
Theoretical Study of Vinyl-Sulfonate Monomers and Their Effect as the Dopants of Polyaniline Dimers
 and
and
Abstract
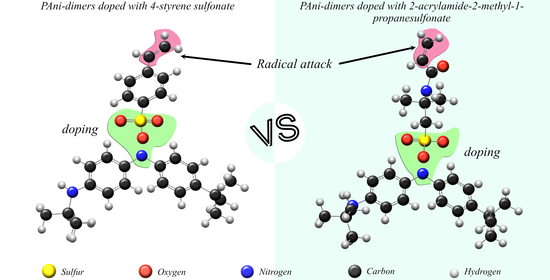
:
1. Introduction
2. Results and Discussion
2.1. Theoretical Structure of Monomers
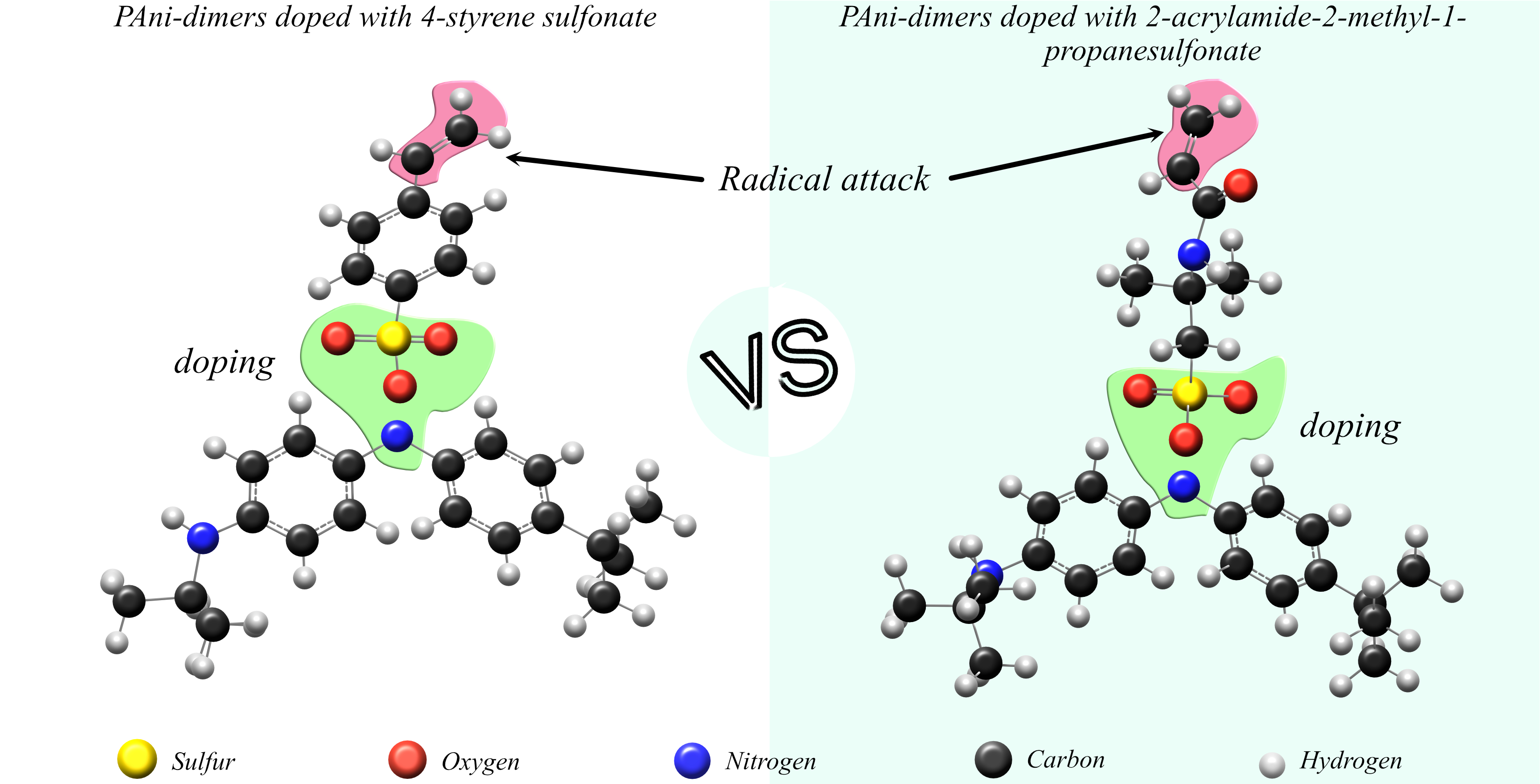
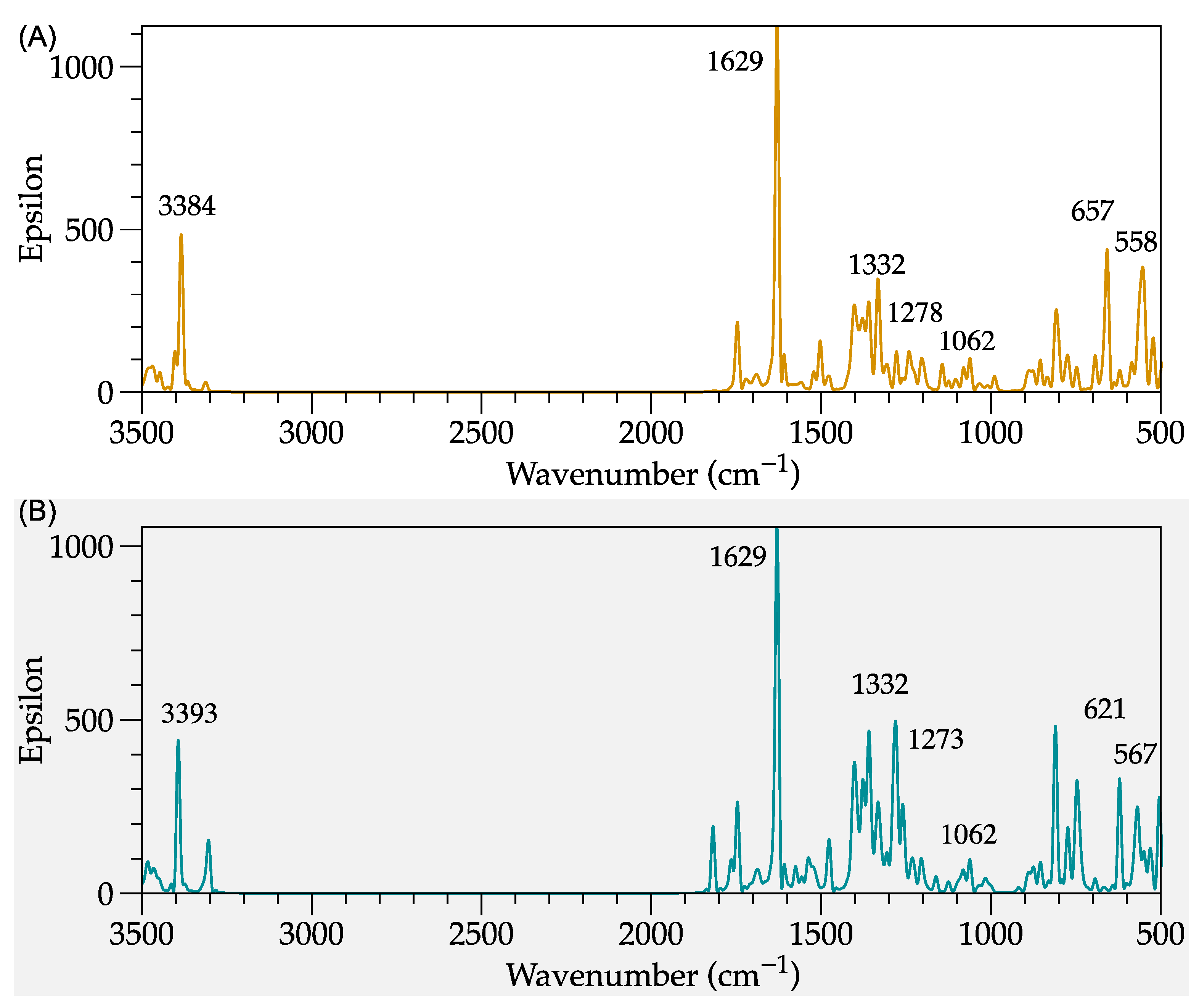
2.1.1. Modeling of IR Spectra
2.1.2. Global and Local Reactivity Descriptors
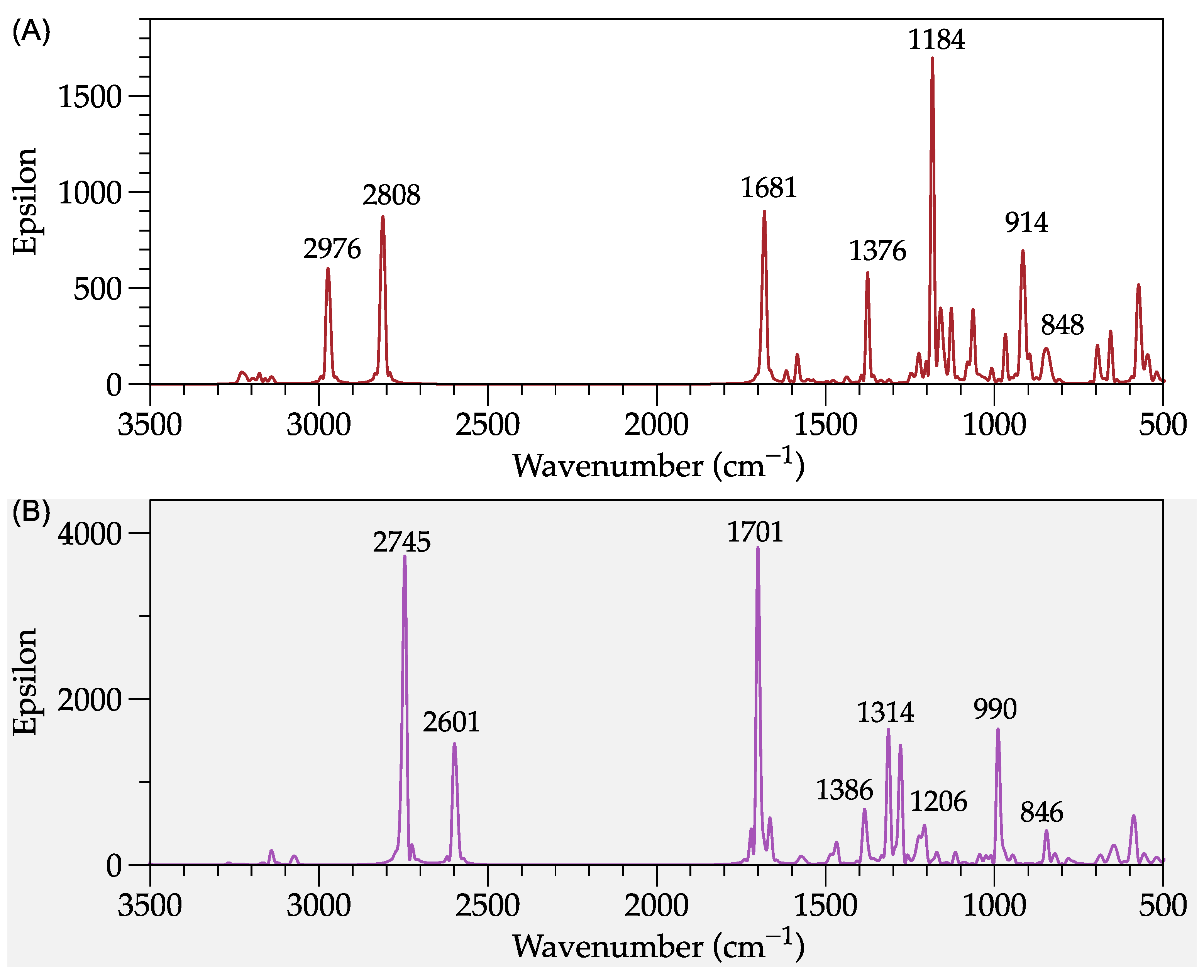
2.2. Structural Simulation of Polyaniline Dimers with Dopants
Reactivity Calculation of PAni-d
2.3. UV-Vis Spectrum
Band Gap
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Zhao, P.; Deng, M.; Yang, Y.; Zhang, J.; Zhang, Y. Synthesis and Self-Assembly of Thermoresponsive Biohybrid Graft Copolymers Based on a Combination of Passerini Multicomponent Reaction and Molecular Recognition. Macromol. Rapid Commun. 2021, 42, 2100424. [Google Scholar] [CrossRef] [PubMed]
- Wójcik, A.J.; Wolski, K.; Zapotoczny, S. Double-stranded surface-grafted polymer brushes with ladder-like architecture. Eur. Polym. J. 2021, 155, 110577. [Google Scholar] [CrossRef]
- Cruz-Medina, R.; Vega-Rios, A.; Hernández-Escobar, C.A.; Estrada-Monje, A.; Rodríguez-Sánchez, I.; Zaragoza-Contreras, E.A. Polystyrene-polyaniline core-shell composite particles using a bifunctional selectively polymerizable monomer as the interfacial linkage. Synth. Met. 2020, 265, 116402. [Google Scholar] [CrossRef]
- Conejo-Dávila, A.; Moya-Quevedo, M.; Chávez-Flores, D.; Vega-Rios, A.; Zaragoza-Contreras, E. Role of the Anilinium Ion on the Selective Polymerization of Anilinium 2-Acrylamide-2-methyl-1-propanesulfonate. Polymers 2021, 13, 2349. [Google Scholar] [CrossRef] [PubMed]
- Conejo-Dávila, A.S.; Hernández-Escobar, C.A.; Vega-Rios, A.; Rodríguez-Sánchez, I.; Estrada-Monje, A.; de León-Gómez, R.E.D.; Zaragoza-Contreras, E.A. Selective polymerization of a new bifunctional monomer via free radical polymerization and oxidative route. Synth. Met. 2020, 259, 116258. [Google Scholar] [CrossRef]
- Alves, G.G.; Lavarda, F.C.; Graeff, C.F.; Batagin-Neto, A. Reactivity of eumelanin building blocks: A DFT study of monomers and dimers. J. Mol. Graph. Model. 2020, 98, 107609. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, J.; Hu, H.; Chen, C.; Li, F.; Bin Ying, W.; Zheng, L.; Zhao, Y.-L.; Wang, J.; Zhang, R.; et al. Acid-triggered, degradable and high strength-toughness copolyesters: Comprehensive experimental and theoretical study. J. Hazard. Mater. 2022, 430, 128392. [Google Scholar] [CrossRef]
- Huang, H.; Yang, L.; Facchetti, A.; Marks, T.J. Organic and Polymeric Semiconductors Enhanced by Noncovalent Conformational Locks. Chem. Rev. 2017, 117, 10291–10318. [Google Scholar] [CrossRef]
- Mehrdad, A.; Parvini, E. Conductometry and Density Functional Theory studies on the interactions of sodium polystyrenesulfonate with 1–butyl–3–methylimidazolium bromide in aqueous solution. J. Mol. Liq. 2017, 243, 324–332. [Google Scholar] [CrossRef]
- Sudharsana, N.; Subramanian, G.; Krishnakumar, V.; Nagalakshmi, R. Growth and characterization of anilinium hydrogen sulfate (AHS) single crystals: An organic nonlinear optical material. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012, 97, 798–805. [Google Scholar] [CrossRef]
- Baker, B.; Cockram, C.J.; Dakein, S.; DeMaria, J.M.; George, D.M.; Malyk, K.R.; Sarno, M.J.F.; Cardenas, A.J.P. Synthesis and characterization of anilinium ionic liquids: Exploring effect of π-π ring stacking. J. Mol. Struct. 2021, 1225, 129122. [Google Scholar] [CrossRef]
- Przybyłek, M.; Gaca, J. Reaction of aniline with ammonium persulphate and concentrated hydrochloric acid: Experimental and DFT studies. Chem. Pap. 2012, 66, 699–708. [Google Scholar] [CrossRef]
- de Souza, V.S.; da Frota, H.O.; Sanches, E.A. Polyaniline-CuO hybrid nanocomposite with enhanced electrical conductivity. J. Mol. Struct. 2018, 1153, 20–27. [Google Scholar] [CrossRef]
- Thirunavukkarasu, M.; Balaji, G.; Muthu, S.; Sakthivel, S.; Prabakaran, P.; Irfan, A. Theoretical conformations studies on 2-Acetyl-gamma-butyrolactone structure and stability in aqueous phase and the solvation effects on electronic properties by quantum computational methods. Comput. Theor. Chem. 2022, 1208, 113534. [Google Scholar] [CrossRef]
- Nifant’ev, I.; Vinogradov, A.; Vinogradov, A.; Ivchenko, P. DFT Modeling of the Alternating Radical Copolymerization and Alder-Ene Reaction between Maleic Anhydride and Olefins. Polymers 2020, 12, 744. [Google Scholar] [CrossRef] [PubMed]
- Orpen, A.G.; Brammer, L.; Allen, F.H.; Watson, D.G.; Taylor, R. Typical interatomic distances: Organometallic compounds and coordination complexes of the d- and f-block metals. In International Tables for Crystallography, Volume C: Mathematical, Physical and Chemical Tables, 3rd ed.; Prince, E., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2006; pp. 812–896. [Google Scholar] [CrossRef]
- Tammer, M.G. Sokrates: Infrared and Raman characteristic group frequencies: Tables and charts. Colloid Polym. Sci. 2004, 283, 235. [Google Scholar] [CrossRef]
- Ji, Y.; Yang, X.; Ji, Z.; Zhu, L.; Ma, N.; Chen, D.; Jia, X.; Tang, J.; Cao, Y. DFT-Calculated IR Spectrum Amide I, II, and III Band Contributions of N-Methylacetamide Fine Components. ACS Omega 2020, 5, 8572–8578. [Google Scholar] [CrossRef]
- Karabacak, M.; Cinar, Z.; Kurt, M.; Sudha, S.; Sundaraganesan, N. FT-IR, FT-Raman, NMR and UV–vis spectra, vibrational assignments and DFT calculations of 4-butyl benzoic acid. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012, 85, 179–189. [Google Scholar] [CrossRef]
- Krasovskii, A.N.; Kalnin’sh, K.K. IR spectra of long-chain alkylsulfonic acids. J. Appl. Spectrosc. 1977, 26, 745–749. [Google Scholar] [CrossRef]
- Das, M.; Devi, N.; Sarma, J.; Devi, N. Preparation, characterization, and water sorption study of 2-acrylamido-2- methylpropane sulfonic acid (AMPS) based hydrogel. J. Chem. Pharm. Res. 2014, 6, 800–806. [Google Scholar]
- Pearson, R.G. Chemical Hardness; Wiley-VCH Verlag GmbH: Weinheim, Germany, 1997; pp. 197–198. [Google Scholar] [CrossRef]
- Bultinck, P.; de Winter, H. Computational Medicinal Chemistry for Drug Discovery; Marcel Dekker, Inc.: New York, NY, USA, 2004. [Google Scholar]
- Roy, R.K. On the Reliability of Global and Local Electrophilicity Descriptors. J. Phys. Chem. A 2004, 108, 4934–4939. [Google Scholar] [CrossRef]
- Sarkar, U.; Padmanabhan, J.; Parthasarathi, R.; Subramanian, V.; Chattaraj, P.K. Toxicity analysis of polychlorinated diben-zofurans through global and local electrophilicities. J. Mol. Struct. THEOCHEM 2006, 758, 119–125. [Google Scholar] [CrossRef]
- Roy, D.; Parthasarathi, R.; Maiti, B.; Subramanian, V.; Chattaraj, P. Electrophilicity as a possible descriptor for toxicity prediction. Bioorganic Med. Chem. 2005, 13, 3405–3412. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.K.; Krishnamurti, S.; Geerlings, P.; Pal, S. Local Softness and Hardness Based Reactivity Descriptors for Predicting Intra- and Intermolecular Reactivity Sequences: Carbonyl Compounds. J. Phys. Chem. A 1998, 102, 3746–3755. [Google Scholar] [CrossRef]
- Kolandaivel, P.; Praveena, G.; Selvarengan, P. Study of atomic and condensed atomic indices for reactive sites of molecules. J. Chem. Sci. 2005, 117, 591–598. [Google Scholar] [CrossRef]
- Deep, A.; Saraf, M.; Neha; Bharadwaj, S.K.; Sharma, A.L. Styrene Sulphonic Acid Doped Polyaniline Based Immunosensor for Highly Sensitive Impedimetric Sensing of Atrazine. Electrochim. Acta 2014, 146, 301–306. [Google Scholar] [CrossRef]
- Zhou, J.; Tzamalis, G.; Zaidi, N.; Comfort, N.; Monkman, A. Effect of thermal aging on electrical conductivity of the 2-acrylamido-2-methyl-1-propanesulfonic acid-doped polyaniline fiber. J. Appl. Polym. Sci. 2001, 79, 2503–2508. [Google Scholar] [CrossRef]
- Matsumura, Y.; Iida, K.; Sato, H. Theoretical study on the ionization of aniline in aqueous solutions. Chem. Phys. Lett. 2013, 584, 103–107. [Google Scholar] [CrossRef]
- Farasat, M.; Shojaei, S.R.; Golzan, M.; Farhadi, K. Theoretical study of the potential energy surface and electric dipole moment of aniline. J. Mol. Struct. 2016, 1108, 341–346. [Google Scholar] [CrossRef]
- Mishra, A.K. DFT study of structural, vibrational and electronic properties of polyaniline pernigraniline model compounds. J. Comput. Sci. 2015, 10, 195–208. [Google Scholar] [CrossRef]
- Ajeel, K.I.; Kareem, Q.S. Synthesis and Characteristics of Polyaniline (PANI) Filled by Graphene (PANI/GR) nano-Films. J. Physics Conf. Ser. 2019, 1234, 012020. [Google Scholar] [CrossRef]
- Lindfors, T.; Ivaska, A. pH sensitivity of polyaniline and its substituted derivatives. J. Electroanal. Chem. 2002, 531, 43–52. [Google Scholar] [CrossRef]
- de Souza, F.G.; Soares, B.G. Methodology for determination of Pani.DBSA content in conductive blends by using UV-Vis spectrometry. Polym. Test. 2006, 25, 512–517. [Google Scholar] [CrossRef]
- Kwon, O.; McKee, M.L. Calculations of Band Gaps in Polyaniline from Theoretical Studies of Oligomers. J. Phys. Chem. B 2000, 104, 1686–1694. [Google Scholar] [CrossRef]
- Pousti, M.; Abbaszadeh, M.; Lashkenari, M.S.; Ghorbani, M. A combined experimental and theoretical studies on molecular structure and vibrational spectra of polyaniline and polyaniline/silver nanocomposite. Synth. Met. 2013, 183, 63–68. [Google Scholar] [CrossRef]
- Almasi, M.; Sheikholeslami, T.F.; Naghdi, M. Band gap study of polyaniline and polyaniline/MWNT nanocomposites with in situ polymerization method. Compos. Part B Eng. 2016, 96, 63–68. [Google Scholar] [CrossRef]
- Capelle, K. A bird’s-eye view of density-functional theory. Braz. J. Phys. 2006, 36, 1318–1343. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Adamo, C.; Cossi, M.; Barone, V. An accurate density functional method for the study of magnetic properties: The PBE0 model. J. Mol. Struct. THEOCHEM 1999, 493, 145–157. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zandler, M.E.; D’Souza, F. The remarkable ability of B3LYP/3-21G(*) calculations to describe geometry, spectral and electrochemical properties of molecular and supramolecular porphyrin–fullerene conjugates. Comptes Rendus Chim. 2006, 9, 960–981. [Google Scholar] [CrossRef]
- Lu, L.; Hu, H.; Hou, H.; Wang, B. An improved B3LYP method in the calculation of organic thermochemistry and reactivity. Comput. Theor. Chem. 2013, 1015, 64–71. [Google Scholar] [CrossRef]
- Mineva, T. Selectivity study from the density functional local reactivity indices. J. Mol. Struct. THEOCHEM 2006, 762, 79–86. [Google Scholar] [CrossRef]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Monomers | Electron Affinity | Ionization Potential | Electronegativity | Hardness | Softness | Philicity |
|---|---|---|---|---|---|---|
| ani-SS | −64.47 | 94.71 | 15.12 | 79.59 | 0.00628 | 1.44 |
| ani-AMPS | −63.33 | 101.34 | 19.00 | 82.33 | 0.00607 | 2.19 |
| Molecule | Electron Affinity | Ionization Potential | Electronegativity | Hardness | Softness | Philicity |
|---|---|---|---|---|---|---|
| PAni-d | −246.17 | −129.33 | −187.75 | 58.42 | 0.00856 | 301.71 |
| PAni-d AMPS | −74.31 | 106.59 | 16.14 | 90.45 | 0.00553 | 1.44 |
| PAni-d SS | −59.95 | 106.08 | 23.07 | 83.01 | 0.00602 | 3.20 |
| Structure | Band Gap, eV |
|---|---|
| PAni-d SS | 3.81 |
| PAni-d AMPS | 4.67 |
| ani-SS | 4.25 |
| ani-AMPS | 4.31 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Sánchez, I.; Conejo-Dávila, A.S.; Estrada-Monje, A.; Vega-Rios, A.; Zaragoza-Contreras, E.A. Theoretical Study of Vinyl-Sulfonate Monomers and Their Effect as the Dopants of Polyaniline Dimers. Molecules 2022, 27, 6353. https://doi.org/10.3390/molecules27196353
Rodríguez-Sánchez I, Conejo-Dávila AS, Estrada-Monje A, Vega-Rios A, Zaragoza-Contreras EA. Theoretical Study of Vinyl-Sulfonate Monomers and Their Effect as the Dopants of Polyaniline Dimers. Molecules. 2022; 27(19):6353. https://doi.org/10.3390/molecules27196353
Chicago/Turabian StyleRodríguez-Sánchez, Isis, Alain S. Conejo-Dávila, Anayansi Estrada-Monje, Alejandro Vega-Rios, and Erasto Armando Zaragoza-Contreras. 2022. "Theoretical Study of Vinyl-Sulfonate Monomers and Their Effect as the Dopants of Polyaniline Dimers" Molecules 27, no. 19: 6353. https://doi.org/10.3390/molecules27196353