3. Materials and Methods
The synthetic manipulations involving air-sensitive compounds were performed in a nitrogen-filled Innovative Technology or Vigor glove box. All solvents were degassed and stored under high-purity nitrogen and activated 4Å molecular sieves. All deuterated solvents were stored under high-purity nitrogen on 3Å molecular sieves. Commercially available reagents (Aldrich, Strem, and Fluka) were used as received. The NMR spectra were recorded on a Bruker Avance 400 MHz spectrometer. 1H and 13C NMR signals are reported in ppm downfield from TMS. All measurements were performed at 22 °C in CDCl3/CD2Cl2 unless stated otherwise. Mass spectra were recorded on a VG-Autospec M-250 instrument. UV and fluorescence spectra were recorded on a Vernier fluorescence/UV-Vis spectrophotometer and Hitachi F-2710 fluorescence spectrophotometer.
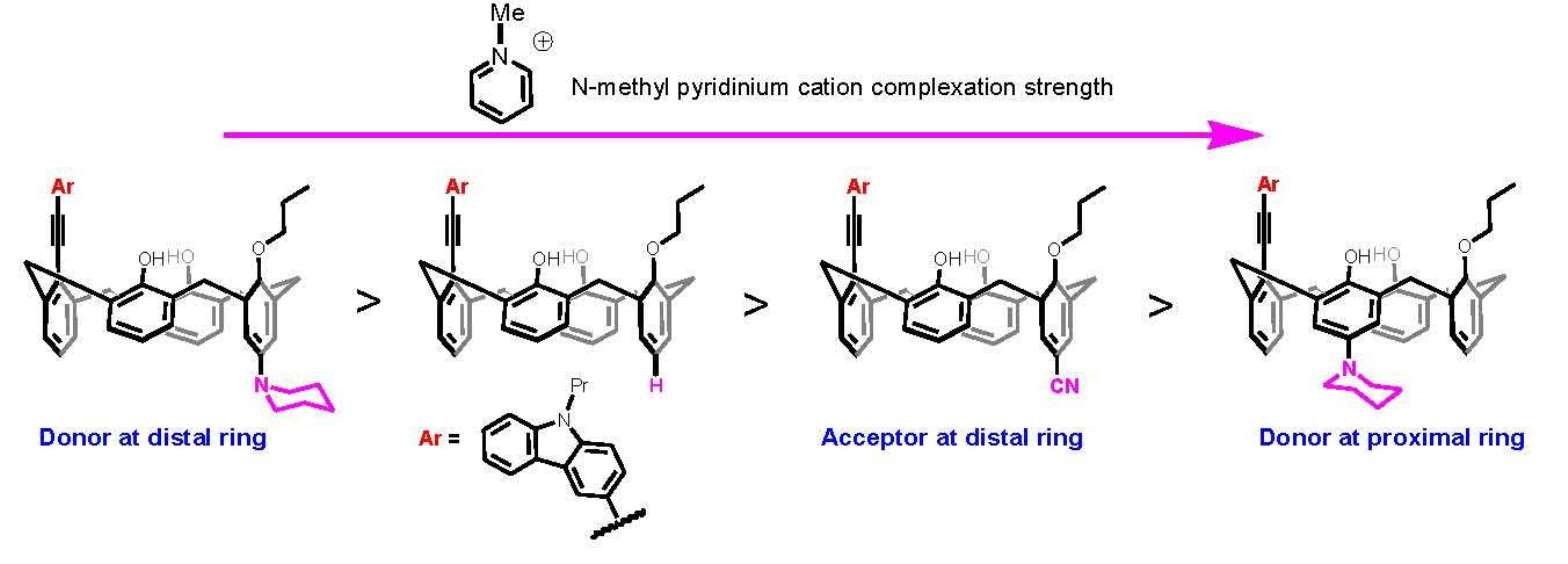
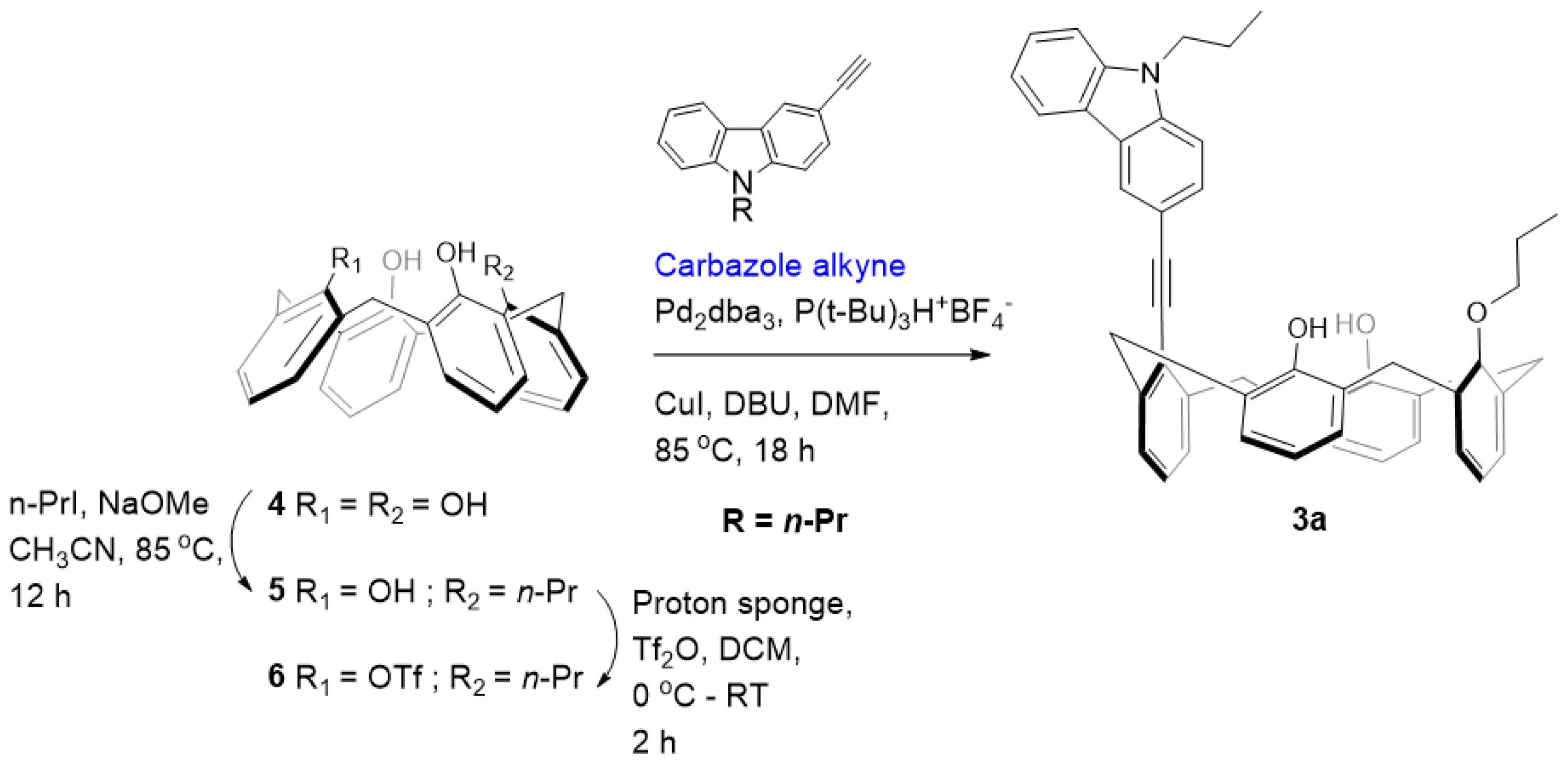
Synthesis of 5: A sample of 4.24 g (10.0 mmol) of calix[4]arene 4 and 0.64 g (11.8 mmol) of NaOCH3 was refluxed in 300 mL of CH3CN for 30 min to monodeprotonate the calix[4]arene completely. To this, 2.4 mL (4.18 g, 24.6 mmol) of n-propyl iodide was added, and the reaction mixture was further refluxed for 12 h. After the completion of the reaction (monitored by TLC), the reaction mixture was neutralized with a few drops of acetic acid, and the solvent was removed to leave an off-white residue. The residue was dissolved in 150 mL CHCl3 and successively washed with H2O and brine. The organic phase was separated, dried over anhydrous MgSO4, and evaporated. The residue was recrystallized from CHCl3 with a slow addition of CH3OH to yield the corresponding monoalkylated calix[4]arene 5. Yield: 3.26 g (70%); White solid; 1H NMR (CDCl3, 400 MHz): δ 10.25 (s, 1H), 9.79 (s, 1H), 9.48 (s, 1H), 7.05–7.13 (m, 6H), 6.93 (t, J = 7.7 Hz, 1H), 6.79 (t, J = 7.0 Hz, 3H), 6.17–6.76 (m, 2H), 4.45 (d, J = 13.4 Hz, 2H), 4.32 (d, J = 13.6 Hz, 2H), 4.18 (t, J = 6.9 Hz, 2H), 3.53 (d, J = 1.7 Hz, 2H), 3.50 (br s, 2H), 2.24 (q, J = 7.3, 14.7 Hz, 2H), 1.34 (t, J = 7.4 Hz, 3H). 13C NMR (CDCl3, 100 MHz): δ 151.58, 150.95, 149.36, 148.91, 134.39, 129.46, 129.11, 128.94, 128.88, 128.56, 128.37, 126.21, 122.38, 122.09, 121.05, 79.16, 32.05, 31.84, 31.56, 23.42, 10.80. ESI-MS calcd for [M+Na]+ C31H30NaO4 489.20, found 489.46.
Synthesis of 6: To a suspension of 1.81 g (3.9 mmol) of mono-propyl ether 5 and 1,8-bis(dimethylamino)naphthalene (proton sponge) (2.16 g, 10.1 mmol) in dry CH2Cl2 (40 mL) at 0 °C, trifluoromethanesulphonic anhydride (1.3 mL, 7.8 mmol) was added under nitrogen. After 2 h of stirring at room temperature, the organic layer was washed twice with HCl 10% and once with water, dried over MgSO4, and evaporated. The residue was subjected to column chromatography purification (CH2Cl2/Hexane 3/10 v/v) giving product 6. Yield: 1.82 g (78%); White solid; 1H NMR (CDCl3, 400 MHz): δ 7.34 (s, 2H), 7.17 (dd, J = 1.4, 7.5 Hz, 2H), 7.09 (dd, J = 1.5, 7.6 Hz, 2H), 7.05 (s, 1H), 7.02 (s, 1H), 6.88–6.92 (m, 3H), 6.79–6.84 (m, 1H), 6.75 (t, J = 7.6 Hz, 2H), 4.50 (d, J = 14.6 Hz, 2H), 4.19 (t, J = 5.5 Hz, 2H), 4.01 (d, J = 13.8 Hz, 2H), 3.58 (d, J = 13.8 Hz, 2H), 3.46 (d, J = 13.8 Hz, 2H), 2.19 (sextet, J = 7.7, 14.2 Hz, 2H), 1.34 (t, J = 7.5 Hz, 3H). 13C NMR (CDCl3, 100 MHz): δ 152.96, 149.95, 143.37, 133.99, 132.66, 129.96, 129.65, 129.07, 128.89, 128.63, 127.53, 127.07, 125.68, 119.63, 80.26, 31.95, 31.72, 23.19, 10.56. 19F NMR: −74.34 (s). ESI-MS calcd for [M+Na]+ C32H29F3NaO6S 621.15, found 621.41.
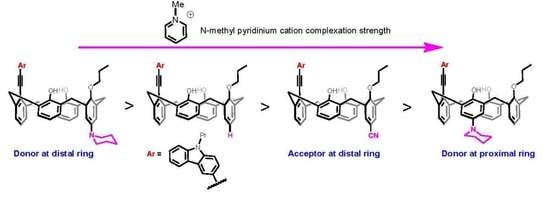
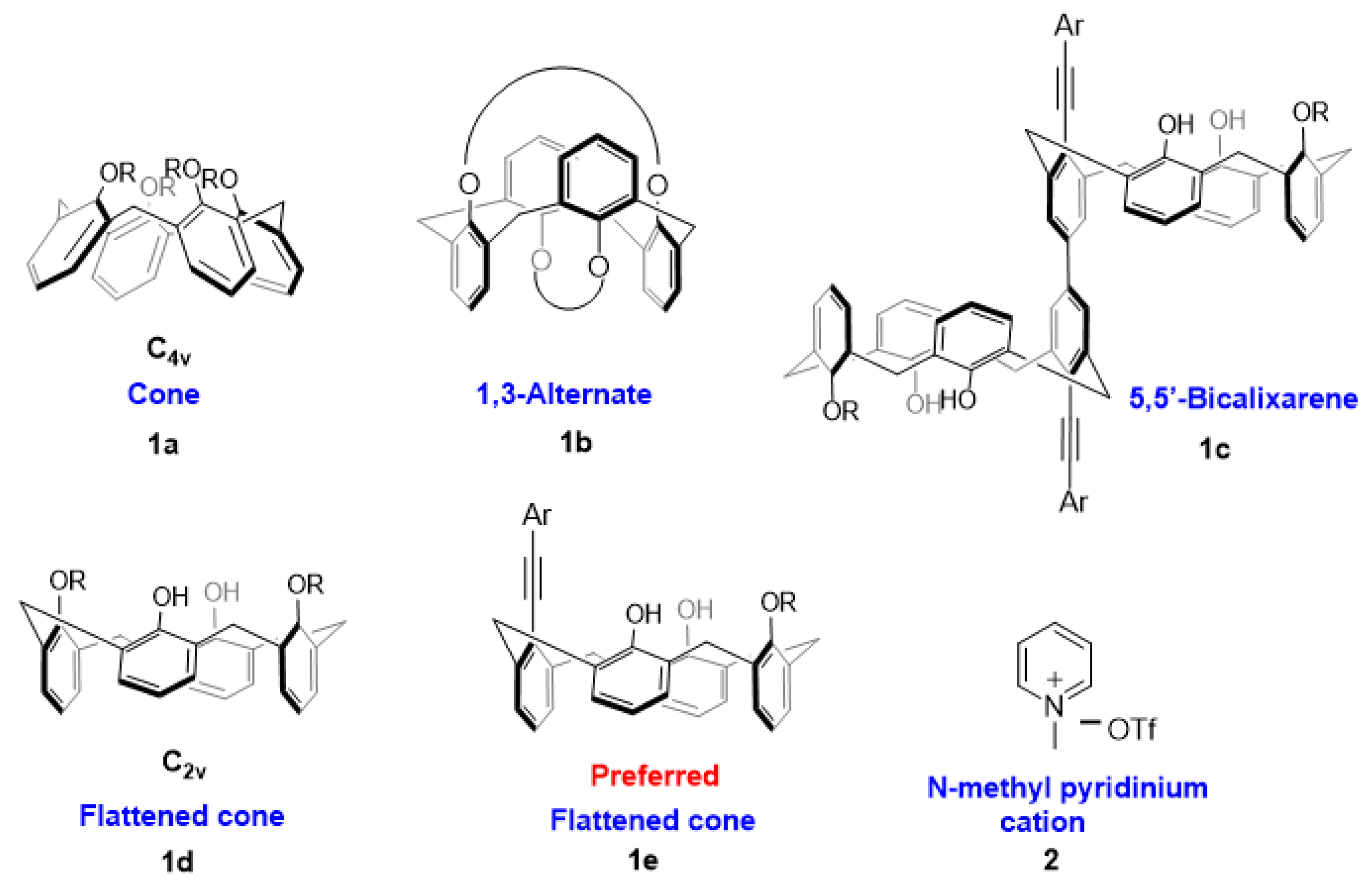
Synthesis of carbazole-appended calix[4]arene 3a: To a mixture of Pd2dba3 (0.05 equiv.) and P(t-Bu)3H+ BF4− (0.2 equiv.) dissolved in 10 mL of dry DMF, CuI (2.5 equiv.), DBU (4 equiv.), carbazole alkyne (5 equiv.) and triflate 6 (0.25 mmol) were added and the mixture was heated at 85 °C in an oil bath for 12 h. The solvent was evaporated, and the resulting crude product was dissolved in CH2Cl2 and washed with brine several times. Drying the CH2Cl2 extract over MgSO4 followed by solvent removal under vacuum gave the crude product. The residue was subjected to column chromatography (CH2Cl2/Hexane 4/10 v/v) to obtain the pure compound 3a. Yield: 0.106 g (62%); White solid; 1H NMR (CD2Cl2, 400 MHz): δ 8.53 (d, J = 0.9 Hz, 1H), 8.19 (d, J = 7.8 Hz, 1H), 7.90 (dd, J = 1.6, 8.5 Hz, 1H), 7.52–7.54 (m, 3H), 7.36 (s, 2H), 7.29–7.33 (m, 1H), 7.22 (dd, J = 1.4, 7.5 Hz, 2H), 7.12 (dd, J = 1.5, 7.6 Hz, 2H), 7.02–7.04 (m, 2H), 6.87–6.96 (m, 4H), 6.76 (t, J = 7.5 Hz, 2H), 4.93 (d, J = 12.1 Hz, 2H), 4.39 (t, J = 8.0 Hz, 2H), 4.17 (d, J = 14.7 Hz, 2H), 3.99 (t, J = 5.3 Hz, 2H), 3.69 (d, J = 13.3 Hz, 2H), 3.51 (d, J = 13.3 Hz, 2H), 1.96–2.05 (m, 2H), 1.83–1.92 (m, 2H), 1.04 (t, J = 7.0 Hz, 3H), 0.87 (t, J = 7.9 Hz, 3H). 13C NMR (CD2Cl2, 100 MHz): δ 153.8, 151.5, 141.7, 141.4, 140.4, 133.2, 129.8, 129.7, 129.5, 129.4, 129.1, 128.5, 128.4, 128.0, 127.7, 127.4, 127.2, 126.1, 125.8, 124.1, 123.0, 122.8, 120.8, 119.4, 114.3, 109.1, 108.8, 98.6, 86.5, 78.7, 44.9, 36.6, 31.9, 23.4, 22.5, 11.9, 10.7. HRMS (ESI-TOF) m/z [M+H]+ calcd for C48H44NO3 682.3321, found 682.3323.
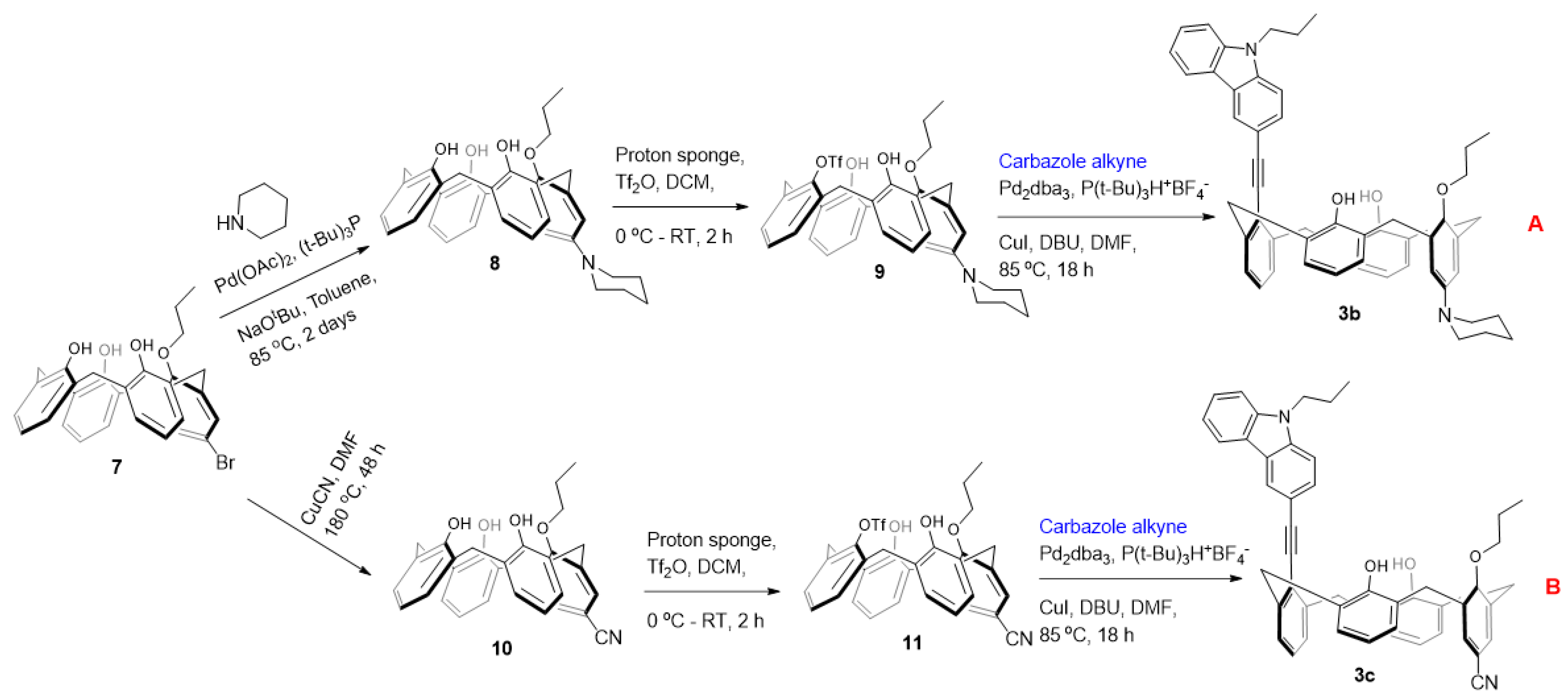
Synthesis of 8: The reaction was carried out under an inert atmosphere of pure nitrogen. To a stirred suspension of Pd(OAc)2 (0.014 g, 0.063 mmol), P(t-Bu)3 (0.019 g, 0.095 mmol) and sodium tert-butoxide (0.121 g, 1.26 mmol), in toluene (15 mL) were added piperidine (0.065 g, 0.75 mmol) and compound 7 (0.343 g, 0.63 mmol). The reaction mixture was then stirred at 85 °C for 48 h. The solvent was evaporated, and the resulting crude product was dissolved in EtOAc (50 mL), washed with water (5 mL × 2), brine, and dried over anhydrous MgSO4. Removal of solvent under reduced pressure and column chromatographic purification with EtOAc/Hexane (2:8 v/v) gave pure compounds 8 in 78% yields (0.270 g). Off-white solid: 1H NMR (CD2Cl2, 400 MHz): δ 9.43 (br s, 2H), 6.99–7.09 (m, 7H), 6.63–6.71 (m, 5H), 4.34 (d, J = 12.6 Hz, 2H), 4.26 (d, J = 12.6 Hz, 2H), 4.08 (t, J = 6.1 Hz, 2H), 3.49 (d, J 12.6 Hz, 2H), 3.39 (d, J = 12.6 Hz, 2H), 3.93–3.96 (m, 4H), 2.16–2.19 (m, 4H), 1.61–1.63 (m 4H), 1.49–1.52 (m, 2H), 1.29 (t, J = 6.8 Hz, 3H). 13C NMR (CD2Cl2, 100 MHz): δ 186.42, 177.34, 160.20, 158.22, 151.17, 150.30, 149.44, 144.46, 134.36, 133.99, 130.69, 129.22, 128.85, 128.76, 128.64, 128.52, 128.36, 128.15, 127.26, 126.01, 124.19, 121.90, 120.86, 120.25, 117.74, 117.41, 117.11, 79.18, 78.60, 78.34, 51.21, 51.04, 32.70, 31.87, 30.70, 30.55, 30.43, 26.16, 24.29, 23.43, 10.68. ESI-MS calcd for [M+H]+ C36H40NO4 550.30, found 550.62.
Synthesis of 9: To a suspension of 8 (0.270 g, 0.49 mmol) and 1,8-bis(dimethylamino)naphthalene (proton sponge) (0.272 g, 1.27 mmol) in dry CH2Cl2 (20 mL) at 0 °C trifluoromethanesulfonic anhydride (0.276 g, 0.16 mL, 0.98 mmol) was added under nitrogen. After 2 h of stirring at room temperature, the organic layer was washed once with 10% HCl and once with water, dried over anhydrous MgSO4, and evaporated. The residue was purified by column chromatography (silica gel, CH2Cl2/Hexane 4/10 v/v) to give the title compound as white solid. Yield: 0.244 g (73%); White solid; 1H NMR (CD2Cl2, 400 MHz): δ 7.65 (br s, 2H), 7.19 (dd, J = 1.5, 7.6 Hz, 2H), 7.14 (dd, J = 1.5, 7.6 Hz, 2H), 7.02 (s, 1H), 6.99 (s, 1H), 6.92–6.94 (m, 1H), 6.75 (t, J = 7.5Hz, 2H), 6.62 (s, 2H), 4.50 (d, J = 12.8 Hz, 2H), 4.17 (t, J = 6.4 Hz, 2H), 3.99 (d, J = 12.8 Hz, 2H), 3.55 (d, J = 12.8 Hz, 2H), 3.48 (d, J = 12.8 Hz, 2H), 2.99 (t, J = 5.3 Hz, 2H), 2.15–2.36 (m, 2H), 1.51–1.67 (m, 4H), 1.52–1.56 (m, 2H), 1.34 (t, J = 7.5 Hz, 3H). 13C NMR (CD2Cl2, 100 MHz): δ 153.03, 150.76, 143.28, 142.51, 134.13, 132.94, 130.04, 129.82, 128.88, 128.79, 128.58, 127.44, 125.93, 120.64, 119.70, 117.47, 117.21, 80.43, 50.44, 32.11, 31.86, 26.02, 24.18, 23.17, 10.36. 19F NMR: −74.74 (s). ESI-MS calcd for [M+H]+ C37H39F3NO6S 682.25, found 682.47.
Synthesis of carbazole-appended calix[4]arene 3b: To a mixture of Pd2dba3 (0.05 equiv.) and P(t-Bu)3H+ BF4- (0.2 equiv.) dissolved in 10 mL of dry DMF, CuI (2.5 equiv.), DBU (4 equiv.), carbazole alkyne (5 equiv.) and triflate 9 (0.25 mmol) were added and the mixture was heated at 85 °C in an oil bath for 12 h. The solvent was evaporated, and the resulting crude product was dissolved in CH2Cl2 and washed with brine several times. Drying the CH2Cl2 extract over anhydrous MgSO4 followed by solvent removal under vacuum gave the crude product. The residue was subjected to column chromatography (CH2Cl2/Hexane 4/10 v/v) to obtain the pure compound. Yield: 0.090 g (47%); Slightly yellow solid; 1H NMR (CD2Cl2, 400 MHz): δ 8.52 (d, J = 1.0 Hz, 1H), 8.20 (dd, J = 4.8, 3.9 Hz, 1H), 7.91 (dd, J = 8.5, 1.6 Hz, 1H), 7.62–7.49 (m, 5H), 7.28–7.34 (m, 1H), 7.22 (dd, J = 7.5, 1.5 Hz, 2H), 7.09 (dd, J = 7.6, 1.6 Hz, 2H), 7.02–6.90 (m, 3H), 6.71 (t, J = 7.5 Hz, 2H), 6.58 (s, 2H), 4.98 (d, J = 12.8 Hz, 2H), 4.39 (t, J = 7.1 Hz, 2H), 4.08 (d, J = 13.5 Hz, 2H), 3.96 (t, J = 6.6 Hz, 2H), 3.69 (d, J = 12.8 Hz, 2H), 3.46 (d, J = 12.9 Hz, 2H), 3.00–2.95 (m, 4H), 2.06–1.95 (m, 2H), 1.90–1.77 (m, 2H), 1.70–1.59 (m, 4H), 1.52–1.54 (m, 2H), 1.05 (t, J = 7.5 Hz, 3H), 0.82 (t, J = 7.4 Hz, 3H). 13C NMR (CD2Cl2, 100 MHz): δ 153.64, 150.28, 143.67, 141.78, 141.10, 140.32, 133.32, 129.53, 129.03, 128.73, 128.51, 128.20, 127.83, 127.23, 127.09, 126.81, 126.09, 123.72, 123.32, 122.90, 122.65, 120.62, 120.18, 119.52, 119.31, 117.28, 114.75, 109.20, 108.97, 97.33, 87.30, 79.50, 50.70, 44.87, 36.33, 32.17, 26.14, 24.26, 23.33, 22.46, 11.64, 10.35. HRMS (ESI-TOF) m/z [M+H]+ calcd for C53H53N2O3 765.4056, found 765.4055.
Synthesis of 10: To a solution of 7 (1.0 g, 1.83 mmol) in DMF (50 mL), CuCN (0.492 g, 5.49 mmol) was added. The resulting heterogeneous mixture was poured into a thick wall glass pressure round bottom flask and then heated at 180 °C for 48 h under vigorous stirring. After cooling, the solvent was completely evaporated under reduced pressure. The resulting sticky residue was extracted twice with hot ethyl acetate (2 × 100 mL). The combined organic phases were then washed twice with brine (2 × 100 mL), dried over anhydrous MgSO4, and then evaporated to dryness (the separated water phase was carefully treated with a solution of sodium hypochlorite to destroy the residuals cyanide ions). Purification of the solid residue by silica column chromatography (CH2Cl2/hexane, v/v 8:2) gave title compound. Yield: 0.603 g (67%); Slightly yellow solid; 1H NMR (CDCl3, 400 MHz): δ 9.43 (s, 1H), 9.13 (s, 2H), 7.41 (s, 2H), 7.14–7.08 (m, 4H), 7.04 (d, J = 7.6 Hz, 2H), 6.75 (m, 3H), 4.43 (d, J = 13.1 Hz, 2H), 4.29 (d, J = 13.8 Hz, 2H), 4.18 (dt, J = 14.3, 7.0 Hz, 2H), 3.54 (d, J = 3.7 Hz, 2H), 3.51 (d, J = 3.0 Hz, 2H), 2.30–2.18 (m, 2H), 0.93 (t, J = 6.9 Hz, 3H). 13C NMR (CDCl3, 100 MHz): δ 157.12, 153.92, 152.75, 143.34, 134.69, 133.88, 133.83, 132.95, 132.80, 132.69, 132.46, 130.99, 130.72, 130.29, 129.83, 129.48, 129.09, 128.73, 127.91, 127.69, 127.39, 126.82, 124.75, 120.56, 120.26, 119.84, 118.09, 117.38, 110.75, 80.54, 31.94, 31.45, 23.22, 23.22, 10.38. ESI-MS calcd for [M−H]− C32H28NO4 490.20, found 490.48.
Synthesis of 11: To a suspension of 0.500 g (1.02 mmol) of mono-propyl ether 10 and 1,8-bis(dimethylamino)naphthalene (proton sponge) (0.567 g, 2.65 mmol) in dry CH2Cl2 (20 mL) at 0 °C, trifluoromethanesulphonic anhydride (0.34 mL, 2.04 mmol) was added under nitrogen. After 2 h of stirring at room temperature, the organic layer was washed twice with 10% HCl and once with water, dried over anhydrous MgSO4, and evaporated. The residue was subjected to column chromatography purification (CH2Cl2/Hexane 3/10 v/v) giving product 11. Yield: 0.439 g (69%); Off-white solid; 1H NMR (CD2Cl2, 400 MHz): δ 7.33 (s, 2H), 7.23 (dd, J = 7.5, 1.3 Hz, 2H), 7.11 (dd, J = 7.6, 1.5 Hz, 2H), 6.96–6.84 (m, 3H), 6.81 (t, J = 7.5 Hz, 2H), 6.73 (s, 2H), 4.49 (d, J = 13.5 Hz, 2H), 4.18 (t, J = 6.5 Hz, 2H), 4.08 (d, J = 14.1 Hz, 2H), 3.61 (d, J = 14.1 Hz, 2H), 3.52 (d, J = 13.5 Hz, 2H), 2.38–2.12 (m, 2H), 1.34 (t, J = 7.4 Hz, 3H). 19F NMR: −74.21 (s). ESI-MS calcd for [M+Na]+ C33H28F3NNaO6S 646.15, found 646.49.
Synthesis of carbazole-appended calix[4]arene 3c: To a mixture of Pd2dba3 (0.05 equiv.) and P(t-Bu)3H+BF4− (0.2 equiv.) dissolved in 10 mL of dry DMF, CuI (2.5 equiv.), DBU (4 equiv.), carbazole alkyne (5 equiv.) and triflate 11 (0.25 mmol) were added and the mixture was heated at 85 °C in an oil bath for 12 h. The solvent was evaporated, and the resulting crude product was dissolved in CH2Cl2 and washed with brine several times. Drying the CH2Cl2 extract over anhydrous MgSO4 followed by solvent removal under vacuum gave the crude product. The residue was subjected to column chromatography (CH2Cl2/Hexane 4/10 v/v) to obtain the pure compound. Yield: 0.053 g (30%); Off-white solid; 1H NMR (CD2Cl2, 400 MHz): δ 8.53 (s, 1H), 8.20 (d, J = 7.7 Hz, 1H), 7.89 (d, J = 7.5 Hz, 1H), 7.54 (t, J = 7.0 Hz, 2H), 7.35–7.23 (m, 5H), 7.13 (d, J = 7.9 Hz, 2H), 7.04 (s, 2H), 6.97 (s, 3H), 6.85–6.65 (m, 3H), 4.89 (d, J = 13.4 Hz, 2H), 4.39 (t, J = 7.0 Hz, 2H), 4.29 (d, J = 13.5 Hz, 2H), 3.95 (t, J = 6.3 Hz, 2H), 3.77 (d, J = 13.5 Hz, 2H), 3.49 (d, J = 13.5 Hz, 2H), 1.98 (m, 2H), 1.85 (m, 2H), 1.04 (t, J = 7.3 Hz, 3H), 0.87 (t, J = 7.4 Hz, 3H). 13C NMR (CD2Cl2, 100 MHz): δ 155.88, 153.60, 141.62, 141.08, 140.60, 135.31, 133.72, 133.48, 133.22, 130.78, 129.47, 129.42, 129.30, 128.84, 128.56, 128.24, 128.06, 127.93, 127.32, 126.63, 126.23, 126.13, 123.92, 122.47, 122.07, 120.61, 120.56, 119.79, 119.43, 118.52, 113.23, 109.24, 109.06, 99.82, 85.68, 80.50, 78.92, 46.66, 44.82, 36.38, 31.27, 23.39, 22.38, 11.56, 10.29. HRMS (ESI-TOF) m/z [M+H]+ calcd for C49H43N2O3 707.3274, found 707.3279.
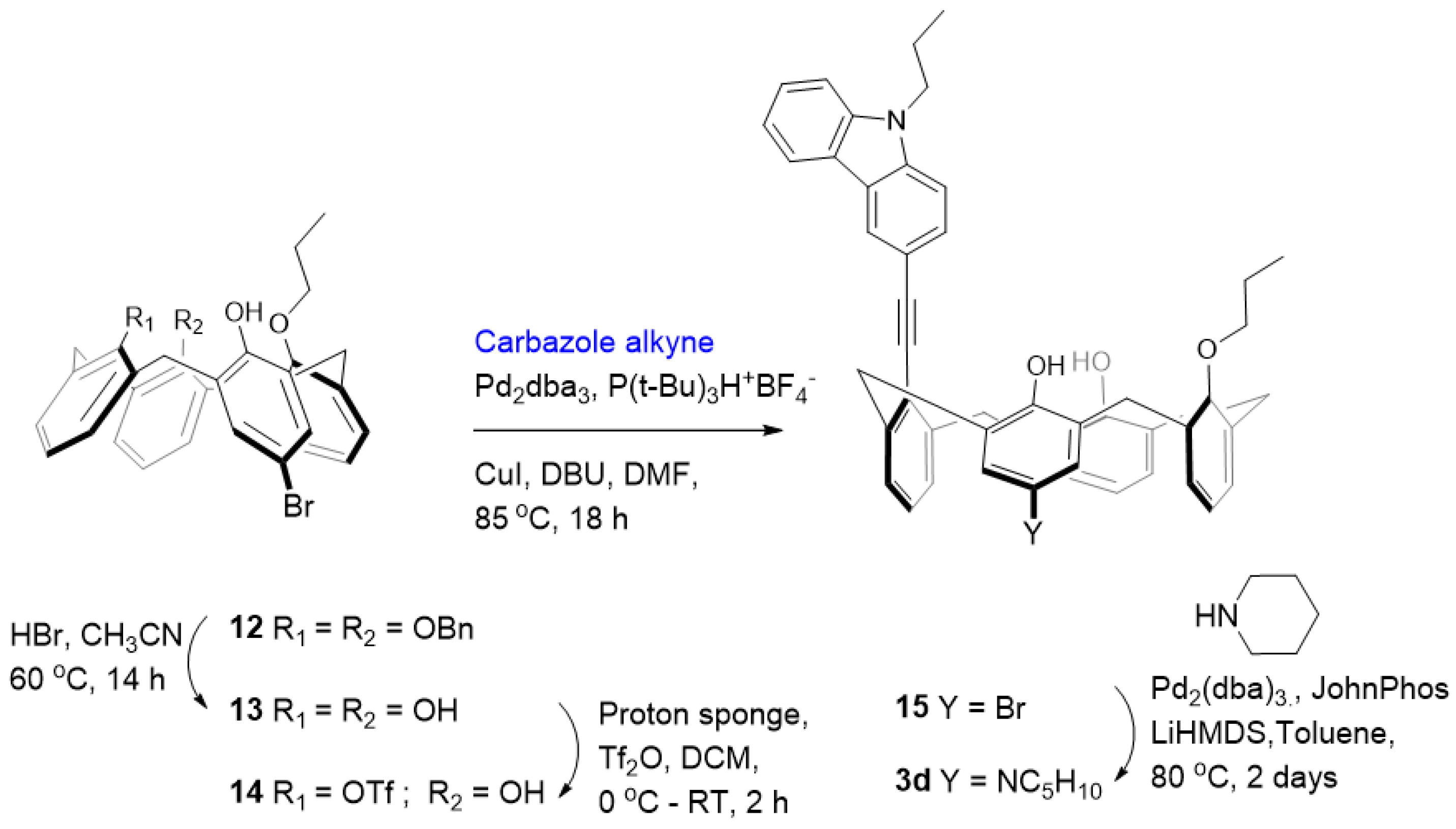
Synthesis of 13: Calixarene 12 [
24] (2.00 g, 2.87 mmol) was suspended in CH
3CN (40 mL), and after addition of 48% HBr (10 mL) the mixture was stirred at 60 °C for 12 h. The resulting suspension was diluted with CH
2Cl
2 and washed twice with water and once with brine. The organic layer dried over MgSO
4 and evaporated. The residue was washed several times with MeOH to remove benzyl alcohol giving the free phenol. The residue was recrystallized from CH
3Cl/MeOH to give pure calixarene 13. Yield: 1.04 g (70%); Pale yellow solid;
1H NMR (CDCl
3, 400 MHz): δ 9.76 (d, J = 1.0 Hz, 1H), 9.58 (d, J = 1.0 Hz, 1H), 9.38 (d, J = 1.1 Hz, 1H), 7.21 (d, J = 1.0 Hz, 2H), 7.16–7.03 (m, 5H), 7.01–6.92 (m, 2H), 6.80–6.70 (m, 2H), 4.48 (d, J = 12.8 Hz, 1H), 4.36–4.21 (m, 3H), 4.20–4.05 (m, 2H), 3.63–3.25 (m, 4H), 2.31–2.13 (m, 2H), 1.32 (td, J = 7.3, 1.3 Hz, 3H).
13C NMR (CDCl
3, 100 MHz): δ 150.56, 150.51, 150.43, 149.38, 134.09, 134.06, 131.20, 131.04, 130.88, 130.69, 129.73, 129.16, 128.90, 128.72, 128.43, 128.37, 128.02, 126.36, 125.59, 125.49, 122.17, 121.47, 119.16, 112.28, 79.24, 31.94, 31.89, 31.86, 3.31, 31.04, 23.38, 10.77. ESI-MS calcd for [M-H]
− C
31H
28BrO
4 543.12, found 543.50.
Synthesis of 14: To a suspension of 1.00 g (1.93 mmol) of mono-propyl ether 13 and 1,8-bis(dimethylamino)naphthalene (proton sponge) (1.08 g, 5.02 mmol) in dry CH2Cl2 (40 mL) at 0 °C, trifluoromethanesulphonic anhydride (0.65 mL, 3.86 mmol) was added under nitrogen. After 2 h of stirring at room temperature, the organic layer was washed twice with 10% HCl and once with water, dried over anhydrous MgSO4, and evaporated. The residue was subjected to column chromatography purification (CH2Cl2/Hexane 3/10 v/v) giving product 14.
Yield: 0.938 g (75%); White solid; 1H NMR (CDCl3, 400 MHz): δ 7.43 (s, 1H), 7.30 (s, 2H), 7.27–7.10 (m, 4H), 6.99 (dd, J = 13.1, 7.5 Hz, 2H), 6.91–6.73 (m, 4H), 4.48 (dd, J = 12.6, 9.5 Hz, 2H), 4.17 (t, J = 6.2 Hz, 2H), 3.99 (t, J = 13.0 Hz, 2H), 3.69–3.37 (m, 4H), 2.29–2.06 (m, 2H), 0.92 (t, J = 6.0 Hz, 3H). 13C NMR (CDCl3, 100 MHz): δ 152.90, 152.18, 149.91, 134.13, 133.11, 132.69, 131.75, 131.28, 131.13, 130.92, 130.32, 130.00, 129.95, 129.63, 128.96, 128.72, 127.62, 127.18, 125.52, 119.72, 111.09, 80.34, 31.93, 31.72, 31.49, 23.18, 22.78, 14.24, 10.53. 19F NMR: −74.34 (s). ESI-MS calcd for [M+Na]+ C32H28BrF3O6SNa 699.06, found 699.52.
Synthesis of 15: To a mixture of Pd2dba3 (0.05 equiv.) and P(t-Bu)3H+ BF4− (0.2 equiv.) dissolved in 10 mL of dry DMF, CuI (2.5 equiv.), DBU (4 equiv.), carbazole alkyne (5 equiv.) and triflate 14 (0.25 mmol) were added and the mixture was heated at 85 °C in an oil bath for 12 h. The solvent was evaporated, and the resulting crude product was dissolved in CH2Cl2and washed with brine several times. Drying the CH2Cl2 extract over anhydrous MgSO4 followed by solvent removal under vacuum gave the crude product. The residue was subjected to column chromatography (CH2Cl2/Hexane 4/10 v/v) to obtain the pure compound. Yield: 0.086 g (45%); Yellow solid; 1H NMR (CDCl3, 400 MHz): δ 8.47 (s, 1H), 8.18 (d, J = 7.8 Hz, 1H), 7.85 (d, J = 8.4 Hz, 1H), 7.64–7.42 (m, 2H), 7.34 (t, J = 9.3 Hz, 1H), 7.26–7.08 (m, 6H), 7.07–6.92 (m, 3H), 6.80 (m, 5H), 4.90 (t, J = 14.0 Hz, 2H), 4.35 (dd, J = 13.6, 6.6 Hz, 2H), 4.26–3.98 (m, 2H), 3.93 (t, J = 6.4 Hz, 2H), 3.66 (dd, J = 23.6, 11.9 Hz, 2H), 3.43 (dd, J = 25.8, 12.1 Hz, 2H), 1.99 (dd, J = 14.5, 7.2 Hz, 2H), 1.82 (dd, J = 14.0, 7.0 Hz, 2H), 1.03 (t, J = 7.5 Hz, 3H), 0.86 (t, J = 7.3 Hz, 3H). 13C NMR (CDCl3, 100 MHz): δ 153.03, 151.69, 142.00, 141.07, 140.80, 140.46, 133.37, 132.40, 131.51, 131.47, 131.04, 130.97, 130.74, 130.35, 130.23, 129.86, 129.78, 129.68, 129.51, 129.40, 129.29, 129.13, 128.49, 128.28, 128.12, 127.84, 127.60, 127.46, 127.41, 127.25, 126.48, 126.15, 125.84, 124.09, 120.82, 120.50, 119.45, 114.08, 110.94, 109.11, 108.83, 99.00, 86.22, 80.37, 78.66, 44.92, 37.58, 37.35, 36.64, 36.55, 31.95, 31.85, 23.42, 23.36, 22.50, 11.94, 10.64. ESI-MS calcd for [M-H]− C48H41BrNO3 758.23, found 758.50.
Synthesis of carbazole-appended calix[4]arene 3d: An oven-dried Schlenk tube was charged with Pd2(dba)3 (0.05 equiv.), JohnPhos (0.1 equiv.), calix halide 15 (0.08 mmol), amine (0.16 mmol) and toluene (2 mL). The reaction was stirred for few minutes and then LiHMDS (0.9–1.1 M in Hexanes) (0.18 mL) was added via syringe. The reaction vessel was sealed and heated at 80 °C with stirring for 48 h. The reaction mixture was then allowed to cool to room temperature, adsorbed on silica, and purified by column chromatography with EtOAc/hexane (2/8 v/v). Yield: 0.013 g (20%); Off-white solid; 1H NMR (CD2Cl2, 400 MHz): δ 8.33 (s, 1H), 8.16 (d, J = 7.8 Hz, 1H), 7.68 (d, J = 8.4 Hz, 1H), 7.58–7.45 (m, 3H), 7.42 (d, J = 6.0 Hz, 2H), 7.30 (t, J = 7.2 Hz, 1H), 7.23–7.13 (m, 2H), 7.10 (s, 1H), 7.02–6.76 (m, 4H), 6.66 (s, 1H), 6.56–6.37 (m, 3H), 4.53 (dt, J = 12.1, 5.9 Hz, 2H), 4.35 (t, J = 7.1 Hz, 2H), 4.15 (d, J = 14.1 Hz, 2H), 4.06 (t, J = 6.0 Hz, 2H), 3.58–3.43 (m, 4H), 3.39–2.83 (m, 4H), 2.09–1.93 (m, 4H), 1.81 (s, 4H), 1.72 (s, 2H), 1.26 (t, J = 7.2 Hz, 3H), 1.03 (t, J = 7.3 Hz, 3H). 13C NMR (CD2Cl2, 100 MHz): δ 153.76, 153.37, 151.62, 149.13, 141.05, 140.08, 138.63, 138.11, 133.07, 132.51, 131.60, 131.09, 130.75, 130.60, 129.40, 129.27, 129.18, 129.06, 128.41, 128.31, 127.67, 127.43, 126.15, 125.56, 125.44, 123.56, 123.48, 122.87, 122.41, 120.47, 119.29, 119.03, 114.01, 113.83, 109.15, 109.04, 88.59, 88.02, 78.62, 44.82, 33.93, 33.75, 31.44, 31.24, 27.71, 25.09, 23.35, 22.38, 13.81, 11.57, 10.43. HRMS (ESI-TOF) m/z [M]+ calcd for C53H53N2O3 765.4056, found 765.4057.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}