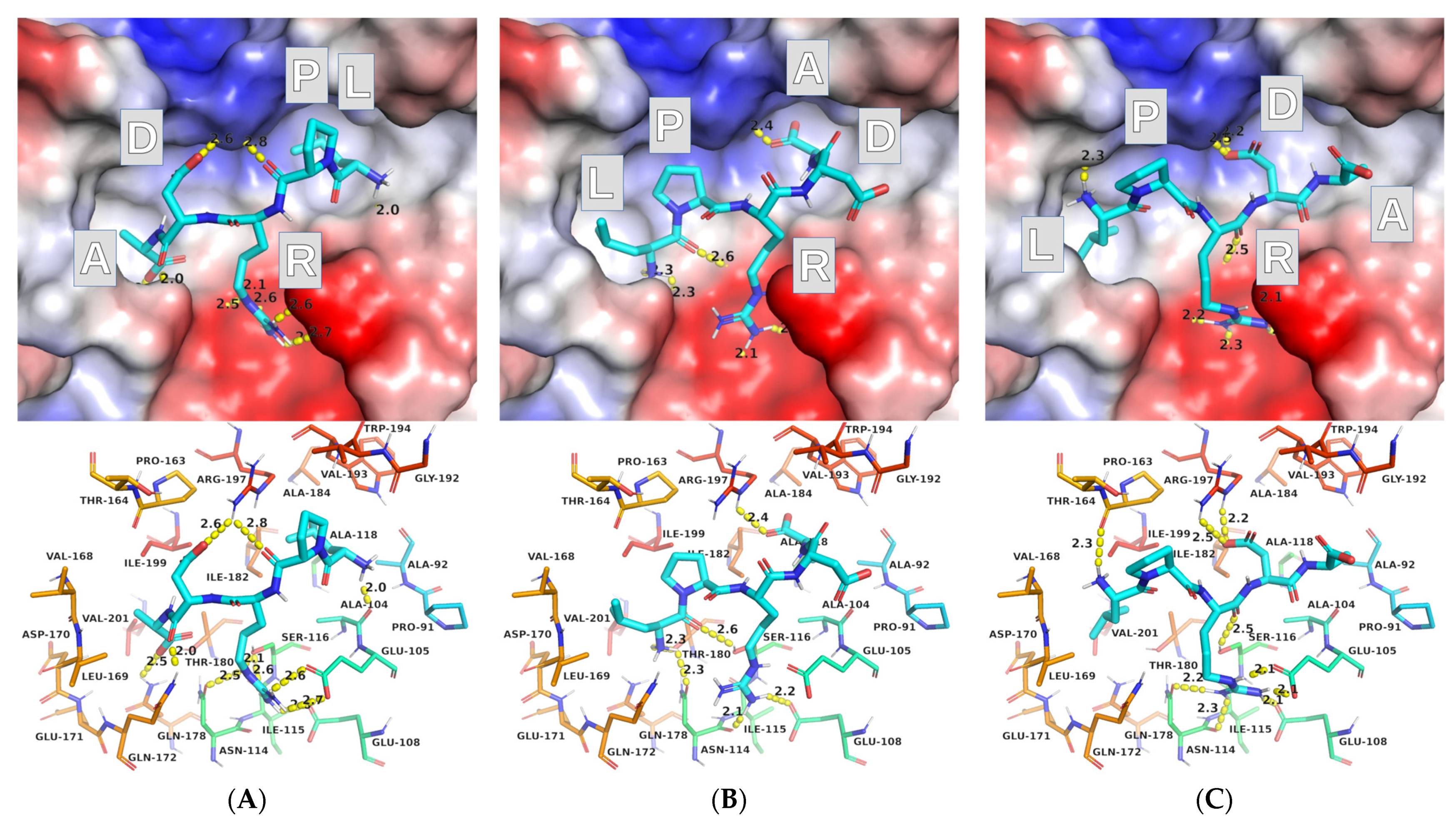
2.2. Molecular Docking
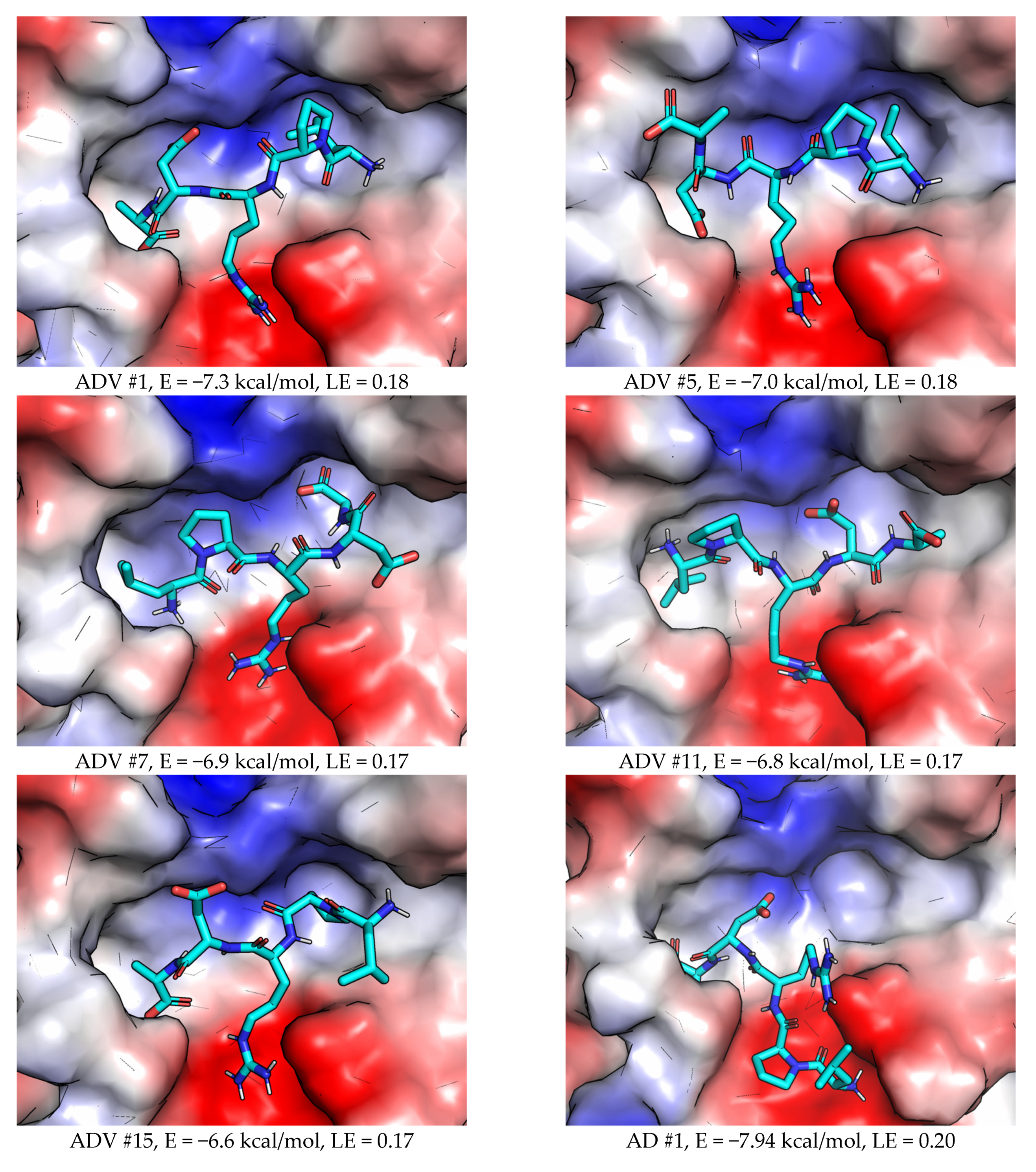
2.2.1. AutoDock Vina
2.2.2. AutoDock
2.3. Protocol Refinement
2.3.1. AutoDock Vina (ADV)
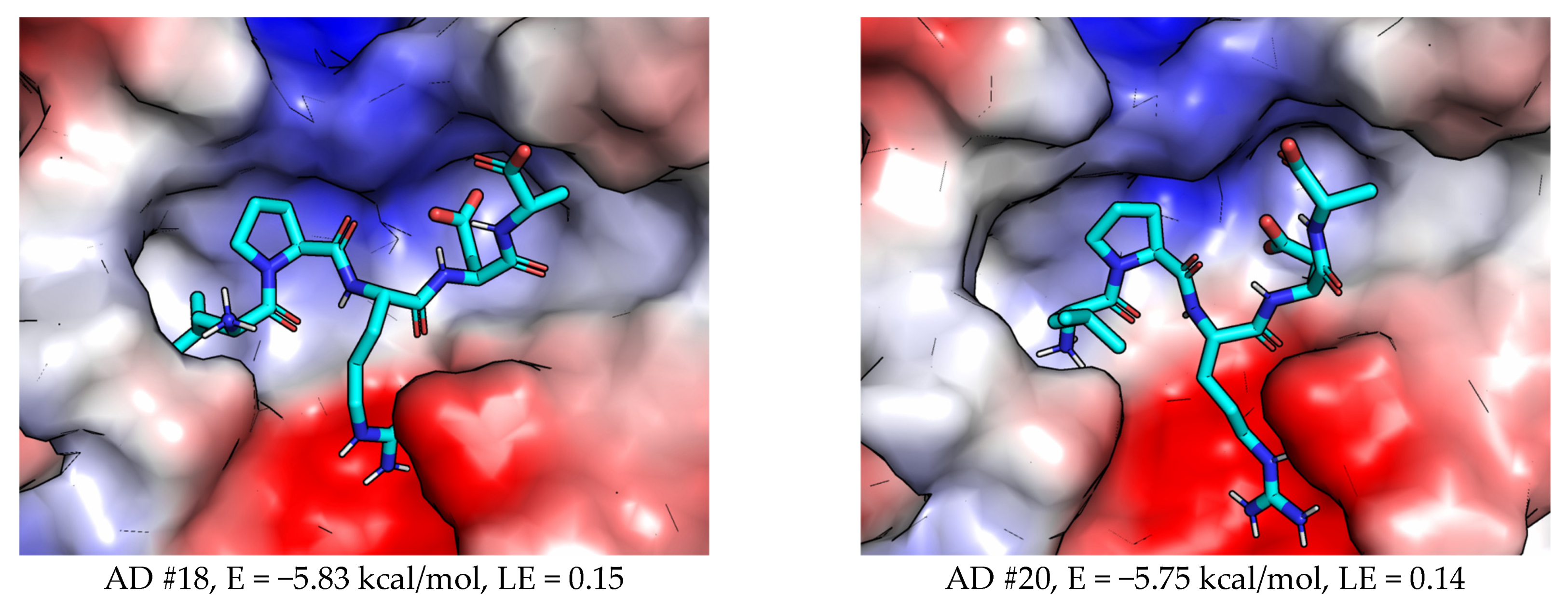
2.3.2. AutoDock (AD)
2.4. Molecular Dynamics (MD)
2.4.1. Docking Poses
2.4.2. SrtA Apo Form Dynamics
2.4.3. Continued Dynamics
3. Discussion
4. Materials and Methods
4.1. Molecular Docking
4.1.1. Target Structure
4.1.2. LPRDA Ligand Preparation
4.1.3. AutoDock Vina
4.1.4. AutoDock
4.1.5. Refined Experiment
4.2. Molecular Dynamics
4.2.1. Complex Preparation
4.2.2. Different Runs Analysis
4.2.3. RMSD and RMSF Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Maresso, A.W.; Schneewind, O. Sortase as a Target of Anti-Infective Therapy. Pharmacol. Rev. 2008, 60, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Alharthi, S.; Alavi, S.E.; Moyle, P.M.; Ziora, Z.M. Sortase A (SrtA) Inhibitors as an Alternative Treatment for Superbug Infections. Drug Discov. Today 2021, 26, 2164–2172. [Google Scholar] [CrossRef] [PubMed]
- Zrelovs, N.; Kurbatska, V.; Rudevica, Z.; Leonchiks, A.; Fridmanis, D. Sorting out the Superbugs: Potential of Sortase A Inhibitors among Other Antimicrobial Strategies to Tackle the Problem of Antibiotic Resistance. Antibiotics 2021, 10, 164. [Google Scholar] [CrossRef] [PubMed]
- Cascioferro, S.; Raffa, D.; Maggio, B.; Raimondi, M.V.; Schillaci, D.; Daidone, G. Sortase A Inhibitors: Recent Advances and Future Perspectives. J. Med. Chem. 2015, 58, 9108–9123. [Google Scholar] [CrossRef] [PubMed]
- Cascioferro, S.; Totsika, M.; Schillaci, D. Sortase A: An Ideal Target for Anti-Virulence Drug Development. Microb. Pathog. 2014, 77, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Kappel, K.; Wereszczynski, J.; Clubb, R.T.; McCammon, J.A. The Binding Mechanism, Multiple Binding Modes, and Allosteric Regulation of Staphylococcus aureus Sortase A Probed by Molecular Dynamics Simulations. Protein Sci. 2012, 21, 1858–1871. [Google Scholar] [CrossRef] [Green Version]
- Pang, X.; Zhou, H.-X. Disorder-to-Order Transition of an Active-Site Loop Mediates the Allosteric Activation of Sortase A. Biophys. J. 2015, 109, 1706–1715. [Google Scholar] [CrossRef] [Green Version]
- Ilangovan, U.; Ton-That, H.; Iwahara, J.; Schneewind, O.; Clubb, R.T. Structure of Sortase, the Transpeptidase That Anchors Proteins to the Cell Wall of Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2001, 98, 6056–6061. [Google Scholar] [CrossRef] [Green Version]
- Shulga, D.A.; Kudryavtsev, K.V. Selection of Promising Novel Fragment Sized S. Aureus SrtA Noncovalent Inhibitors Based on QSAR and Docking Modeling Studies. Molecules 2021, 26, 7677. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Cai, S.; Gu, G.; Guo, Z.; Long, Z. Recent Progress in the Development of Sortase A Inhibitors as Novel Anti-Bacterial Virulence Agents. RSC Adv. 2015, 5, 49880–49889. [Google Scholar] [CrossRef]
- Kudryavtsev, K.V.; Fedotcheva, T.A.; Shimanovsky, N.L. Inhibitors of Sortases of Gram-Positive Bacteria and Their Role in the Treatment of Infectious Diseases (Review). Pharm. Chem. J. 2021, 55, 751–756. [Google Scholar] [CrossRef]
- Nitulescu, G.; Margina, D.; Zanfirescu, A.; Olaru, O.T.; Nitulescu, G.M. Targeting Bacterial Sortases in Search of Anti-Virulence Therapies with Low Risk of Resistance Development. Pharmaceuticals 2021, 14, 415. [Google Scholar] [CrossRef]
- Jaudzems, K.; Kurbatska, V.; Jekabsons, A.; Bobrovs, R.; Rudevica, Z.; Leonchiks, A. Targeting Bacterial Sortase A with Covalent Inhibitors: 27 New Starting Points for Structure-Based Hit-to-Lead Optimization. ACS Infect. Dis. 2020, 6, 186–194. [Google Scholar] [CrossRef]
- Rentero Rebollo, I.; McCallin, S.; Bertoldo, D.; Entenza, J.M.; Moreillon, P.; Heinis, C. Development of Potent and Selective S. Aureus Sortase A Inhibitors Based on Peptide Macrocycles. ACS Med. Chem. Lett. 2016, 7, 606–611. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, H.; Pan, J.; Dong, J.; Zhou, X.; Niu, X.; Deng, X. Oligopeptide Targeting Sortase A as Potential Anti-Infective Therapy for Staphylococcus aureus. Front. Microbiol. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Kudryavtsev, K.V.; Bentley, M.L.; McCafferty, D.G. Probing of the Cis-5-Phenyl Proline Scaffold as a Platform for the Synthesis of Mechanism-Based Inhibitors of the Staphylococcus aureus Sortase SrtA Isoform. Bioorg. Med. Chem. 2009, 17, 2886–2893. [Google Scholar] [CrossRef] [Green Version]
- Bozhkova, S.A.; Gordina, E.M.; Labutin, D.V.; Sokolov, M.N.; Kudryatsev, K.V. Synthetic Low-Molecular-Mass Compounds as Potential Inhibitors of Staphylococcus aureus Adhesion in Experiment. Pharm. Chem. J. 2022, 55, 1269–1275. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic Peptides: Current Applications and Future Directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Song, L.F.; Merz, K.M. Evolution of Alchemical Free Energy Methods in Drug Discovery. J. Chem. Inf. Model. 2020, 60, 5308–5318. [Google Scholar] [CrossRef]
- Zara, L.; Efrém, N.-L.; van Muijlwijk-Koezen, J.E.; de Esch, I.J.P.; Zarzycka, B. Progress in Free Energy Perturbation: Options for Evolving Fragments. Drug Discov. Today Technol. 2021, 40, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Chodera, J.D.; Mobley, D.L.; Shirts, M.R.; Dixon, R.W.; Branson, K.; Pande, V.S. Alchemical Free Energy Methods for Drug Discovery: Progress and Challenges. Curr. Opin. Struct. Biol. 2011, 21, 150–160. [Google Scholar] [CrossRef] [Green Version]
- Suree, N.; Liew, C.K.; Villareal, V.A.; Thieu, W.; Fadeev, E.A.; Clemens, J.J.; Jung, M.E.; Clubb, R.T. The Structure of the Staphylococcus aureus Sortase-Substrate Complex Reveals How the Universally Conserved LPXTG Sorting Signal Is Recognized. J. Biol. Chem. 2009, 284, 24465–24477. [Google Scholar] [CrossRef] [Green Version]
- Zhulenkovs, D.; Rudevica, Z.; Jaudzems, K.; Turks, M.; Leonchiks, A. Discovery and Structure-Activity Relationship Studies of Irreversible Benzisothiazolinone-Based Inhibitors against Staphylococcus aureus Sortase A Transpeptidase. Bioorg. Med. Chem. 2014, 22, 5988–6003. [Google Scholar] [CrossRef]
- Zong, Y.; Bice, T.W.; Ton-That, H.; Schneewind, O.; Narayana, S.V.L. Crystal Structures of Staphylococcus aureus Sortase A and Its Substrate Complex. J. Biol. Chem. 2004, 279, 31383–31389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ugur, I.; Schatte, M.; Marion, A.; Glaser, M.; Boenitz-Dulat, M.; Antes, I. Ca2+ Binding Induced Sequential Allosteric Activation of Sortase A: An Example for Ion-Triggered Conformational Selection. PLoS ONE 2018, 13, e0205057. [Google Scholar] [CrossRef]
- Fischer, A.; Smieško, M.; Sellner, M.; Lill, M.A. Decision Making in Structure-Based Drug Discovery: Visual Inspection of Docking Results. J. Med. Chem. 2021, 64, 2489–2500. [Google Scholar] [CrossRef] [PubMed]
- Naik, M.T.; Suree, N.; Ilangovan, U.; Liew, C.K.; Thieu, W.; Campbell, D.O.; Clemens, J.J.; Jung, M.E.; Clubb, R.T. Staphylococcus aureus Sortase a Transpeptidase: Calcium Promotes Sorting Signal Binding by Altering the Mobility and Structure of an Active Site Loop. J. Biol. Chem. 2006, 281, 1817–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobitz, A.W.; Kattke, M.D.; Wereszczynski, J.; Clubb, R.T. Sortase Transpeptidases: Structural Biology and Catalytic Mechanism, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2017; Volume 109. [Google Scholar]
- Bissantz, C.; Kuhn, B.; Stahl, M. A Medicinal Chemist’s Guide to Molecular Interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef]
- Bentley, M.L.; Lamb, E.C.; McCafferty, D.G. Mutagenesis Studies of Substrate Recognition and Catalysis in the Sortase A Transpeptidase from Staphylococcus aureus. J. Biol. Chem. 2008, 283, 14762–14771. [Google Scholar] [CrossRef] [Green Version]
- Fuhrmann, J.; Rurainski, A.; Lenhof, H.-P.; Neumann, D. A New Lamarckian Genetic Algorithm for Flexible Ligand-Receptor Docking. J. Comput. Chem. 2010, 31, 1911–1918. [Google Scholar] [CrossRef]
- Best, R.B. Computational and Theoretical Advances in Studies of Intrinsically Disordered Proteins. Curr. Opin. Struct. Biol. 2017, 42, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Uzelac, I.; Gottfries, J.; Eriksson, L.A. Exploration of Multiple Sortase A Protein Conformations in Virtual Screening. Sci. Rep. 2016, 6, 20413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small Molecules, Big Targets: Drug Discovery Faces the Protein–Protein Interaction Challenge. Nat. Rev. Drug Discov. 2016, 15, 533–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldfield, C.J.; Dunker, A.K. Intrinsically Disordered Proteins and Intrinsically Disordered Protein Regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef]
- Chong, S.-H.; Chatterjee, P.; Ham, S. Computer Simulations of Intrinsically Disordered Proteins. Annu. Rev. Phys. Chem. 2017, 68, 117–134. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Crippen, G.M. Atomic Physicochemical Parameters for Three-Dimensional-Structure-Directed Quantitative Structure-Activity Relationships. 2. Modeling Dispersive and Hydrophobic Interactions. J. Chem. Inf. Comput. Sci. 1987, 27, 21–35. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A High-Throughput and Highly Parallel Open Source Molecular Simulation Toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.Y.; Berryman, J.T.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cisneros, G.A., III; Cruzeiro, V.W.D.; et al. Amber 2022; University of California: San Francisco, CA, USA, 2022. [Google Scholar]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, Efficient Generation of High-Quality Atomic Charges. AM1-BCC Model: II. Parameterization and Validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Merck Molecular Force Field. II. MMFF94 van Der Waals and Electrostatic Parameters for Intermolecular Interactions. J. Comput. Chem. 1996, 17, 520–552. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Shirts, M.R.; Klein, C.; Swails, J.M.; Yin, J.; Gilson, M.K.; Mobley, D.L.; Case, D.A.; Zhong, E.D. Lessons Learned from Comparing Molecular Dynamics Engines on the SAMPL5 Dataset. J. Comput. Aided Mol. Des. 2017, 31, 147–161. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB | Source (Number of Modes) | Ligand | Ca2+ | Bonding | Comment |
|---|---|---|---|---|---|
| 2KID [22] | NMR (20) | Cbz-LPAT* | + | Covalent | - |
| 6R1V [13] | NMR (20) | JPT | - | Covalent | - |
| 1IJA [8] | NMR (25) | - | - | - | - |
| 2MLM [23] | NMR (20) | 2W7 | - | Covalent | Strong variation of loop coordinates. |
| 1T2P [24] | X-ray (1) | - | - | - | Three differing subunits. Crystallographic water. |
| 1T2O [24] | X-ray (1) | - | - | - | C184A mutant. Three differing subunits. Crystallographic water. Two Met residues away from the binding site are replaced with Se-analogs. |
| 1T2W [24] | X-ray (1) | LPETG | - | Non-covalent | C184A mutant. Three differing subunits. Crystallographic water. OXT atom (C-end) of the ligand has an unrealistic position. |
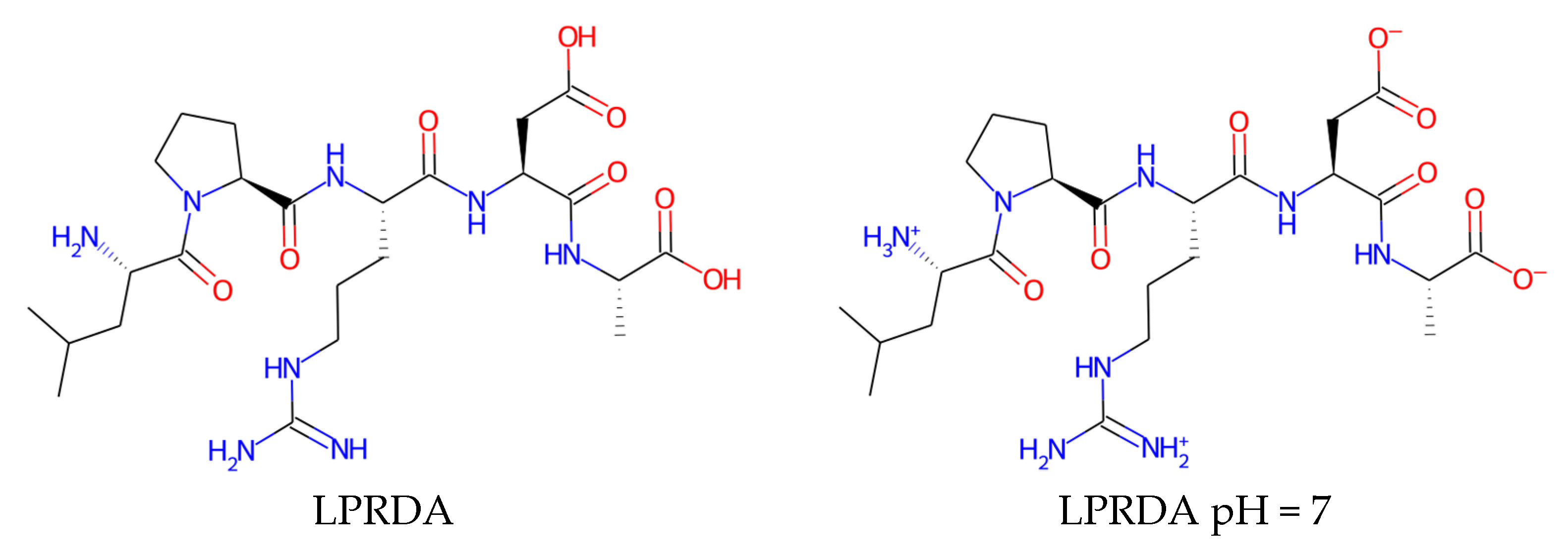
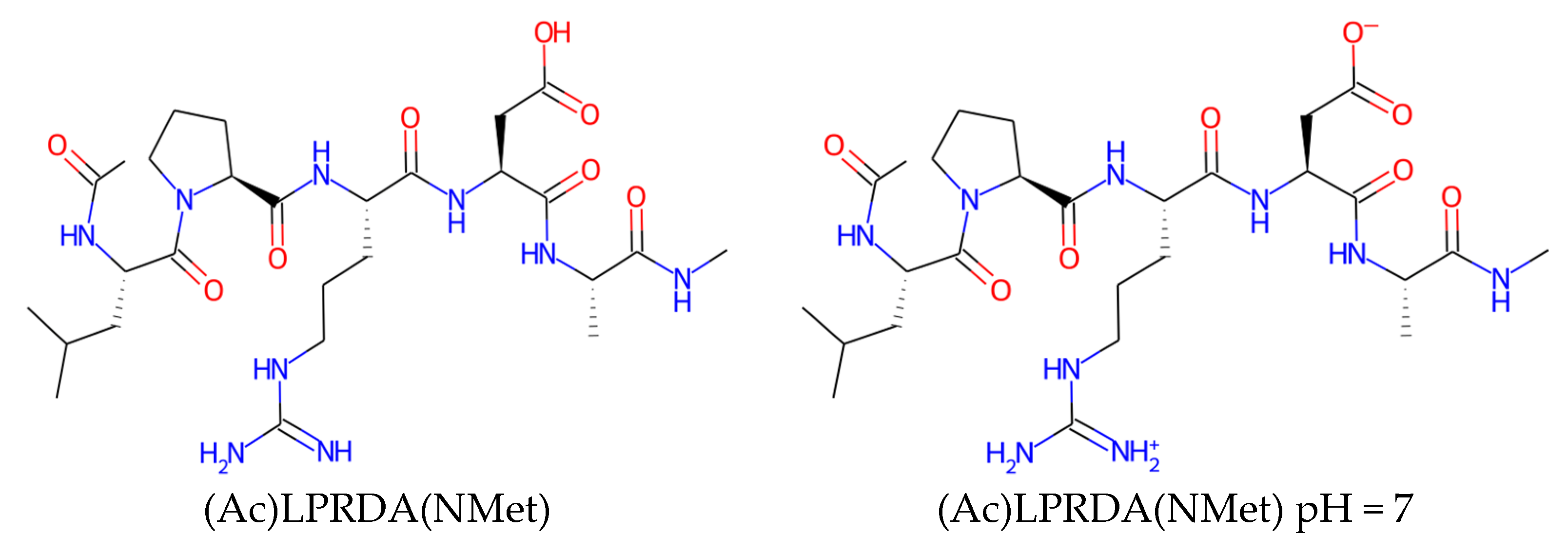
| Name | Form | cLogP, OB * | cLogP, RDKit | HBD/HBA 1 | TPSA | MW | #Rotors, OB 1 |
|---|---|---|---|---|---|---|---|
| LPRDA | neutral | 0.65 | −2.35 | 9/16 | 270.13 | 570.6 | 21 |
| LPRDA | pH = 7 | −5.36 | −7.56 | 7/14 | 279.15 | 570.6 | 21 |
| (Ac)LPRDA(NMet) | neutral | 0.57 | −2.51 | 9/17 | 265.01 | 625.7 | 24 |
| (Ac)LPRDA(NMet) | pH = 7 | −2.68 | −5.67 | 8/16 | 269.58 | 625.7 | 24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shulga, D.A.; Kudryavtsev, K.V. Theoretical Studies of Leu-Pro-Arg-Asp-Ala Pentapeptide (LPRDA) Binding to Sortase A of Staphylococcus aureus. Molecules 2022, 27, 8182. https://doi.org/10.3390/molecules27238182
Shulga DA, Kudryavtsev KV. Theoretical Studies of Leu-Pro-Arg-Asp-Ala Pentapeptide (LPRDA) Binding to Sortase A of Staphylococcus aureus. Molecules. 2022; 27(23):8182. https://doi.org/10.3390/molecules27238182
Chicago/Turabian StyleShulga, Dmitry A., and Konstantin V. Kudryavtsev. 2022. "Theoretical Studies of Leu-Pro-Arg-Asp-Ala Pentapeptide (LPRDA) Binding to Sortase A of Staphylococcus aureus" Molecules 27, no. 23: 8182. https://doi.org/10.3390/molecules27238182