
Thermodynamics and Spectroscopy of Halogen- and Hydrogen-Bonded Complexes of Haloforms with Aromatic and Aliphatic Amines
Abstract
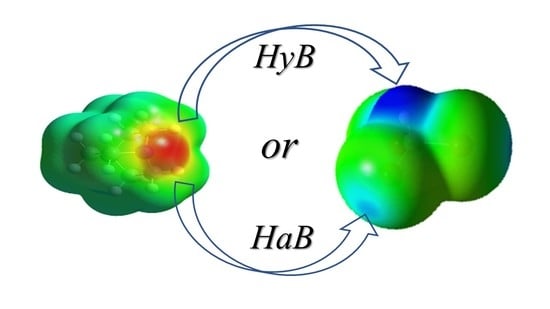
:
1. Introduction
2. Results and Discussion
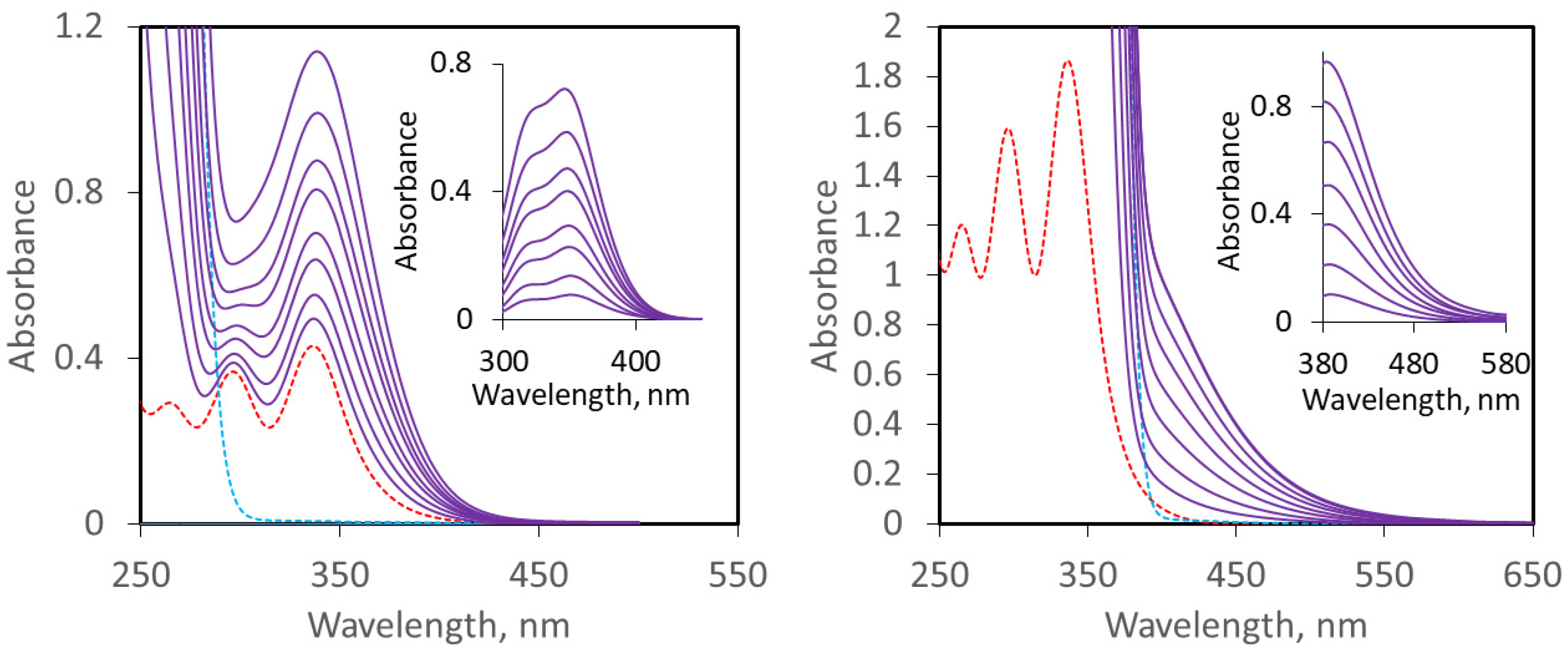
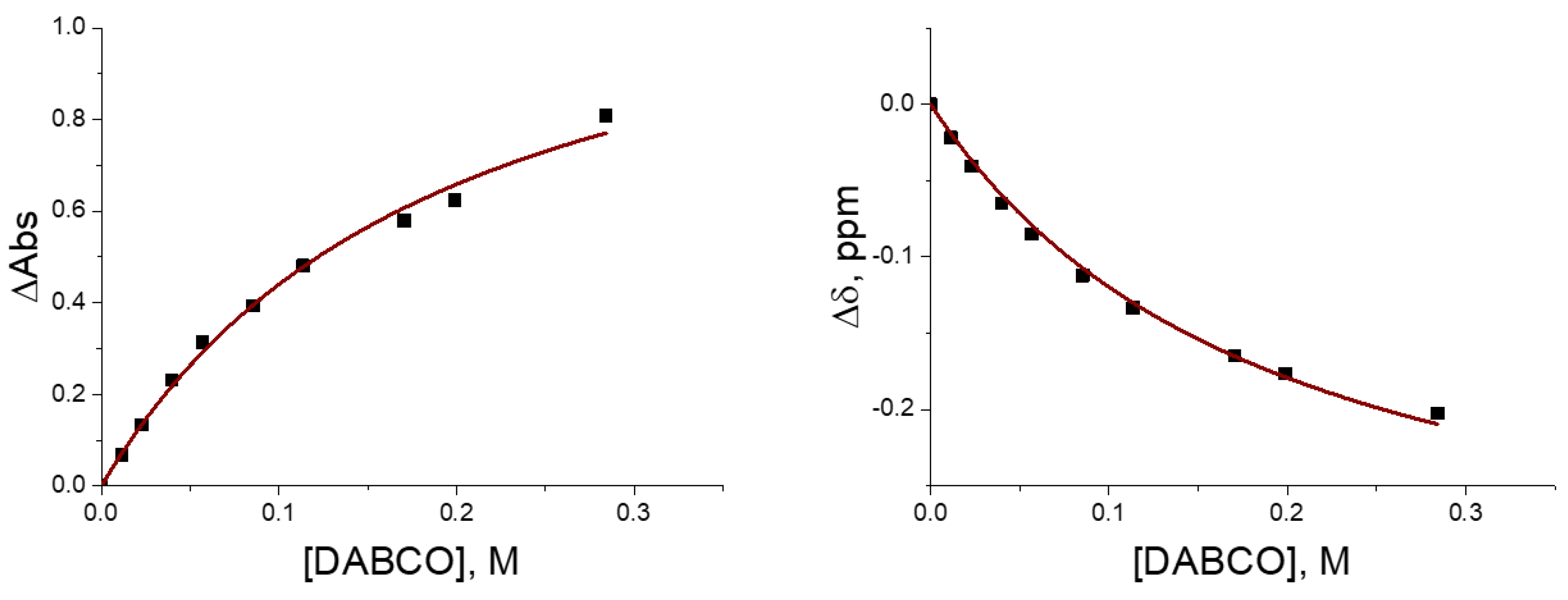
2.1. UV–Vis Study of Interaction of CHX3 with Amines
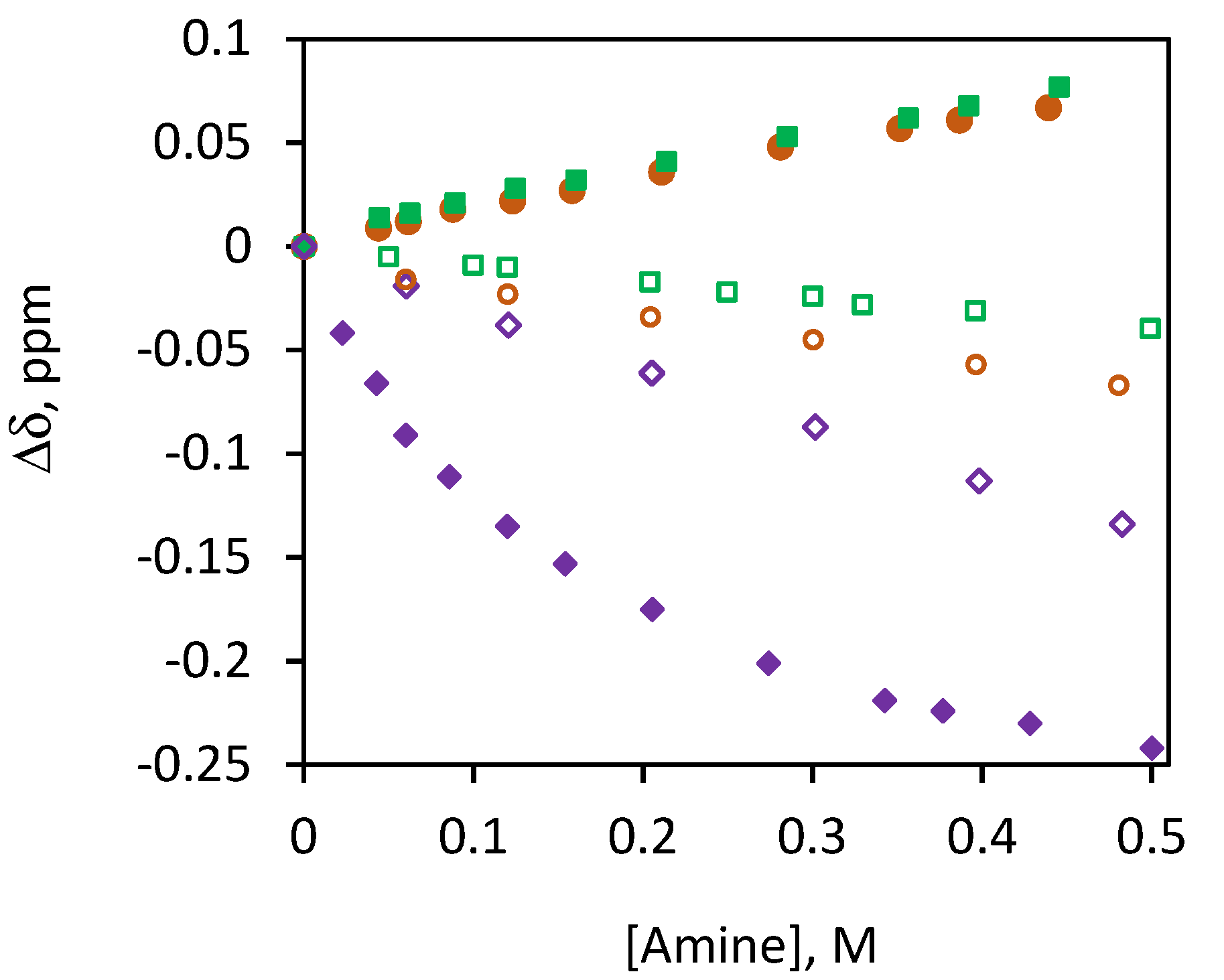
2.2. 1H NMR Study of the Interaction of CHX3 with Amines
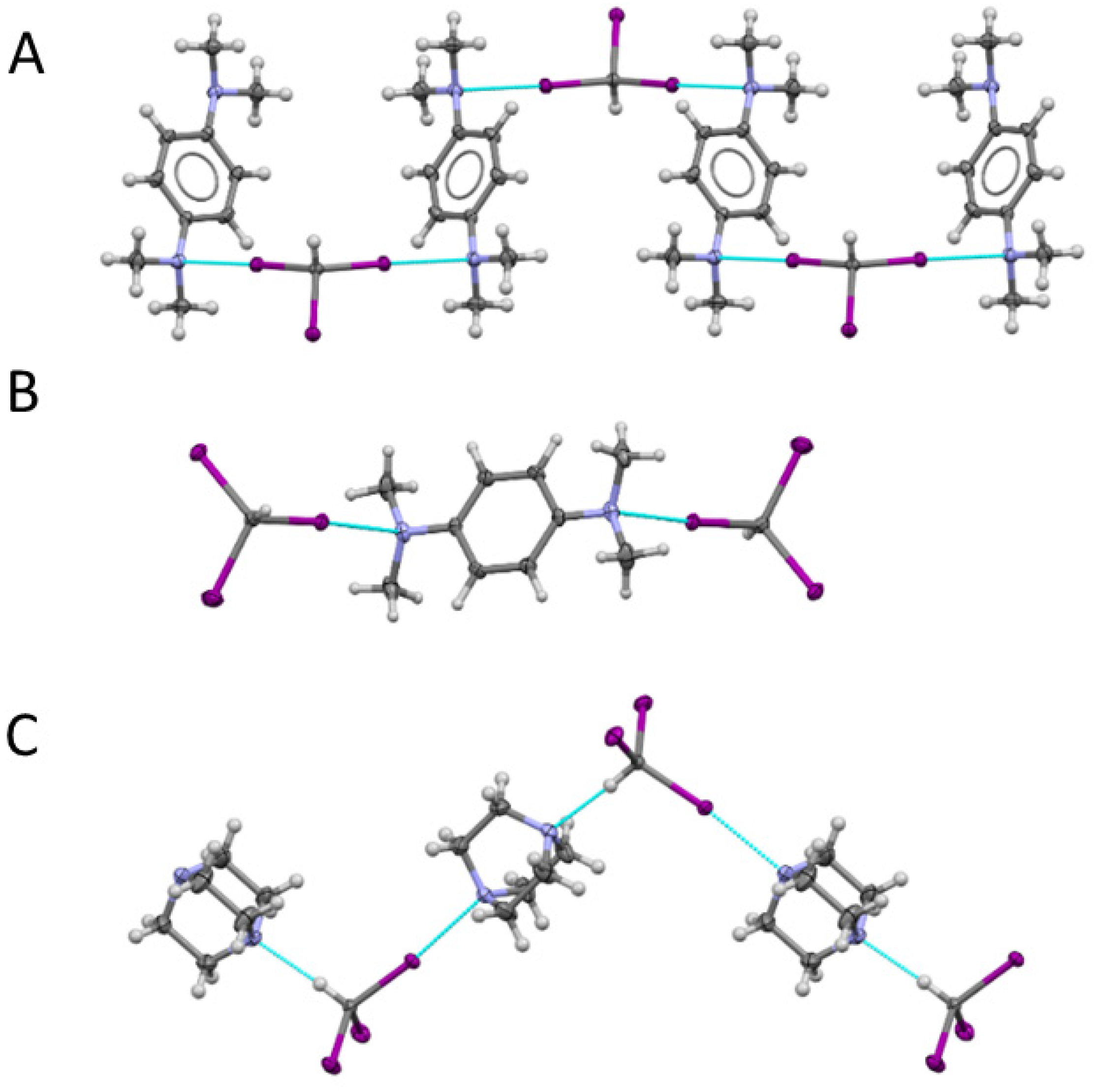
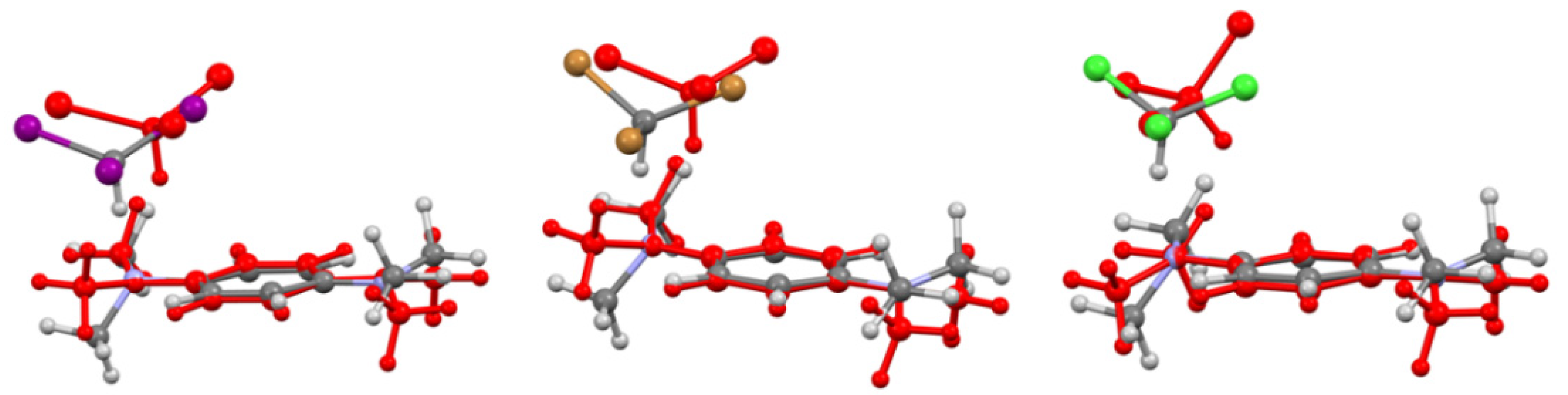
2.3. X-ray Structural Analysis of Co-crystals of Iodoform with TMPD or DABCO
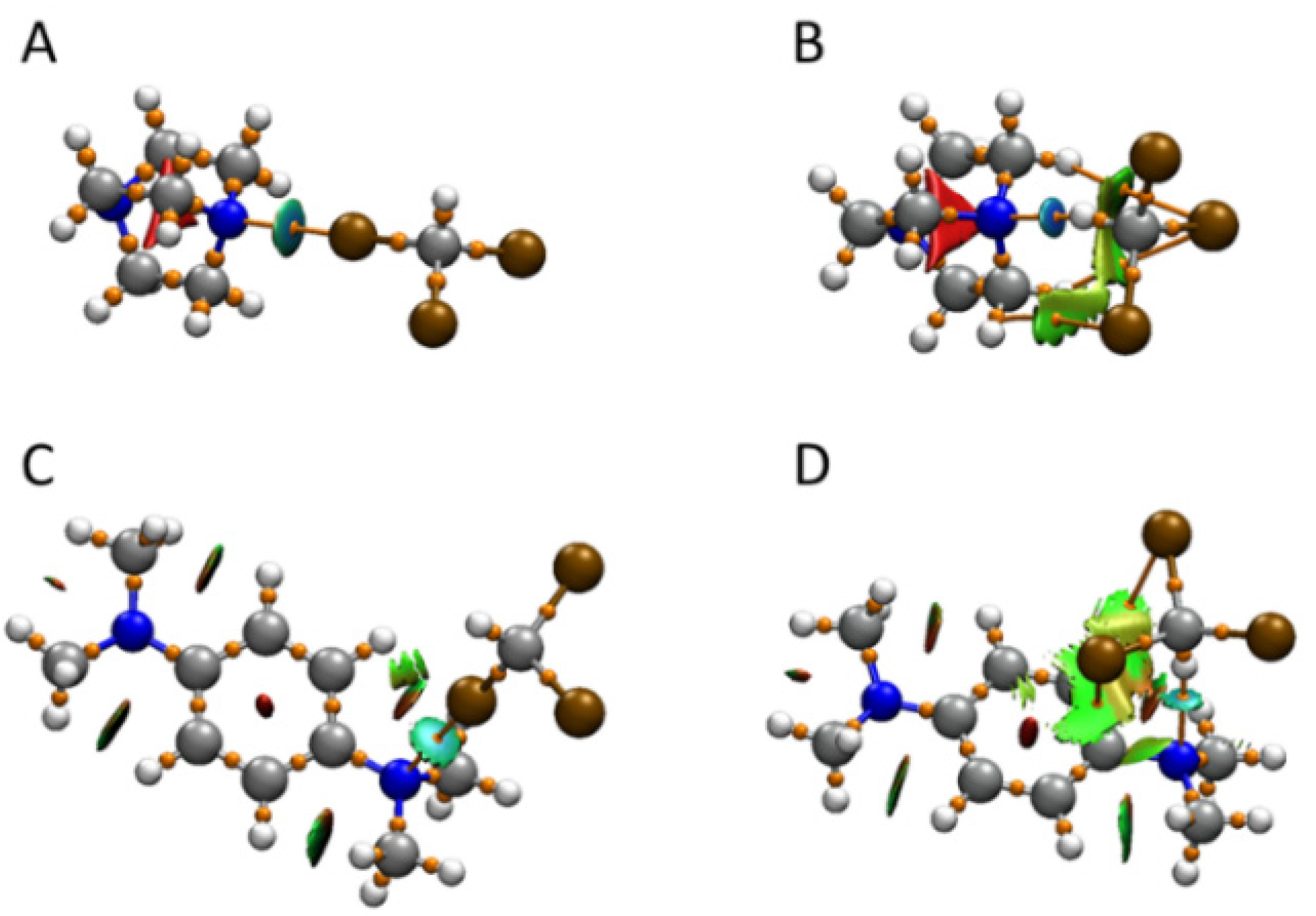
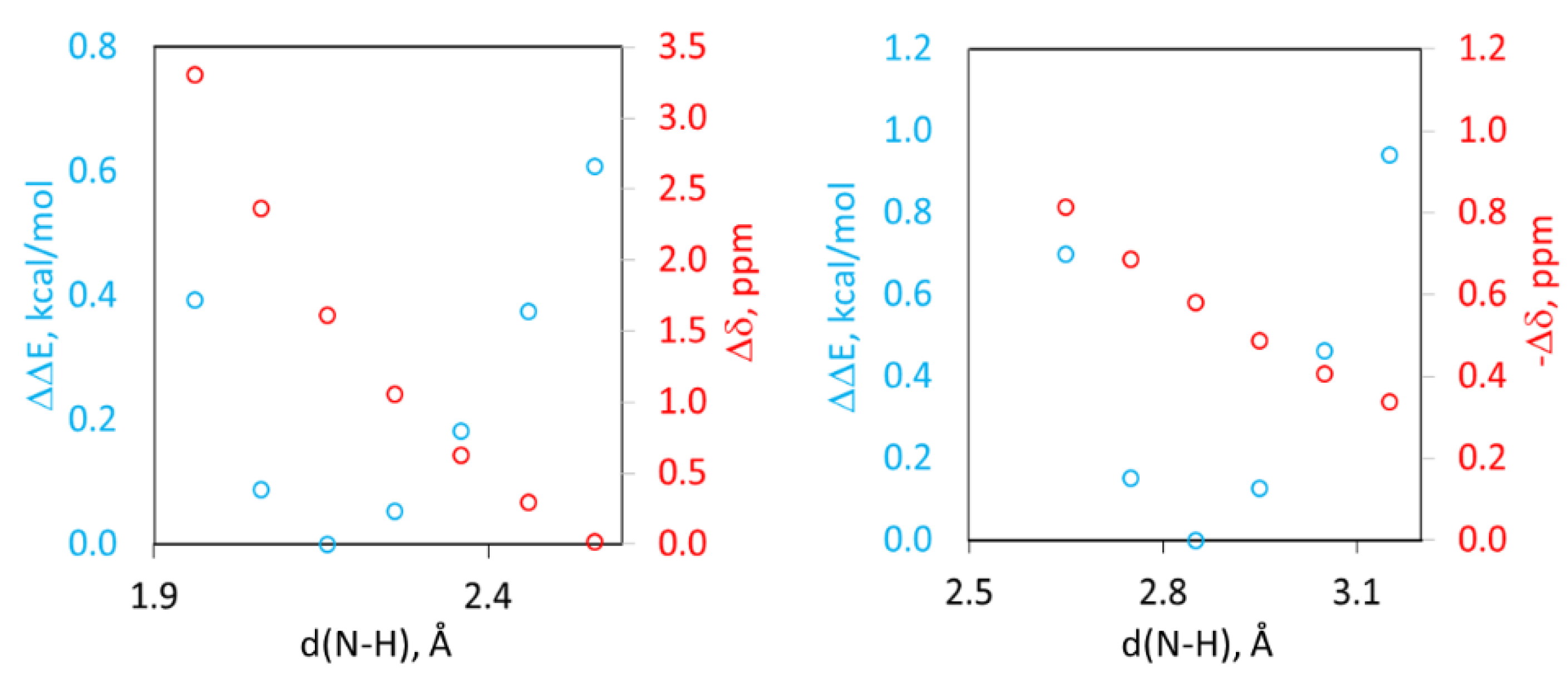
2.4. Computational Analysis of XB and HB Complexes
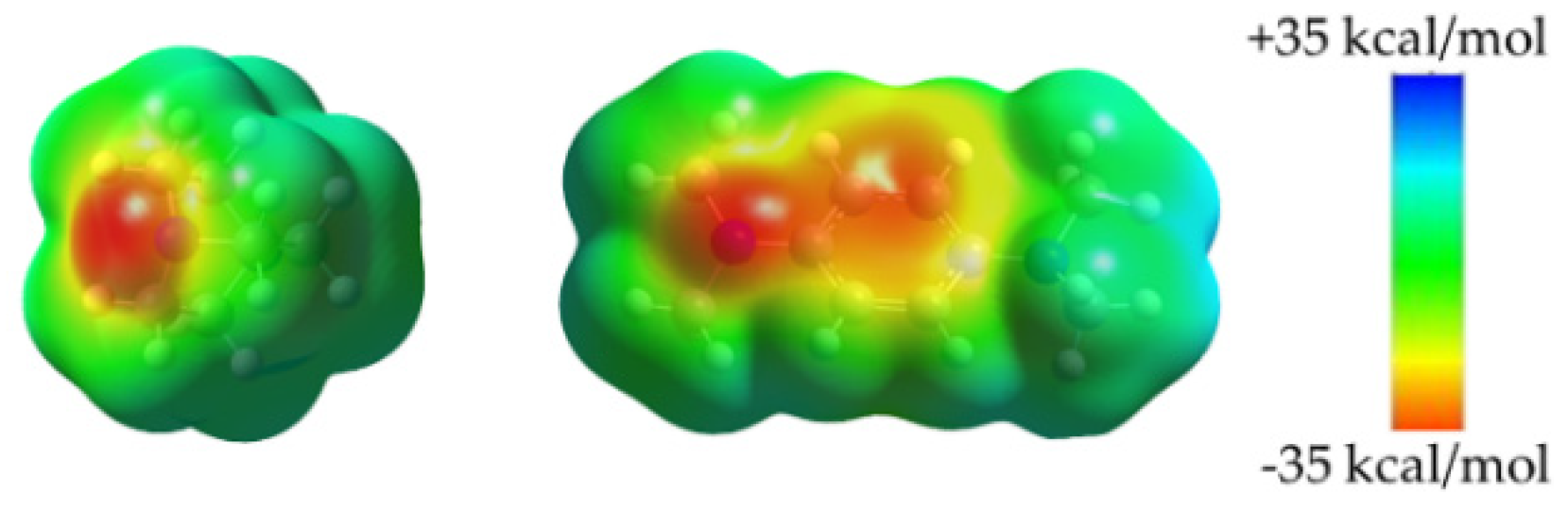
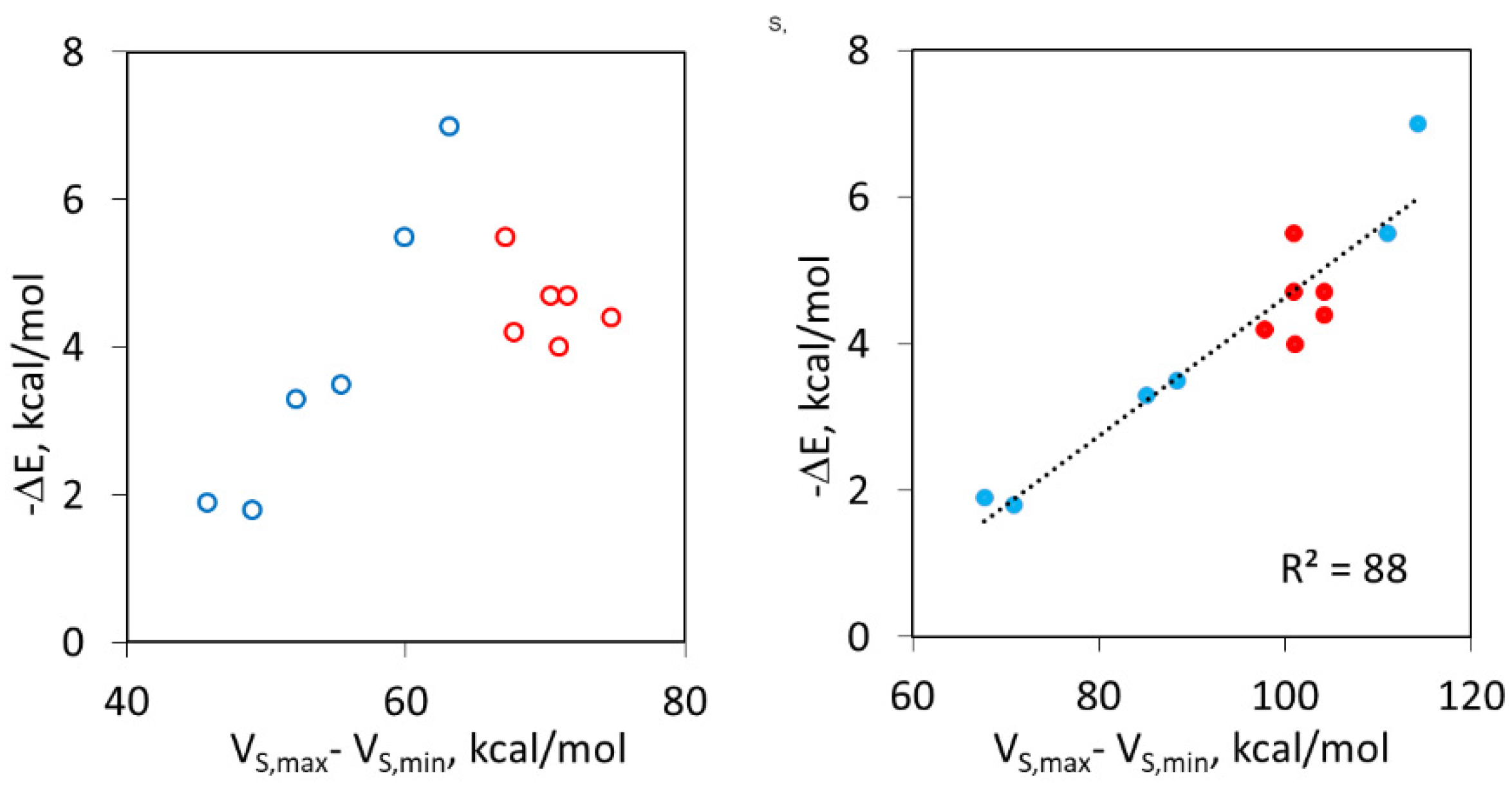
2.5. Unified Correlation of Strength of the XB and HB Complexes with the Surface Electrostatic Potentials in the Polarized Molecules
2.6. Differentiation of XB and HB Complexes Based on Their UV–Vis and NMR Characteristics
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Vinogradov, S.N.; Linnell, R.H. Hydrogen Bonding; Van Nostrand Reinhold: New York, NY, USA, 1971. [Google Scholar]
- Scheiner, S. Hydrogen Bonding: A Theoretical Perspective; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Metrangolo, P.; Neukirch, H.; Pilati, T.; Resnati, G. Halogen bonding based recognition processes: A world parallel to hydrogen bonding. Acc. Chem. Res. 2005, 38, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. The pnicogen bond: Its relation to hydrogen, halogen, and other noncovalent bonds. Acc. Chem. Res. 2013, 46, 280–288. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed]
- Gilday, L.C.; Robinson, S.W.; Barendt, T.A.; Langton, M.J.; Mullaney, B.R.; Beer, P.D. Halogen bonding in supramolecular chemistry. Chem. Rev. 2015, 115, 7118–7195. [Google Scholar] [CrossRef] [PubMed]
- Pennington, W.T.; Resnati, G.; Taylor, M.S. Halogen bonding: From self-assembly to materials and biomolecules. CrystEngComm 2013, 15, 3057. [Google Scholar] [CrossRef]
- Awwadi, F.F.; Taher, D.; Haddad, S.F.; Turnbull, M.M. Competition between hydrogen and halogen bonding interactions: Theoretical and crystallographic studies. Cryst. Growth Des. 2014, 14, 1961–1971. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Panikkattu, S.; Chopade, P.D.; Desper, J. Competing hydrogen-bond and halogen-bond donors in crystal engineering. CrystEngComm 2013, 15, 3125–3136. [Google Scholar] [CrossRef]
- Posavec, L.; Nemec, V.; Stilinović, V.; Cinčić, D. Halogen and hydrogen bond motifs in ionic cocrystals derived from 3-halopyridinium halogenides and perfluorinated iodobenzenes. Cryst. Growth Des. 2021, 21, 6044–6050. [Google Scholar] [CrossRef]
- Zapata, F.; Caballero, A.; Molina, P.; Alkorta, I.; Elguero, J. Open bis(triazolium) structural motifs as a benchmark to study combined hydrogen- and halogen-bonding interactions in oxoanion recognition processes. J. Org. Chem. 2014, 79, 6959–6969. [Google Scholar] [CrossRef] [Green Version]
- Aakeröy, C.B.; Fasulo, M.; Schultheiss, N.; Desper, J.; Moore, C. Structural competition between hydrogen bonds and halogen bonds. J. Am. Chem. Soc. 2007, 129, 13772–13773. [Google Scholar] [CrossRef] [PubMed]
- Watson, B.; Grounds, O.; Borley, W.; Rosokha, S.V.; Resolving halogen, vs. hydrogen bonding dichotomy in solutions: Intermolecular complexes of trihalomethanes with halide and pseudohalide anions. Phys. Chem. Chem. Phys. 2018, 2, 21999–22007. [Google Scholar] [CrossRef] [PubMed]
- von der Heiden, D.; Vanderkooy, A.; Erdélyi, M. Halogen bonding in solution: NMR spectroscopic approaches. Coord. Chem. Rev. 2020, 407, 213147. [Google Scholar] [CrossRef]
- Lu, J.; Scheiner, S. Effects of halogen, chalcogen, pnicogen, and tetrel bonds on IR and NMR Spectra. Molecules 2019, 24, 2822. [Google Scholar] [CrossRef]
- Green, R.D.; Martin, J.S. Anion-molecule complexes in solution. I. Nuclear magnetic resonance and infrared studies of halide ion-trihalomethane association. J. Am. Chem. Soc. 1968, 90, 3659–3668. [Google Scholar] [CrossRef]
- Bertrán, J.F.; Rodríguez, M. Detection of halogen bond formation by correlation of proton solvent shifts. 1. Haloforms in n-electron donor solvents. Org. Magn. Reson. 1979, 12, 92–94. [Google Scholar] [CrossRef]
- Schulz, N.; Sokkar, P.; Engelage, E.; Schindler, S.; Erdélyi, M.; Sanchez-Garcia, E.; Huber, S.M. The interaction modes of haloimidazolium salts in solution. Chem. Eur. J. 2018, 24, 3464–3473. [Google Scholar] [CrossRef]
- Nayak, S.K.; Terraneo, G.; Piacevoli, Q.; Bertolotti, F.; Scilabra, P.; Brown, J.T.; Rosokha, S.V.; Resnati, G. Molecular bases for anesthesia: Halothane as a halogen and hydrogen bonds donor. Angew. Chem. Int. Ed. 2019, 58, 12456–12459. [Google Scholar] [CrossRef]
- Corradi, E.; Meille, S.V.; Messina, M.T.; Metrangolo, P.; Resnati, G. Halogen bonding versus hydrogen bonding in driving self-assembly processes. Angew. Chem. Int. Ed. 2000, 39, 1782–1786. [Google Scholar] [CrossRef]
- Liantonio, R.; Luzzati, S.; Metrangolo, P.; Pilati, T.; Resnati, G. Perfluorocarbon-hydrocarbon self-assembly. Part 16: Anilines as new electron donor modules for halogen bonded infinite chain formation. Tetrahedron 2002, 58, 4023–4029. [Google Scholar] [CrossRef]
- Raatikainen, K.; Rissanen, K. Interaction between amines and N-haloimides: A new motif for unprecedentedly short Br⋯N and I⋯N halogen bonds. CrystEngComm 2011, 13, 6972–6977. [Google Scholar] [CrossRef]
- Weinberger, C.; Hines, R.; Zeller, M.; Rosokha, S.V. Continuum of covalent to intermolecular bonding in the halogen-bonded complexes of 1,4-diazabicyclo[2.2.2]octane with bromine-containing electrophiles. Chem. Commun. 2018, 54, 8060–8063. [Google Scholar] [CrossRef] [PubMed]
- Riel, A.M.S.; Rowe, R.K.; Ho, E.N.; Carlsson, A.-C.C.; Rappé, A.K.; Berryman, O.B.; Ho, P.S. Hydrogen bond enhanced halogen bonds: A synergistic interaction in chemistry and biochemistry. Acc. Chem. Res. 2019, 52, 2870–2880. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Hydrogen and halogen bonds are ruled by the same mechanisms. Phys. Chem. Chem. Phys. 2013, 15, 7249–7259. [Google Scholar] [CrossRef] [PubMed]
- Wolters, L.P.; Bickelhaupt, F.M. Halogen bonding versus hydrogen bonding: A molecular orbital perspective. ChemistryOpen 2012, 1, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xu, T.; van Mourik, T.; Früchtl, H.; Kirk, S.R.; Jenkins, S. Halogen and hydrogen bonding in halogenabenzene/NH3 complexes compared using next-generation QTAIM. Molecules 2019, 24, 2875. [Google Scholar] [CrossRef]
- Nepal, B.; Scheiner, S. Competitive halide binding by halogen versus hydrogen bonding: Bis-triazole pyridinium. Chem. Eur. J. 2015, 21, 13330–13335. [Google Scholar] [CrossRef]
- Benesi, H.A.; Hildebrand, J.H. A spectrophotometric investigation of the interaction of iodine with aromatic hydrocarbons. J. Am. Chem. Soc. 1949, 71, 2703–2707. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Bondi, A. Van der Waals volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef] [PubMed]
- Clark, T. Halogen bonds and σ-holes. Faraday Discuss. 2017, 203, 9–27. [Google Scholar] [CrossRef] [PubMed]
- Popelier, P.L.A. The QTAIM Perspective of Chemical Bonding. In The Chemical Bond: Fundamental Aspects of Chemical Bonding; John Wiley & Sons: Hoboken, NJ, USA, 2014; pp. 271–308. [Google Scholar]
- Miller, D.K.; Loy, C.; Rosokha, S.V. Examining a transition from supramolecular halogen bonding to covalent bonds: Topological analysis of electron densities and energies in the complexes of bromosubstituted electrophiles. ACS Omega 2021, 6, 23588–23597. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Bruker Apex3 v2016.9-0, SAINT V8.37A. Bruker AXS Inc.: Madison, WI, USA, 2016.
- SHELXTL Suite of Programs, version 6.14; Bruker AXS Inc.: Madison, WI, USA, 2003.
- Sheldrick, G. Crystal Structure Refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Hübschle, C.; Sheldrick, G.; Dittrich, B. ShelXle: A Qt Graphical User Interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
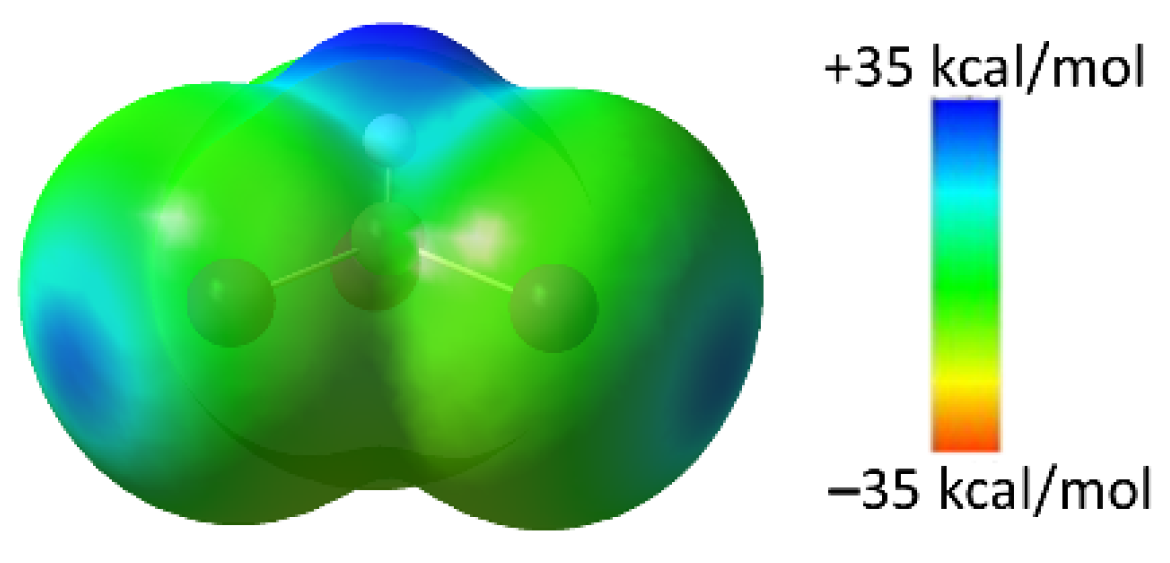{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CHX3 | B | XB Complexes | HB Complexes | ||||
|---|---|---|---|---|---|---|---|
| ΔE, kcal/mol | dX-N, Å | RXN a | ΔE, kcal/mol | dH-N, Å | RHN a | ||
| CHI3 | DABCO | −7.0 | 2.694 | 0.72 | −4.7 | 2.051 | 0.75 |
| TMPD | −5.5 | 2.845 | 0.76 | −5.5 | 2.163 | 0.79 | |
| CHBr3 | DABCO | −3.5 | 2.802 | 0.79 | −4.4 | 2.043 | 0.78 |
| TMPD | −3.3 | 2.872 | 0.81 | −4.7 | 2.095 | 0.76 | |
| CHCl3 | DABCO | −1.8 | 2.931 | 0.89 | −4.0 | 2.095 | 0.76 |
| TMPD | −1.9 | 2.892 | 0.88 | −4.2 | 2.183 | 0.79 | |
| CHX3 | B | XB Complexes | HB Complexes | ||
|---|---|---|---|---|---|
| ρ(r) × 102 | H(r) × 103 | ρ(r) × 102 | H(r) × 103 | ||
| CHI3 | DABCO | 3.65 | −3.70 | 2.76 | −1.11 |
| TMPD | 2.70 | −1.11 | 2.15 | 0.44 | |
| CHBr3 | DABCO | 2.39 | 0.51 | 2.81 | −1.31 |
| TMPD | 2.07 | 0.76 | 2.16 | 0.34 | |
| CHCl3 | DABCO | 1.52 | 1.61 | 2.53 | −0.37 |
| TMPD | 1.66 | 1.51 | 2.05 | 0.57 | |
| CHX3 | B | XB Complexes | HB Complexes | ||||
|---|---|---|---|---|---|---|---|
| λmax, nm | ε × 10−3, M−1 cm−1 | Δδ, ppm b | λmax, nm | ε × 10−3, M−1cm−1 | Δδ, ppm b | ||
| CHI3 | DABCO | 311 | 9.0 | −1.539 | 302 | 1.4 | 1.847 |
| TMPD | 335 | 7.2 | −0.669 | 295 | 4.5 | 1.506 | |
| CHBr3 | DABCO | 259 | 7.5 | −0.452 | 212 | 2.8 | 2.502 |
| TMPD | 285 | 8.0 | −0.437 | 245 | 20.0 | 1.503 | |
| CHCl3 | DABCO | 216 | 3.3 | −0.230 | 220 | 250 | 2.119 |
| TMPD | 290 | 3.0 | −0.344 | 247 | 20.1 | 1.353 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adeniyi, E.; Grounds, O.; Stephens, Z.; Zeller, M.; Rosokha, S.V. Thermodynamics and Spectroscopy of Halogen- and Hydrogen-Bonded Complexes of Haloforms with Aromatic and Aliphatic Amines. Molecules 2022, 27, 6124. https://doi.org/10.3390/molecules27186124
Adeniyi E, Grounds O, Stephens Z, Zeller M, Rosokha SV. Thermodynamics and Spectroscopy of Halogen- and Hydrogen-Bonded Complexes of Haloforms with Aromatic and Aliphatic Amines. Molecules. 2022; 27(18):6124. https://doi.org/10.3390/molecules27186124
Chicago/Turabian StyleAdeniyi, Emmanuel, Olivia Grounds, Zachary Stephens, Matthias Zeller, and Sergiy V. Rosokha. 2022. "Thermodynamics and Spectroscopy of Halogen- and Hydrogen-Bonded Complexes of Haloforms with Aromatic and Aliphatic Amines" Molecules 27, no. 18: 6124. https://doi.org/10.3390/molecules27186124