
Current Status of Quantum Chemical Studies of Cyclodextrin Host–Guest Complexes



Abstract
:
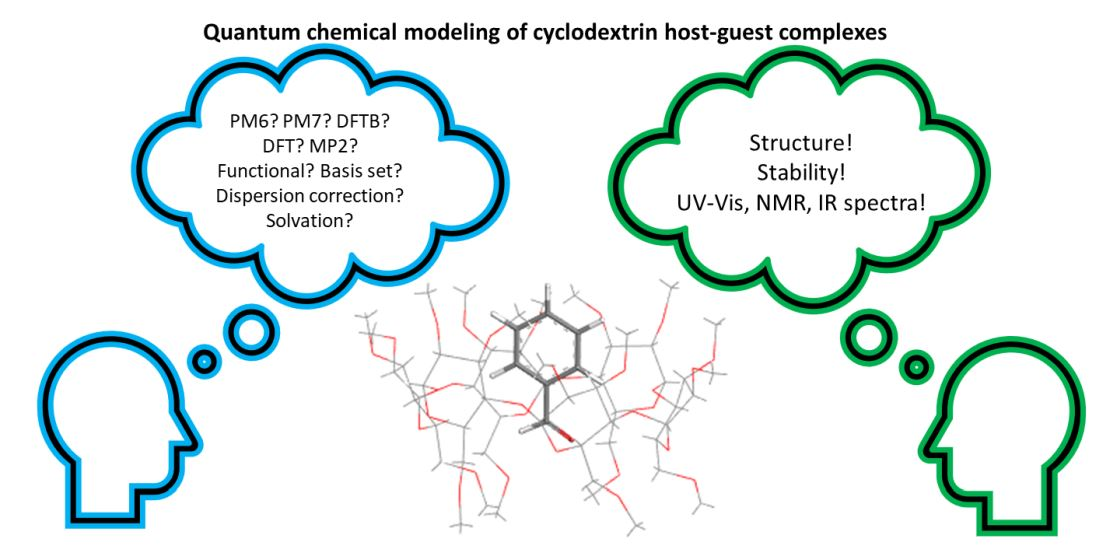
1. Introduction
2. Applied Calculational Methods and Parameters
2.1. Choice of QC Method
2.2. General Remarks
- Calculations of CD other than in the form of typical complexes
- Software
- ONIOM
- Molecular dynamics
2.3. Semi-Empirical Methods
2.4. Density Functional Tight Binding (DFTB)
2.5. Density Functional Theory (DFT)
- Functionals
- Dispersion correction
- Basis set
2.6. Solvent
2.7. Møller–Plesset Perturbation Theory 2 (MP2)
3. Preparation of Structures, Post-Processing Methods, and Some Examples
3.1. Preparation of the CD Complexes for the QC Calculations
3.2. Description of QC Results
3.3. Analyzed CD Complexes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | CD | Guest | Functional | Basis Set | Environment | DFT Application | Ref. |
|---|---|---|---|---|---|---|---|
| A (potential) drugs | |||||||
| 1 | β | (s)-2-Isopropyl-1-(o-nitrophenyl) Sulfonyl) Aziridine | B3LYP, WB97X-D, B97D3 | 6-31G(d) | gas, water | [110] | |
| 2 | β | boron-based aromatic systems | BLYP-D3(BJ) | def2-SVP | vacuum, CPCM | geo. opt., natural bond orbital calculations (NBO), complexation energy | [100] |
| 3 | α, β, γ | alprazolam | B3LYP, M06L | def-TZVP | vacuum | geo. opt. in gas, NMR spectra | [21] |
| 4 | β | lenalidomide | B3LYP, M06-2X | 6-31G(d,p) | PCM | [111] | |
| 5 | β | dexamethasone | BLYP-D4 | def2-TZVP | gas, water | geo. opt., complexation energy | [89] |
| 6 | β | 2,2′-Bipyridine | B3LYP, wB97XD | 6-31G(d) | PCM (eight solvents) | geo. opt., UV–Vis spectrum, HOMO-LUMO | [82] |
| 7 | β | 2,2′-Dipyridylamine | B3LYP | 6-311++G(d,p) | PCM | [112] | |
| 8 | vardenafil hydrochloride | B3LYP | 6-311G(2d,2p) | vacuum | geo. opt., FT-IR | [113] | |
| 9 | amino-CD | doxorubicin | B3LYP | 6-31G | vacuum | geo. opt., complexation energy, HOMO-LUMO, dipole moment, chemical potential, electrophilicity | [114] |
| 10 | β | 5-fluorouracil | B3LYP-D3 | 6-31+G(d,p) | vacuum, PCM | geo. opt., complexation energy, harmonic frequency calculations | [85] |
| 11 | HP-β | 2-methyl mercapto phenothiazine | B97-D3, BP86-D3 | 6-31G(d,p) | gas, CPCM | geo. opt., vibrational spectra, NBO, QTAIM, HOMO-LUMO | [115] |
| 12 | β | vemurafenib | ωB97XD | 6-31+G(d) | vacuum, PCM | Geo. opt., vibrational spectra, MD, NBO, TD, HOMO-LUMO | [116] |
| 13 | β | procaine hydrochloride | B3LYP, M06-2X, WB97XD | 6-31G(d,p) | gas, PCM | Geo. opt., NBO | [83] |
| 14 | β, SBE-β | fluorometholone, cholesterol | M06-2X | 6-31G** | PCM | Geo opt., interaction energy | [117] |
| 15 | α, β, γ | chlordecone | M06-2X-D3 | 6-31G(d,p) | SMD | Geo. opt., QTAIM | [118] |
| 16 | β, methyl-β | nicotine | M06-2X | 6-31G(d,p) | n.i.p. | Geo., opt., complexation enrgy | [34] |
| 17 | β | 8-Anilinonaphthalene-1-sulfonate | B3LYP, M06-2X, WB97X-D | 6-31G(d) | gas, water | Geo. opt., interaction energy, NMR, TD, NBO | [84] |
| 18 | β | benzocaine | B3LYP, CAM-B3LYP, M05-2X, M06-2X | 6-31G(d,p) | PCM | Geo. opt., QTAIM, NBO, NMR, HOMO-LUMO, TD | [53] |
| 19 | β | aryl pentazole | M06-2X | 6-31+G(d,p) | PCM | Geo. opt. | [119] |
| 20 | β | 2,4D, dicamba pesticides | PBE1PBE (PBE0), B97-D, M06-2X | 6-31G(d,p) | gas, SMD | Geo. opt. | [120] |
| 21 | Monochlorotriazinyl-β | permethrin, cyppermethrin | BLYP (geo. opt.); BLYP-D3, B3LYP-D3, M06-2X-D3 (UV–Vis) | def2-SV(P) (geo. opt.); TZVP (UV–Vis) | COSMO | Geo. opt. | [22] |
| 22 | β | dopamine | B3LYP, MPW1PW91, M05-2X, M06-2X, ωB97X-D | 3-21G* | CPCM | Geo. opt., complexation energy, QTAIM, NBO | [61] |
| 23 | α | benzoate derivatives | M06L (geo. opt.); M06-2X//M06-L (SP) | 6-31+G(d,p) | gas | Geo. opt. | [121] |
| 24 | α, β, γ | cholic, deoxycholic acid | B97-D, M06-2X, B3LYP | 6-31G(d) | PCM | Geo. opt., interaction energy | [122] |
| 25 | α | benzoate derivatives | M06-2X//M06-L, M06-2X//BLYP, BLYP, M06-2X | 6-31+G(d,p) | gas | Geo. opt., interaction energy | [123] |
| 26 | γ | cetirizine | B3LYP | def-TZVP | n.i.p. | Geo.opt., interaction energy, HOMO-LUMO, DOS, NMR | [124] |
| 27 | succinyl-β | uranium | M06-2X | 6-31G(d,p) | SMD | Geo. opt. | [125] |
| 28 | β-CD, DM -β | thymidine-carbonate | B3LYP-GD2 | 6-31G(d,p) | PCM | Geo. opt., complexation energy, TD, HOMO-LUMO, NMR | [126] |
| 29 | β | glycyl-L-phenylalanine | B3LYP | 3-21G(d) | PCM | Geo. opt., interaction energy, HOMO-LUMO | [127] |
| 30 | β | sodium salicylate | B3LYP | 6-31G(d) | gas, PCM | Geo. opt., solvation energy, relative stabilization energy, complexation energy, change of volume | [128] |
| 31 | β | benzyl isothiocyanthe | B97-D3 | def2-SVP | vacuum | Geo. opt., complexation energy, HOMO-LUMO, NBO, NMR | [23] |
| 32 | α | iodine solution | CAM-B3LYP | 6-31*G | PCM | Geo. opt., absorption spectra, HOMO-LUMO | [129] |
| 33 | β | meta-aminophenol | M06-2X | 6-31G(d,p) | IEFPCM | Geo. opt., complexation energy, HOMO-LUMO, TD, NBO | [130] |
| 35 | β | L-glutamine | B97-D3 | 6-31G(d) | n.i.p. | Geo. opt., complexation energy, TD, NBO, QTAIM | [131] |
| 36 | β | R and S ibuprofen | M062X | 6-31G(d,p) (geo. opt.); 6-311++G(d,p) (SP) | gas, SMD | Geo. opt., solvation energy | [132] |
| 37 | α, β | thioureides | B97-D3 | 6–31G(d,p) | Geo. opt., interaction energy | [133] | |
| 38 | β | mepivacaine | B97-D3 | 6-31G(d,p) | gas, SMD | Geo. opt., interaction energy, TD | [134] |
| 39 | β | L-metheonine | WB97-D3 | 6-31G(d) | PCM | geo. opt., interaction energy, QTAIM, TD, NMR | [98] |
| 40 | β | prazosin, losartan | B3LYP | 6–311+G(d,p) | gas | Geo. opt. | [135] |
| 41 | β | olsalazine | B3LYP, WB97-D3, CAM-B3LYP (UV-vis) | 6-31+G(d) | PCM | Geo. opt., ADMP | [62] |
| 42 | β | aspirin | B3LYP-D3 | cc-pVDZ | gas | Geo. opt., qTAIM, NBO | [136] |
| 43 | β | quinine | B3PW91 | 6-311++G(d,p) | PCM | Geo. opt. | [137] |
| 44 | β | erlotinib | B3LYP | 6-31+G* | n.i.p. | Geo. opt., harmonic frequencies, HOMO-LUMO | [138] |
| 45 | γ | rocuronium, vecuronium | B3LYP | 6–31+G(d,p) | n.i.p. | Geo. opt., NBO, HOMO-LUMO | [139] |
| 46 | α, β, γ | cathinone | M05-2X | 6-31G(d) | gas, CPCM (water, chloroform, methanol) | Geo. opt., QTAIM, NBO, IR spectra, TD | [140] |
| 47 | α | CO2 | B3LYP | G-31G* | PCM | NMR | [141] |
| 48 | β | flutafemic acid | B3LYP, M05-2X | 6-31G(d) | vacuum, water | Geo. opt., complexation energy, TD, NMR | [142] |
| 49 | 2-HP-β | Cu (II) and Fe (III) complexes of quercetin, morin, primuletin | B3LYP | 6-311++G** | n.i.p. | Geo. opt., complexation energy, HOMO-LUMO | [25] |
| 50 | β | 6-thioguanine, 6-mercaptopurine | B3LYP | 6-31+g(d,p) | IEFPCM (DMSO) | Geo. opt., interaction energy, TD | [37] |
| 51 | β | N-(2-chloroethyl),N -nitroso,N′,N′-dicyclohexylsulfamid | B3LYP | 6-31G(d) | PCM (DMSO) | Geo. opt., NBO, QTAIM | [143] |
| 52 | β | benzaldehyde | B97-D | 6-31G(d,p) (geo. opt.); 6-311++G(2d,p) (SP) | gas, SMD | Geo. opt., interaction energy, TD | [144] |
| 53 | α | chitibiose | M06-2X | 6-311++G** | n.i.p. | Geo. opt., NBO, QTAIM | [145] |
| 54 | α | hydrated and nonhydrated IIA/IIB group metal cations | M06-2X | 6-31G(d,p) | gas, PCM | Geo. opt., interaction energy, TD | [146] |
| 55 | β | nabumetone | WB97X-D, B97-D, B3LYP, M05-2X, M06-2X | 6-31G(d) | IEFPCM | Geo. opt., NBO, QTAIM | [40] |
| 56 | β | propranolol | B3LYP, ωB97XB (ONIOM) | 6-31+G(d) | gas, IEFPCM, explicit solvent effect: explicit water molecules inside of the complex | Geo. opt., interaction energy, ADMP, TD | [59] |
| 57 | functionalized CDs | 8-hydroxyquinoline ligands | B3LYP | 6-31G** | n.i.p. | Geo. opt. | [147] |
| 58 | β | pentoxifilline | M06-2X | 6-31g(d,p) | gas | Geo. opt., NBO, HOMO-LUMO | [101] |
| 59 | β | p-nitropenthyl acetate | B3LYP | 6-31G(d,p) | n.i.p. | Geo. opt., interaction energy, NBO, HOMO-LUMO | [148] |
| 60 | β | norfloxacin | B97D (geo. opt.), B3LYP (SP, NMR) | 6-31G(d,p) | IEFPCM | Geo. opt., interaction and stabilization energy, NMR, TD | [149] |
| A1. Antidepressants | |||||||
| 61 | β | paroxetine | B3LYP (geo. opt.); B97D (SP) | 6-31+G* for H, N, O and 4-31G for C | vacuum | Geo. opt., interaction energy, TD | [109] |
| 62 | 2,6-DM-β | mianserin | B3LYP-GD2 (geo. opt.); M05-GD3, M06-GD3, M062X-GD3, ωB97XD, mPW1PW91, M11 (SP) | 6-31G(d,p) | PCM, vacuum | Geo. opt., interaction energy, NMR | [150] |
| 63 | β | sertraline HCl, fluoxetine HCl | B3LYP | 6-31+G* for H, N, O and 4-31G for C | gas | Geo. opt., interaction energy | [151] |
| 64 | β | protriptyline, maprotiline | B3LYP | 6-31+G* for H, N, O and 4-31G for C | vacuum | Geo. opt., interaction and stabilization energy | [152] |
| 65 | β | clomipramine, doxepin | B3LYP | 31+G(d) for H, N, O, Cl, and 4-31G for C | gas | Geo. opt., interaction energy | [153] |
| 66 | β | desipramine, imipramine | B3LYP | 6-31þG(d) for H, N, O and 4- 31G for C | gas, implicit solvent (water) | Geo. opt., interaction and stabilization energy | [108] |
| 67 | β | amitryptyline, nortryptiline | n.i.p. | 6-31+G* for H, N, O, Cl and 4-31G for C | vacuum, SMD | Geo. opt., interaction and stabilization energy | [154] |
| B. Plant derivatives | |||||||
| 68 | HP-β | thymoquinone | B3LYP-D2, B3LYP-D3 | 6-31G(d,p) | PCM | Geo. opt., NBO, QTAIM, HOMO-LUMO, NMR | [155] |
| 69 | α | β-carotene | B3LYP | cc-pVDZ | vacuum | Geo. opt., interaction energy, Raman spectra | [156] |
| 70 | γ | 3-hydroxyflavone | PBE0 | def2-SV | PCM | Geo. opt., HOMO-LUMO, IT spectra | [157] |
| 71 | β | vanillina | B3LYP, ωB97xD, M06- 2X | 6-311G(d,p) | vacuum, CPCM | Geo. opt., interaction energy, NMR, HOMO-LUMO, NBO, QTAIM, UV–Vis | [107] |
| 72 | β | alfa-terpineol | B3LYP (for UV–Vis), B3LYP/CAM, M062X, WB97-D3 | 6-311G(d,p) | vacuum, CPCM | Geo. opt., complexation energy, NBO, QTAIM, TD, UV-vis | [158] |
| 73 | TM-β, β | naringenin | B3LYP, M06-2X, wB97X-D | 6-31G(d) | vacuum | Geo. opt., interaction energy, NBO, QTAIM, NMR, HOMO-LUMO | [159] |
| 74 | 2,6-DMβ, 2HP-β, 2,6-DH-β, β | eucalyptol | M06-2X | 6-31G(d,p) | Geo. opt., interaction energy | [160] | |
| 75 | β | fisetin | M06-2X | 6-31G(d,p) | gas, PCM | Geo. opt., interaction energy | [161] |
| 76 | β | gallic acid | B97-D3 | 6-31G*, for GIAO: 6-311++g** | gas, solvent | Geo. opt., HOMO-LUMO, NBO, NMR | [162] |
| 77 | β | gabapentin | B3LYP-D3 | 6- 31G(d) | vacuum, PCM | Geo. opt., interaction energy, NBO, HOMO-LUMO | [163] |
| 78 | Β, γ | tropane alkaloids | B3LYP | 6-31+G(d,p) | PCM | Geo. opt., interaction energy, NMR | [164] |
| 79 | β | coumarins | EDF2 | 6-311G(d,p) | PCM | Geo. opt. | [165] |
| 80 | 2-HP-β | quercetin | B3LYP | 6-31G* | Geo. opt. | [166] | |
| 81 | β | carvacrol, thymol | B3LYP | 6-31G, 6-31+G(d) | SMD | Geo. opt., interaction energy, NBO, HOMO-LUMO | [167] |
| 82 | β | thymol | B3LYP, PBEPBE, CAM-B3LYP | 6-31G(d,p) | PCM | Geo. opt., interaction energy, UV–Vis | [168] |
| 83 | β | carvacrol | B3LYP, M05-2X | 6-31G(d) | PCM | Geo. opt., HOMO-LUMO, NBO | [42] |
| C. Functionalized food | |||||||
| 84 | β | (−)-gallocatechin, (−)-catechin gallate, (−)-gallocatechin gallate | B3LYP | 6-31+G* for H, O and 4-31G for C | gas | Geo. opt., interaction and stabilization energy | [102,103] |
| 85 | β | (−)-epigallocatechin, (−)-epigallocatechin gallate | B3PW91 | cc-pVDZ | gas | Geo. opt., interaction energy | [104] |
| 86 | β | catechol derivatives: protocatechuic aldehyde, protocatechuic acid | B3LYP | 6-31+G* for H, O and 4-31G for C | gas (geo. opt.), implicit solvent (TD) | Geo. opt., interaction energy, TD | [169] |
| 87 | β | oleuropein, hydroxytyrosol, tyrosol | n.i.p. | 6-31+G* for H, O and 4-31G for C | gas | Geo. opt., interaction energy, TD | [170] |
| 88 | β | chlorogenic, caffeic, quinic acids | B3LYP | 6-31+G* for H, O and 4-31G for C | gas | Geo. opt., interaction energy, TD | [105,106] |
| D. CD as a chiral selector | |||||||
| 89 | β | D- and L-penicillamine | B3LYP-D3 (geo. opt.); M062X-D3, xB97X-D, B3LYP-D3 (interaction energy) | 6-31G(d, p) (geo. opt.); G-311+G(d,p) (interaction energy) | water | Geo. opt., interaction energy | [171] |
| 90 | metal-ion coupled β | D- and L-penicillamine | DFT, M062X | 6-31G(d,p) | vacuum | Geo. opt. | [172] |
| 91 | β | R- and S-propranolol | B3LYP | 6-311+G(d,p) | vacuum | Geo. opt., vibrational spectra | [173] |
| 92 | per-M β | D- and L-isoleucine | B3LYP (geo. opt.), wB97X-D (IR) | 6-31G*, 6-311G** | gas | Geo. opt., interaction energy, IR spectra, TD | [174] |
| 93 | per-M β | D- and L-alanine | B3LYP, wB97X-D, M06-2X | 6-31G**, 6-311G** | Geo. opt., IR spectra | [175] | |
| 94 | 2,3,6-TM-β | cis-(2S,4R) and -(2R,4S) ketoconazole | B3LYP | 6-311G(d,p) | gas (geo. opt.), PCM (SP) | Geo. opt., interaction energy | [36] |
| 95 | 2-HP-β | abacavir enentiomers | PBE | 6-31G* | PCM | Geo. opt., interaction energy | [176] |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aiassa, V.; Garnero, C.; Longhi, M.R.; Zoppi, A. Cyclodextrin Multicomponent Complexes: Pharmaceutical Applications. Pharmaceutics 2021, 13, 1099. [Google Scholar] [CrossRef] [PubMed]
- Poulson, B.G.; Alsulami, Q.A.; Sharfalddin, A.; El Agammy, E.F.; Mouffouk, F.; Emwas, A.-H.; Jaremko, L.; Jaremko, M. Cyclodextrins: Structural, Chemical, and Physical Properties, and Applications. Polysaccharides 2022, 3, 1. [Google Scholar] [CrossRef]
- Hădărugă, N.G.; Bandur, G.N.; David, I.; Hădărugă, D.I. A review on thermal analyses of cyclodextrins and cyclodextrin complexes. Environ. Chem. Lett. 2019, 17, 349–373. [Google Scholar] [CrossRef]
- Szejtli, J.; Szente, L. Elimination of bitter, disgusting tastes of drugs and foods by cyclodextrins. Eur. J. Pharm. Biopharm. 2005, 61, 115–125. [Google Scholar] [CrossRef]
- European Medicines Agency. Available online: https://www.ema.europa.eu/en (accessed on 29 May 2022).
- U.S. Food & Drug Administration (FDA). Available online: http://www.fda.gov/cder/guidance/index.htm (accessed on 29 May 2022).
- Pharmaceutical and Medical Devices Agencty. Available online: https://www.pmda.go.jp/english/index.html (accessed on 29 May 2022).
- Tran, D.N.; Colesnic, D.; de Beaumais, S.A.; Pembouong, G.; Portier, F.; Queijo, A.; Tato, J.V.; Zhang, Y.; Ménand, M.; Bouteiller, L.; et al. Cyclodextrin-adamantane conjugates, self-inclusion and aggregation versus supramolecular polymer formation. Org. Chem. Front. 2014, 1, 703–706. [Google Scholar] [CrossRef]
- Carrazana, J.; Jover, A.; Meijide, F.; Soto, A.V.H.; Tato, J.V. Complexation of Adamantyl Compounds by β-Cyclodextrin and Monoaminoderivatives. J. Phys. Chem. B 2005, 109, 9719–9726. [Google Scholar] [CrossRef]
- Jug, M.; Mura, P.A. Grinding as Solvent-Free Green Chemistry Approach for Cyclodextrin Inclusion Complex Preparation in the Solid State. Pharmaceutics 2018, 10, 189. [Google Scholar] [CrossRef] [Green Version]
- Wdowiak, K.; Rosiak, N.; Tykarska, E.; Żarowski, M.; Płazińska, A.; Płaziński, W.; Cielecka-Piontek, J. Amorphous Inclusion Complexes: Molecular Interactions of Hesperidin and Hesperetin with HP-Β-CD and Their Biological Effects. Int. J. Mol. Sci. 2022, 23, 4000. [Google Scholar] [CrossRef]
- Mazurek, A.H.; Szeleszczuk, Ł.; Gubica, T. Application of Molecular Dynamics Simulations in the Analysis of Cyclodextrin Complexes. Int. J. Mol. Sci. 2021, 22, 9422. [Google Scholar] [CrossRef]
- Dehghani, A.; Bahlakeh, G.; Ramezanzadeh, B.; Mofidabadi, A.H.J.; Mostafatabar, A.H. Benzimidazole loaded β-cyclodextrin as a novel anti-corrosion system; Coupled experimental/computational assessments. J. Colloid Interface Sci. 2021, 603, 716–727. [Google Scholar] [CrossRef]
- Roschi, E.; Gellini, C.; Ricci, M.; Sanchez-Cortes, S.; Focardi, C.; Neri, B.; Otero, J.C.; López-Tocón, I.; Smulevich, G.; Becucci, M. Surface-Enhanced Raman Spectroscopy for Bisphenols Detection: Toward a Better Understanding of the Analyte–Nanosystem Interactions. Nanomaterials 2021, 11, 881. [Google Scholar] [CrossRef] [PubMed]
- Paulino, P.H.S.; Silva, C.F.; De Almeida, B.D.; Guimarães, L.; Nascimento, C.S. A theoretical study of poly(p-phenylenes) and their cyclodextrin-based insulated molecular wires. Comput. Theor. Chem. 2021, 1197, 113157. [Google Scholar] [CrossRef]
- Dehghani, A.; Bahlakeh, G.; Ramezanzadeh, B. Synthesis of a non-hazardous/smart anti-corrosion nano-carrier based on beta-cyclodextrin-zinc acetylacetonate inclusion complex decorated graphene oxide (β-CD-ZnA-MGO). J. Hazard. Mater. 2020, 398, 122962. [Google Scholar] [CrossRef] [PubMed]
- Tomer, A.; Kusema, B.T.; Paul, J.-F.; Przybylski, C.; Monflier, E.; Pera-Titus, M.; Ponchel, A. Cyclodextrin-assisted low-metal Ni-Pd/Al2O3 bimetallic catalysts for the direct amination of aliphatic alcohols. J. Catal. 2018, 368, 172–189. [Google Scholar] [CrossRef]
- Quan, X.; Yi, S.; Wang, X. Theoretical study of an anti-Markovnikov addition reaction catalyzed by β-cyclodextrin. J. Mol. Model. 2018, 24, 77. [Google Scholar] [CrossRef]
- Mazurek, A.H.; Szeleszczuk, Ł.; Pisklak, D.M. Periodic DFT Calculations—Review of Applications in the Pharmaceutical Sciences. Pharmaceutics 2020, 12, 415. [Google Scholar] [CrossRef]
- Van De Streek, J.; Neumann, A.M. Validation of molecular crystal structures from powder diffraction data with dispersion-corrected density functional theory (DFT-D). Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2014, B70, 1020–1032. [Google Scholar] [CrossRef]
- Imtiaz, S.; Ali, S.M. Atom accurate structure determination of alprazolam/cyclodextrin inclusion complexes by ROESY and computational approaches. J. Indian Chem. Soc. 2022, 99, 100299. [Google Scholar] [CrossRef]
- Lu, H.; Wang, Y.; Xie, X.; Chen, F.; Li, W. Molecular dynamics simulation and TDDFT study of the structures and UV–vis absorption spectra of MCT-β-CD and its inclusion complexes. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 149, 564–570. [Google Scholar] [CrossRef]
- Bouhadiba, A.; Rahali, S.; Belhocine, Y.; Allal, H.; Nouar, L.; Rahim, M. Structural and energetic investigation on the host/guest inclusion process of benzyl isothiocyanate into β-cyclodextrin using dispersion-corrected DFT calculations. Carbohydr. Res. 2020, 491, 107980. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, R.; Lu, Z.; Ai, Y. Experimental and theoretical studies of spherical β-cyclodextrin modified titanium dioxide composites for uranium removal. Ecol. Eng. 2020, 149, 105835. [Google Scholar] [CrossRef]
- Jabeen, E.; Janjua, N.K.; Ahmed, S.; Murtaza, I.; Ali, T.; Masood, N.; Rizvi, A.S.; Murtaza, G. DFT predictions, synthesis, stoichiometric structures and anti-diabetic activity of Cu (II) and Fe (III) complexes of quercetin, morin, and primuletin. J. Mol. Struct. 2017, 1150, 459–468. [Google Scholar] [CrossRef]
- Yuan, S.; Shi, W.; Li, B.; Wang, J.; Jiao, H.; Li, Y.-W. Theoretical ONIOM2 Study on Pyridine Adsorption in the Channels and Intersection of ZSM-5. J. Phys. Chem. A 2005, 109, 2594–2601. [Google Scholar] [CrossRef]
- Christensen, A.; Kubař, T.; Cui, Q.; Elstner, M. Semiempirical Quantum Mechanical Methods for Noncovalent Interactions for Chemical and Biochemical Applications. Chem. Rev. 2016, 116, 5301–5337. [Google Scholar] [CrossRef]
- Ferrero, R.; Pantaleone, S.; Delle Piane, M.; Caldera, F.; Corno, M.; Trotta, F.; Brunella, V. On the Interactions of Melatonin/β-Cyclodextrin Inclusion Complex: A Novel Approach Combining Efficient Semiempirical Extended Tight-Binding (xTB) Results with Ab Initio Methods. Molecules 2021, 26, 5881. [Google Scholar] [CrossRef] [PubMed]
- Vidal, R.B.P.; Ibañez, G.A.; Escandar, G.M. Spectrofluorimetric study of phenolic endocrine disruptors in cyclodextrin media. RSC Adv. 2015, 5, 20914–20923. [Google Scholar] [CrossRef]
- Li, X.-S.; Liu, L.; Mu, T.-W.; Guo, Q.-X. A Systematic Quantum Chemistry Study on Cyclodextrins. Mon. Chem. 2000, 131, 849–855. [Google Scholar] [CrossRef]
- Fatiha, M.; Khatmi, D.E.; Largate, L. Theoretical approach in the study of the inclusion processes of sulconazole with β-cyclodextrin. J. Mol. Liq. 2010, 154, 1–5. [Google Scholar] [CrossRef]
- Setiadji, S.; Sundari, C.D.D.; Ramdhani, M.A.; Umam, A.B.K.; Ivansyah, A.L. Theoretical Investigation of Inclusion Complex between Omeprazole Enantiomers and Carboxymethyl-β-Cyclodextrin. IOP Conf. Ser. Mater. Sci. Eng. 2018, 288, 012138. [Google Scholar] [CrossRef]
- Saroj, M.K.; Payal, R.; Jain, S.; Sharma, N.; Rastogi, R.C. Investigation of indole chalcones encapsulation in β-cyclodextrin: Determination of stoichiometry, binding constants and thermodynamic parameters. J. Incl. Phenom. Macrocycl. Chem. 2018, 90, 306–320. [Google Scholar] [CrossRef]
- Nurhidayah, E.S.; Martoprawiro, M.; Zulfikar, M.A. PM3 and ONIOM2 modelling of inclusion complex of ibuprofen enantiomers with dimethyl-β-Cyclodextrin. J. Chem. Tech. Metall. 2019, 54, 673–678. [Google Scholar]
- Srihakulung, O.; Triamchaisri, N.; Toochinda, P.; Lawtrakul, L. Theoretical study on ferrocenyl hydrazones inclusion complexes with β-cyclodextrin and its three methylated derivatives. J. Incl. Phenom. Macrocycl. Chem. 2020, 98, 79–91. [Google Scholar] [CrossRef]
- Arsad, S.R.; Maarof, H.; Ibrahim, W.A.W.; Aboul-Enein, H.Y. Theoretical and Molecular Docking Study of Ketoconazole on Heptakis(2,3,6-tri-O -methyl)-β-cyclodextrin as Chiral Selector. Chirality 2016, 28, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Bensouilah, N.; Fisli, H.; Bensouilah, H.; Zaater, G.; Abdaoui, M.; Boutemeur-Kheddis, B. Host-guest complex of N-(2-chloroethyl), N-nitroso,N′,N′-dicyclohexylsulfamid with β-cyclodextrin: Fluorescence, QTAIM analysis and structure-chemical reactivity. J. Mol. Struct. 2017, 1146, 179–190. [Google Scholar] [CrossRef]
- Ivansyah, A.L.; Martoprawiro, M. Buchari Computational modeling of inclusion complex of r/s-omeprazole with β-cyclodextrin using oniom2 method. J. Phys. Conf. Ser. 2017, 812, 012070. [Google Scholar] [CrossRef]
- Yang, Z.; Huang, L.; Yao, X.; Ji, H. Host-guest complexes of estragole with β-cyclodextrin: An experimental and theoretical investigation. Flavour Fragr. J. 2017, 32, 102–111. [Google Scholar] [CrossRef]
- Bensouilah, N.; Boutemeur-Kheddis, B.; Meddour, I.; Abdaoui, M. Host-guest complex of nabumetone: β-cyclodextrin: Quantum chemical study and QTAIM analysis. J. Incl. Phenom. Macrocycl. Chem. 2017, 87, 191–206. [Google Scholar] [CrossRef]
- Reis, V.S.; Santos, E.S.; Bonsolhos, D.N.F.; Guimarães, L.; De Almeida, W.G.; Nascimento, C.S. Theoretical study on the formation process of Cross-Linked β-Cyclodextrin molecular tubes. Chem. Phys. Lett. 2017, 677, 13–18. [Google Scholar] [CrossRef]
- Abdelmalek, L.; Fatiha, M.; Leila, N.; Mouna, C.; Nora, M.; Djameleddine, K. Computational study of inclusion complex formation between carvacrol and β-cyclodextrin in vacuum and in water: Charge transfer, electronic transitions and NBO analysis. J. Mol. Liq. 2016, 224, 62–71. [Google Scholar] [CrossRef]
- Al Azzam, K.M.; Muhammad, E. Host-guest Inclusion Complexes between Mitiglinide and the Naturally Occurring Cyclodextrins α, β, and γ: A Theoretical Approach. Adv. Pharm. Bull. 2015, 5, 289–291. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Yao, X.; Xiao, Z.; Chen, H.; Ji, H. Preparation and release behaviour of the inclusion complexes of phenylethanol withβ-cyclodextrin. Flavour Fragr. J. 2016, 31, 206–216. [Google Scholar] [CrossRef]
- Nazarov, V.B.; Avakyan, V.G.; Bagrii, E.I.; Vershinnikova, T.G.; Alfimov, M.V. Long-lived phosphorescence of arenes in complexes with cyclodextrins 2. Room-temperature phosphorescence of ternary complexes of naphthalene and phenanthrene with β-cyclodextrin and adamantane derivatives in the presence of oxygen. Russ. Chem. Bull. 2005, 54, 2752–2756. [Google Scholar] [CrossRef]
- Bouzit, H.; Stiti, M.; Abdaoui, M. Spectroscopic and molecular modelling investigations of supramolecular complex of β-cyclodextrin with N-[(4-sulfonamidophenyl)ethyl]-5-(1,2-dithiolan-3-yl)pentanamide. J. Incl. Phenom. Macrocycl. Chem. 2016, 86, 121–134. [Google Scholar] [CrossRef]
- Laspidou, C.S.; Archimandritis, A.S.; Papadimitriou, T.; Kormas, K.A.; Yannakopoulou, K.; Lazarou, Y.G. Theoretical investigation of microcystin-LR, microcystin-RR and nodularin-R complexation with α-, β-, and γ-cyclodextrin as a starting point for the targeted design of efficient cyanotoxin traps. Sustain. Chem. Pharm. 2016, 3, 25–32. [Google Scholar] [CrossRef]
- Suliman, F.O.; Elbashir, A.A.; Schmitz, O.J. Study on the separation of ofloxacin enantiomers by hydroxyl-propyl-β-cyclodextrin as a chiral selector in capillary electrophoresis: A computational approach. J. Incl. Phenom. Macrocycl. Chem. 2015, 83, 119–129. [Google Scholar] [CrossRef]
- Mizera, M.; Lewandowska, K.; Miklaszewski, A.; Cielecka-Piontek, J. Machine Learning Approach for Determining the Formation of β-Lactam Antibiotic Complexes with Cyclodextrins Using Multispectral Analysis. Molecules 2019, 24, 743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djilani, I.; Madi, F.; Nouar, L.; Haiahem, S.; Rahim, M.; Khatmi, D.E.; Bouhadiba, A. Theoretical investigation to characterize the inclusion complex of α-lipoic acid and β-cyclodextrin. Comptes Rendus Chim. 2015, 18, 170–177. [Google Scholar] [CrossRef]
- Bouhadiba, A.; Belhocine, Y.; Rahim, M.; Djilani, I.; Nouar, L.; Khatmi, D.E. Host-guest interaction between tyrosine and β-cyclodextrin: Molecular modeling and nuclear studies. J. Mol. Liq. 2017, 233, 358–363. [Google Scholar] [CrossRef]
- Sambrook, M.R.; Vincent, J.C.; Ede, J.A.; Gass, I.A.; Cragg, P.I. Experimental and computational study of the inclusion complexes of β-cyclodextrin with the chemical warfare agent soman (GD) and commonly used simulants. RSC Adv. 2017, 7, 38069–38076. [Google Scholar] [CrossRef] [Green Version]
- Yahia, H.A.; Yahia, O.A.; Khatmi, D.; Belghiche, R.; Bouzitouna, A. Quantum chemical investigations on hydrogen bonding interactions established in the inclusion complex β-cyclodextrin/benzocaine through the DFT, AIM and NBO approaches. J. Incl. Phenom. Macrocycl. Chem. 2017, 89, 353–365. [Google Scholar] [CrossRef]
- Ceborska, M.; Kędra-Królik, K.; Kowalska, A.A.; Koźbiał, M. Comparative study of molecular recognition of folic acid subunits with cyclodextrins. Carbohydr. Polym. 2018, 184, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Appell, M.; Evans, K.O.; Jackson, M.A.; Compton, D.L. Determination of ochratoxin A in grape juice and wine using nanosponge solid phase extraction clean-up and liquid chromatography with fluorescence detection. J. Liq. Chromatogr. Relat. Technol. 2018, 41, 949–954. [Google Scholar] [CrossRef]
- López-Méndez, L.J.; Rojas-Aguirre, Y.; Vázquez-Lima, H.; Cassani, J.; Enríquez, R.G.; Rojo-Domínguez, A.; Guadarrama, P. On the conformational search of a βCD dendritic derivative: NMR and theoretical calculations working together reveal a donut-like amphiphilic structure. J. Mol. Struct. 2020, 1204, 127535. [Google Scholar] [CrossRef]
- Sahra, K.; Dinar, K.; Seridi, A.; Kadri, M. Investigation on the inclusion of diclofenac with β-cyclodextrin: A molecular modeling approach. Struct. Chem. 2015, 26, 61–69. [Google Scholar] [CrossRef]
- Srihakulung, O.; Maezono, R.; Toochinda, P.; Kongprawechnon, W.; Intarapanich, A.; Lawtrakul, A.L. Host-Guest Interactions of Plumbagin with β-Cyclodextrin, Dimethyl-β-Cyclodextrin and Hydroxypropyl-β-Cyclodextrin: Semi-Empirical Quantum Mechanical PM6 and PM7 Methods. Sci. Pharm. 2018, 86, 20. [Google Scholar] [CrossRef] [Green Version]
- Bani-Yaseen, A.W. Computational molecular perspectives on the interaction of propranolol with β-cyclodextrin in solution: Towards the drug-receptor mechanism of interaction. J. Mol. Liq. 2017, 227, 280–290. [Google Scholar] [CrossRef]
- Prabhu, A.A.M.; Fatiha, M.; Leila, N.; Raj, T.A.; Navarro-González, I.; Periago, M.J.; Yáñez-Gascón, M.J.; Pérez-Sánchez, H. Investigation of 3D Contour Map and Intermolecular Interaction of Dopamine with β-Cyclodextrin and 2-Hydroxypropyl-β-cyclodextrin. J. Solut. Chem. 2018, 47, 409–429. [Google Scholar] [CrossRef]
- Iyengar, S.S.; Schlegel, B.H.; Voth, G.A. Atom-Centered Density Matrix Propagation (ADMP): Generalizations Using Bohmian Mechanics. J. Phys. Chem. A 2003, 107, 7269–7277. [Google Scholar] [CrossRef] [Green Version]
- Al-Jaber, A.S.; Bani-Yaseen, A.D. On the encapsulation of Olsalazine by β-cyclodextrin: A DFT-based computational and spectroscopic investigations. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 214, 531–536. [Google Scholar] [CrossRef]
- Silva, D.A.; Xavier, M.J.; Dutra, J.D.L.; Gimenez, I.F.; Freire, R.O.; da Costa, N.B. Prediction of correct intermolecular interactions in host-guest systems involving cyclodextrins. J. Mol. Struct. 2020, 1205, 127517. [Google Scholar] [CrossRef]
- Lula, I.; Gomes, M.F.; Piló-Veloso, D.; Noronha, A.L.O.; Duarte, H.A.; Santos, R.A.S.; Sinisterra, R.D. Spironolactone and its Complexes with β-cyclodextrin: Modern NMR Characterization and Structural DFTB-SCC Calculations. J. Incl. Phenom. Macrocycl. Chem. 2006, 56, 293–302. [Google Scholar] [CrossRef]
- Lula, I.; Denadai, L.; Resende, J.M.; de Sousa, F.B.; de Lima, G.F.; Pilo-Veloso, D.; Heine, T.; Duarte, H.A.; Santos, R.A.; Sinisterra, R.D. Study of angiotensin-(1–7) vasoactive peptide and its β-cyclodextrin inclusion complexes: Complete sequence-specific NMR assignments and structural studies. Peptides 2007, 28, 2199–2210. [Google Scholar] [CrossRef] [PubMed]
- Lukin, O.; Dolgonos, G.; Leszczynski, J. A comprehensive test of computational approaches for evaluation of cyclodextrin complexes. Self-inclusion in monosubstituted β-cyclodextrins—A case study. Tetrahedron 2017, 73, 5302–5306. [Google Scholar] [CrossRef]
- Schnupf, U.; Momany, F.A. DFT Energy Optimization of a Large Carbohydrate: Cyclomaltohexaicosaose (CA-26). J. Phys. Chem. B 2011, 116, 6618–6627. [Google Scholar] [CrossRef] [PubMed]
- Hanpaibool, C.; Chakcharoensap, T.; Hijikata, Y.; Irle, S.; Wolschann, P.; Kungwan, N.; Pongsawasdi, P.; Ounjai, P.; Rungrotmongkol, T. Theoretical analysis of orientations and tautomerization of genistein in β-cyclodextrin. J. Mol. Liq. 2018, 265, 16–23. [Google Scholar] [CrossRef]
- Yunta, M. Using Molecular Modelling to Study Interactions between Molecules with Biological Activity. IntechOpen 2012. [Google Scholar] [CrossRef] [Green Version]
- Lu, L. Can B3LYP be improved by optimization of the proportions of exchange and correlation functionals? Int. J. Quantum Chem. 2015, 115, 502–509. [Google Scholar] [CrossRef]
- Ivanov, P. Performance of some DFT functionals with dispersion on modeling of the translational isomers of a solvent-switchable [2]rotaxane. J. Mol. Struct. 2016, 1107, 31–38. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. How Accurate Are the Minnesota Density Functionals for Noncovalent Interactions, Isomerization Energies, Thermochemistry, and Barrier Heights Involving Molecules Composed of Main-Group Elements? J. Chem. Theory Comput. 2016, 12, 4303–4325. [Google Scholar] [CrossRef] [Green Version]
- Valero, R.; Costa, R.; Moreira, I.D.P.R.; Truhlar, D.; Illas, F. Performance of the M06 family of exchange-correlation functionals for predicting magnetic coupling in organic and inorganic molecules. J. Chem. Phys. 2008, 128, 114103. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Jin, X.; Yu, H.S.; Truhlar, D.G.; He, X. Revised M06-L functional for improved accuracy on chemical reaction barrier heights, noncovalent interactions, and solid-state physics. Proc. Natl. Acad. Sci. USA 2017, 114, 8487–8492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardirossian, N.; Head-Gordon, M. ωB97X-V: A 10-parameter, range-separated hybrid, generalized gradient approximation density functional with nonlocal correlation, designed by a survival-of-the-fittest strategy. Phys. Chem. Chem. Phys. 2014, 16, 9904–9924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Bučko, T.; Lebègue, S.; Hafner, J.; Ángyán, J.G. Tkatchenko-Scheffler van der Waals correction method with and without self-consistent screening applied to solids. Phys. Rev. B 2013, 87, 064110. [Google Scholar] [CrossRef]
- Grimme, S.; Steinmetz, M. Effects of London dispersion correction in density functional theory on the structures of organic molecules in the gas phase. Phys. Chem. Chem. Phys. 2013, 15, 16031–16042. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Alkan, M.; Gordon, M.S. Many-Body Dispersion. Chem. Rev. 2020, 120, 12343–12356. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Salma, A.; Fatiha, M.; Leila, N. Effect of solvent on absorption and emission spectra of 2,2′-Bipyridine and its inclusion complexinto β-cyclodextrin: DFT and TD-DFT study. Comput. Theor. Chem. 2021, 1206, 113481. [Google Scholar] [CrossRef]
- Azayez, M.; Fergoug, T.; Meddah-Araibi, N.; Zelmat, C.; Bouhadda, Y. Theoretical Investigation of the Complexation Reaction of Procaine-hydrochloride by β-cyclodextrin. Phys. Chem. Res. 2020, 8, 155–165. [Google Scholar] [CrossRef]
- Safia, H.; Ismahan, L.; Abdelkrim, G.; Mouna, C.; Leila, N.; Fatiha, M. Density functional theories study of the interactions between host β-Cyclodextrin and guest 8-Anilinonaphthalene-1-sulfonate: Molecular structure, HOMO, LUMO, NBO, QTAIM and NMR analyses. J. Mol. Liq. 2019, 280, 218–229. [Google Scholar] [CrossRef]
- Buczek, A.; Staś, A.; Hebenstreit, C.; Maller, C.; Broda, M.A.; Kupka, T.; Kelterer, A. Interaction of 5-fluorouracil with β-cyclodextrin: A density functional theory study with dispersion correction. Int. J. Quantum Chem. 2021, 121, e26487. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cancès, E. The IEF version of the PCM solvation method: An overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct. 1999, 464, 211–226. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Belhocine, Y.; Rahali, S.; Allal, H.; Assaba, I.M.; Ghoniem, M.G.; Ali, F.A.M. A Dispersion Corrected DFT Investigation of the Inclusion Complexation of Dexamethasone with β-Cyclodextrin and Molecular Docking Study of Its Potential Activity against COVID-19. Molecules 2021, 26, 7622. [Google Scholar] [CrossRef]
- Oqmhula, K.; Hongo, K.; Maezono, R.; Ichibha, T. Ab Initio Evaluation of Complexation Energies for Cyclodextrin-Drug Inclusion Complexes. ACS Omega 2020, 5, 19371–19376. [Google Scholar] [CrossRef]
- Sambrook, M.R.; Gass, I.A.; Cragg, P.J. Spectroscopic and inclusion properties of G-series chemical warfare agents and their simulants: A DFT study. Supramol. Chem. 2017, 30, 206–217. [Google Scholar] [CrossRef] [Green Version]
- Sure, R.; Grimme, S. Comprehensive Benchmark of Association (Free) Energies of Realistic Host–Guest Complexes. J. Chem. Theory Comput. 2015, 11, 3785–3801. [Google Scholar] [CrossRef]
- Lopes, J.F.; Nascimento, C.S.; Anconi, C.P.A.; Santos, H.F.; Almeida, W.B. Inclusion complex thermodynamics: The β-cyclodextrin and sertraline complex example. J. Mol. Graph. Model. 2015, 62, 11–17. [Google Scholar] [CrossRef]
- CCDC, Cambridge Crystallographic Data Centre. Available online: https://www.ccdc.cam.ac.uk/ (accessed on 29 May 2022).
- Autodock. Available online: https://autodock.scripps.edu/ (accessed on 29 May 2022).
- Available online: https://www.schrodinger.com/products/maestro (accessed on 29 May 2022).
- Biovia. Available online: https://www.3ds.com/ (accessed on 29 May 2022).
- Nora, M.; Ismahan, L.; Abdelkrim, G.; Mouna, C.; Leila, N.; Fatiha, M.; Nada, B.; Brahim, H. Interactions in inclusion complex of β-cyclodextrin/l-Metheonine: DFT computational studies. J. Incl. Phenom. Macrocycl. Chem. 2020, 96, 43–54. [Google Scholar] [CrossRef]
- Pan, A.; Kar, T.; Rakshit, A.K.; Moulik, S.P. Enthalpy–Entropy Compensation (EEC) Effect: Decisive Role of Free Energy. J. Phys. Chem. B 2016, 120, 10531–10539. [Google Scholar] [CrossRef] [PubMed]
- Rahali, S.; Belhocine, Y.; Allal, H.; Bouhadiba, A.; Assaba, I.M.; Seydou, M. A DFT Investigation of The Host-Guest Interactions Between Boron-Based Aromatic Systems and β-Cyclodextrin. Res. Sq. 2022, 1–2. [Google Scholar] [CrossRef]
- Morais, C.A.S.; Silva, B.L.; Denadai, A.M.L.; Lopes, J.F.; De Sousa, F.B. Structural and thermodynamic investigation of pentoxifylline-cyclodextrin inclusion complex. Chem. Phys. Lett. 2017, 682, 43–48. [Google Scholar] [CrossRef]
- Aree, T.; Jongrungruangchok, S. β-Cyclodextrin encapsulation elevates antioxidant capacity of tea: A closing chapter on non-epicatechins, atomistic insights from X-ray analysis, DFT calculation and DPPH assay. Carbohydr. Polym. 2018, 194, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Aree, T.; Jongrungruangchok, S. Enhancement of antioxidant activity of green tea epicatechins in β-cyclodextrin cavity: Single-crystal X-ray analysis, DFT calculation and DPPH assay. Carbohydr. Polym. 2016, 20, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Ohata, T.; Yukawa, M.; Tsutsumi, H.; Fujisawa, M.; Aki, H. Calculation study on complex formation of catechins with β-cyclodextrin using density function theory. J. Incl. Phenom. Macrocycl. Chem. 2021, 100, 99–107. [Google Scholar] [CrossRef]
- Aree, T. Understanding structures and thermodynamics of β-cyclodextrin encapsulation of chlorogenic, caffeic and quinic acids: Implications for enriching antioxidant capacity and masking bitterness in coffee. Food Chem. 2019, 293, 550–560. [Google Scholar] [CrossRef]
- Aree, T. Inclusion complex of β-cyclodextrin with coffee chlorogenic acid: New insights from a combined crystallographic and theoretical study. Acta Crystallogr. Sect. C Struct. Chem. 2019, 75, 15–21. [Google Scholar] [CrossRef]
- Meryem, G.; Rabah, K.; Fatiha, M.; Leila, N.; Aziz, B.A.; Imane, D.; Rachid, M. Computational investigation of vanillin@βéta-cyclodextrin inclusion complex: Electronic and intermolecular analysis. J. Mol. Liq. 2021, 321, 114839. [Google Scholar] [CrossRef]
- Aree, T. β-Cyclodextrin Inclusion Complexation With Tricyclic Antidepressants Desipramine and Imipramine: A Structural Chemistry Perspective. J. Pharm. Sci. 2020, 109, 3086–3094. [Google Scholar] [CrossRef] [PubMed]
- Aree, T. Inclusion Scenarios and Conformational Flexibility of the SSRI Paroxetine as Perceived from Polymorphism of β-Cyclodextrin–Paroxetine Complex. Pharmaceuticals 2022, 15, 98. [Google Scholar] [CrossRef] [PubMed]
- Keniche, A.; Slimani, M.Z.; Miranda, J.I.; Aizpurua, J.M.; Mulengi, J.K. NMR Investigation of the complexation of (S)-2-isopropyl-1-(o-nitrophenyl)sulfonyl)aziridine with β-cyclodextrin. Mediterr. J. Chem. 2013, 2, 620–631. [Google Scholar] [CrossRef]
- Harati, H.; Morsali, A.; Bozorgmehr, M.R.; Beyramabadi, S.A. β-cyclodextrin-lenalidomide anticancer drug delivery nanosystem: A quantum chemical approach. J. Mol. Liq. 2021, 344, 117762. [Google Scholar] [CrossRef]
- Rohman, M.A.; Phanrang, P.T.; Chamlagai, D.; Mitra, S. Deciphering Spectroscopic and Structural Insights into the Photophysical Behavior of 2,2′-Dipyridylamine: An Efficient Environment Sensitive Fluorescence Probe. J. Phys. Chem. A 2021, 125, 6964–6975. [Google Scholar] [CrossRef]
- Wiergowska, G.; Ludowicz, D.; Wdowiak, K.; Miklaszewski, A.; Lewandowska, K.; Cielecka-Piontek, J. Combinations of Freeze-Dried Amorphous Vardenafil Hydrochloride with Saccharides as a Way to Enhance Dissolution Rate and Permeability. Pharmaceuticals 2021, 14, 453. [Google Scholar] [CrossRef]
- Akhondi, M.; Jamalizadeh, E.; Mohebbi, A. MD and DFT calculations on the structural variations of amino-cyclodextrin as a pH-sensitive carrier for smart carriage and release of Doxorubicin. J. Mol. Struct. 2021, 1230, 129855. [Google Scholar] [CrossRef]
- Mezari, Y.; Nouar, L.; Madi, F.; Guendouzi, A.; Djellala, I.; Lafifi, I.; Merdes, R.; Bouhadiba, A.; Houari, B. Theoretical investigation of inclusion complex of 2-methyl mercapto phenothiazine with hydroxy propyl β-cyclodextrin by DFT approaches. Bulg. Chem. Comm. 2021, 53, 196–210. [Google Scholar] [CrossRef]
- Bani-Yaseen, A.D. The supramolecular host-guest complexation of Vemurafenib with β-cyclodextrin and cucurbit[7]uril as drug photoprotecting systems: A DFT/TD-DFT study. Comput. Theor. Chem. 2020, 1191, 113026. [Google Scholar] [CrossRef]
- Jafari, G.; Raissi, H.; Hashemzadeh, H. Molecular insight into the interaction of fluorometholone and cholesterol molecules with β-cyclodextrin and sulfobutylether-β-cyclodextrin. Comput. Theor. Chem. 2022, 1208, 113554. [Google Scholar] [CrossRef]
- Gamboa-Carballo, J.J.; Ferino-Pérez, A.; Rana, V.K.; Levalois-Grützmacher, J.; Gaspard, S.; Montero-Cabrera, L.A.; Haza, U.J.J. Theoretical Evaluation of the Molecular Inclusion Process between Chlordecone and Cyclodextrins: A New Method for Mitigating the Basis Set Superposition Error in the Case of an Implicit Solvation Model. J. Chem. Inf. Model. 2020, 60, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-Z.; Liu, X.-F.; Zhang, R.-B.; Pang, S.-P. Joint experimental and theoretical studies of the surprising stability of the aryl pentazole upon noncovalent binding to β-cyclodextrin. Phys. Chem. Chem. Phys. 2017, 19, 31236–31244. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.A.; Borges, W.M.D.S.; Peraro, C.R.; Anconi, C.P.A. Theoretical inclusion of deprotonated 2,4-D and dicamba pesticides in ß-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2016, 86, 343–349. [Google Scholar] [CrossRef]
- Li, Z.; Couzijn, E.P.A.; Zhang, X. Intrinsic Properties of α-Cyclodextrin Complexes with Benzoate Derivatives in the Gas Phase: An Experimental and Theoretical Study. J. Phys. Chem. B 2012, 116, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Mori, Y.; Takano, K. Theoretical Study on Intermolecular Interactions in Complexes of Cyclodextrins with Bile Acids: DFT and Ab Initio Fragment Molecular Orbital Calculations. Bull. Chem. Soc. Jpn. 2014, 87, 258–266. [Google Scholar] [CrossRef]
- Li, Z.; Couzijn, E.P.A.; Zhang, X. A quantitative study of intrinsic non-covalent interactions within complexes of α-cyclodextrin and benzoate derivatives. Chem. Commun. 2012, 48, 9864–9866. [Google Scholar] [CrossRef] [Green Version]
- Muzaffar, S.; Imtiaz, S.; Ali, S.M. Demonstrating accuracy of the proposed protocol for structure elucidation of cyclodextrin inclusion complexes by validation using DFT studies. J. Mol. Struct. 2020, 1217, 128419. [Google Scholar] [CrossRef]
- Li, N.; Yang, L.; Ji, X.; Ren, J.; Gao, B.; Deng, W.-Q.; Wang, Z. Bioinspired succinyl-β-cyclodextrin membranes for enhanced uranium extraction and reclamation. Environ. Sci. Nano 2020, 7, 3124–3135. [Google Scholar] [CrossRef]
- Ignaczak, A.; Orszański, Ł.; Adamiak, M.; Olejniczak, A.B. Comparative DFT study of inclusion complexes of thymidine-carborane conjugate with β-cyclodextrin and heptakis(2,6-O-dimethyl)-β-cyclodextrin in water. J. Mol. Liq. 2020, 315, 113767. [Google Scholar] [CrossRef]
- Ramos, M.L.; Dias, D.C.; Justino, L.L.G.; Verissimo, L.M.P.; Valente, A.J.M.; Esteso, M.A.; Ribeiro, A.C.F.; Leaist, D.G.; Pina, J.; Cabral, A.; et al. Interactions between glycyl-L-phenylalanine and β-cyclodextrin from diffusion, spectroscopic and computational studies. J. Mol. Liq. 2020, 315, 113704. [Google Scholar] [CrossRef]
- Deosarkar, S.D.; Sawale, R.T.; Pinjari, R.V.; Kalyankar, T.M. Interactions of sodium salicylate and β-cyclodextrin in water: A volumetric, ultraacoustic and optical study. J. Mol. Liq. 2020, 130, 113151. [Google Scholar] [CrossRef]
- Okuda, M.; Hiramatsu, T.; Yasuda, M.; Ishigaki, M.; Ozaki, Y.; Hayashi, M.; Tominaga, K.; Chatani, E. Theoretical Modeling of Electronic Structures of Polyiodide Species Included in α-Cyclodextrin. J. Phys. Chem. B 2020, 124, 4089–4096. [Google Scholar] [CrossRef] [PubMed]
- Majhi, K.; Bandyopadhyay, P.; Khatun, R.; Sinha, S. Prediction of the most preferable rotamer of meta-aminophenol in β-cyclodextrin cavity in aqueous medium by using spectroscopic and DFT computational studies. J. Incl. Phenom. Macrocycl. Chem. 2020, 97, 77–86. [Google Scholar] [CrossRef]
- Ismahan, L.; Leila, N.; Fatiha, M.; Abdelkrim, G.; Mouna, C.; Nada, B.; Brahim, H. Computational study of inclusion complex of l-Glutamine/beta-Cycldextrin: Electronic and intermolecular interactions investigations. J. Mol. Struct. 2020, 1206, 127740. [Google Scholar] [CrossRef]
- Pereva, S.; Nikolova, V.; Sarafska, T.; Angelova, S.; Spassov, T.; Dudev, T. Inclusion complexes of ibuprofen and β-cyclodextrin: Supramolecular structure and stability. J. Mol. Struct. 2020, 1205, 127575. [Google Scholar] [CrossRef]
- Stoicescu, C.S.; Neacşu, A.D.; Bădiceanu, C.D.; Munteanu, G. Inclusion complexes of some thiourea derivatives in cyclodextrins. J. Incl. Phenom. Macrocycl. Chem. 2019, 96, 275–283. [Google Scholar] [CrossRef]
- Paulino, P.H.S.; Sousaab, S.M.R.; Da Silva, H.C.; De Almeida, W.B.; Ferrari, J.L.; Guimarãesa, L.; Nascimento, C.S., Jr. A theoretical investigation on the encapsulation process of mepivacaine into β-cyclodextrin. Chem. Phys. Lett. 2020, 740, 137060. [Google Scholar] [CrossRef]
- Wu, J.; Ma, H.; Bu, X.; Zhu, L.; Hao, B.; Zhao, B.; Tian, Y. SERS determination of the antihypertensive drugs prazosin and losartan by using silver nanoparticles coated with β-cyclodextrin. Mikrochim. Acta 2019, 186, 801. [Google Scholar] [CrossRef]
- Bezzina, B.; Djemil, R.; Bensouilah, N. Quantitative and qualitative analyses of intermolecular interactions in neutral/deprotonated aspirin@β-CD inclusion complexes: QTAIM and NBO analyses. Theor. Chim. Acta 2019, 138, 43. [Google Scholar] [CrossRef]
- Wójcik, J.; Ejchart, A.; Nowakowski, M. Shape adaptation of quinine in cyclodextrin cavities: NMR studies. Phys. Chem. Chem. Phys. 2019, 21, 6925–6934. [Google Scholar] [CrossRef]
- Bakirhan, N.K.; Tok, T.T.; Ozkan, S.A. The redox mechanism investigation of non-small cell lung cancer drug: Erlotinib via theoretical and experimental techniques and its host–guest detection by β-Cyclodextrin nanoparticles modified glassy carbon electrode. Sens. Actuators B Chem. 2019, 278, 172–180. [Google Scholar] [CrossRef]
- Li, L.; Zhou, Y.; Wang, Z.; Wu, C.; Li, Z.; Sun, C.; Sun, T. Theoretical studies on the mechanism of sugammadex for the reversal of aminosteroid-induced neuromuscular blockade. J. Mol. Liq. 2018, 265, 450–456. [Google Scholar] [CrossRef]
- Khavani, M.; Kalantarinezhad, R.; Izadyar, M. A joint QM/MD study on α-, β- and γ-cyclodextrins in selective complexation with cathinone. Supramol. Chem. 2017, 30, 687–696. [Google Scholar] [CrossRef]
- Eftaiha, A.F.; Qaroush, A.K.; Alsoubani, F.; Pehl, T.M.; Troll, C.; Rieger, B.; Al-Maythalony, B.A.; Assaf, K.I. A green sorbent for CO2 capture: α-cyclodextrin-based carbonate in DMSO solution. RSC Adv. 2018, 8, 37757–37764. [Google Scholar] [CrossRef] [Green Version]
- Belhocine, Y.; Bouhadiba, A.; Rahim, M.; Nouar, L.; Djilani, I.; Khatmi, D.I. Inclusion Complex Formation of β-Cyclodextrin with the Nonsteroidal Anti-inflammatory Drug Flufenamic Acid: Computational Study. Macroheterocycles 2018, 11, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Sierpe, R.; Noyong, M.; Simon, U.; Aguayo, D.; Huerta, J.; Kogan, M.J.; Yutronic, N. Construction of 6-thioguanine and 6-mercaptopurine carriers based on βcyclodextrins and gold nanoparticles. Carbohydr. Polym. 2017, 177, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Anconi, C.P.A.; Santos, T.M.R.; Souza, A.C.; Borges, W.M.S.; Sales, A.L.R. Host–guest intermolecular hydrogen bonds and stability in aqueous media: The benzaldehyde/β-CD case study. J. Incl. Phenom. Macrocycl. Chem. 2017, 89, 137–142. [Google Scholar] [CrossRef]
- Cao, B.; Du, J.; Cao, Z.; Sun, X.; Sun, H.; Fu, H. DFT study on the dissolution mechanisms of α-cyclodextrin and chitobiose in ionic liquid. Carbohydr. Polym. 2017, 169, 227–235. [Google Scholar] [CrossRef]
- Angelova, S.; Nikolova, V.; Molla, N.; Dudev, T. Factors Governing the Host–Guest Interactions between IIA/IIB Group Metal Cations and α-Cyclodextrin: A DFT/CDM Study. Inorg. Chem. 2017, 56, 1981–1987. [Google Scholar] [CrossRef]
- Oliveri, V.; Pietropaolo, A.; Sgarlata, C.; Vecchio, G. Zinc Complexes of Cyclodextrin-bearing 8-Hydroxyquinoline Ligands: A Comparative Study. Chem. Asian J. 2016, 12, 110–115. [Google Scholar] [CrossRef]
- Cheng, Y.; Wang, X.; Chang, D.; Li, W. DFT study on the effects of catalysis by β-cyclodextrin in the reaction of p-nitrophenyl acetate. J. Mol. Model. 2017, 23, 21. [Google Scholar] [CrossRef] [PubMed]
- Maia, P.P.; de Sousa, S.M.R.; De Almeida, W.B.; Guimaraes, L.; Nascimento, C.S. Computational investigation on the host–guest inclusion process of norfloxacin into β-cyclodextrin. J. Mol. Model. 2016, 22, 220. [Google Scholar] [CrossRef] [PubMed]
- Ignaczak, A.; Orszański, Ł. In Search of the Most Stable Molecular Configuration of Heptakis(2,6-O-dimethyl)-β-cyclodextrin and Its Complex with Mianserin: A Comparison of the B3LYP-GD2 and M062X-GD3 Results. J. Phys. Chem. B 2021, 125, 13077–13087. [Google Scholar] [CrossRef] [PubMed]
- Aree, T. Advancing insights on β-cyclodextrin inclusion complexes with SSRIs through lens of X-ray diffraction and DFT calculation. Int. J. Pharm. 2021, 609, 121113. [Google Scholar] [CrossRef] [PubMed]
- Aree, T. Distinctive Supramolecular Features of β-Cyclodextrin Inclusion Complexes with Antidepressants Protriptyline and Maprotiline: A Comprehensive Structural Investigation. Pharmaceuticals 2021, 14, 812. [Google Scholar] [CrossRef]
- Aree, T. Supramolecular Complexes of β-Cyclodextrin with Clomipramine and Doxepin: Effect of the Ring Substituent and Component of Drugs on Their Inclusion Topologies and Structural Flexibilities. Pharmaceuticals 2020, 13, 278. [Google Scholar] [CrossRef]
- Aree, T. β-Cyclodextrin encapsulation of nortriptyline HCl and amitriptyline HCl: Molecular insights from single-crystal X-ray diffraction and DFT calculation. Int. J. Pharm. 2020, 575, 118899. [Google Scholar] [CrossRef]
- Rayene, K.; Imane, D.; Abdelaziz, B.; Leila, N.; Fatiha, M.; Abdelkrim, G.; Bouzid, G.; Ismahan, L.; Brahim, H.; Rabah, O. Molecular modeling study of structures, Hirschfield surface, NBO, AIM, RDG, IGM and 1HNMR of thymoquinone/hydroxypropyl-β-cyclodextrin inclusion complex from QM calculations. J. Mol. Struct. 2022, 1249, 131565. [Google Scholar] [CrossRef]
- Macernis, M.; Bockuviene, A.; Gruskiene, R.; Krivorotova, T.; Sereikaite, J. Raman study for β-ring positioning in β-Carotene complexes with Cyclodextrins and Chitooligosaccharides. J. Mol. Struct. 2021, 1226, 129362. [Google Scholar] [CrossRef]
- Kerdpol, K.; Daengngern, R.; Sattayanon, C.; Namuangruk, S.; Rungrotmongkol, T.; Wolschann, P.; Kungwan, N.; Hannongbua, S. Effect of Water Microsolvation on the Excited-State Proton Transfer of 3-Hydroxyflavone Enclosed in γ-Cyclodextrin. Molecules 2021, 26, 843. [Google Scholar] [CrossRef]
- Bouchemela, H.; Madi, F.; Nouar, L. DFT investigation of host–guest interactions between α-Terpineol and β-cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2019, 95, 247–258. [Google Scholar] [CrossRef]
- Seridi, L.; Boufelfel, A. Naringenin encapsulation in β-CD and in heptakis(2,6-di-O-methyl)-β-CD:NMR, NBO and QTAIM analysis. J. Incl. Phenom. Macrocycl. Chem. 2018, 90, 287–304. [Google Scholar] [CrossRef]
- Nutho, B.; Nunthaboot, N.; Wolschann, P.; Kungwan, N.; Rungrotmongkol, T. Metadynamics supports molecular dynamics simulation-based binding affinities of eucalyptol and beta-cyclodextrin inclusion complexes. RSC Adv. 2017, 7, 50899–50911. [Google Scholar] [CrossRef] [Green Version]
- Nutho, B.; Khuntawee, W.; Rungnim, C.; Pongsawasdi, P.; Wolschann, P.; Karpfen, A.; Kungwan, N.; Rungrotmongkol, T. Binding mode and free energy prediction of fisetin/β-cyclodextrin inclusion complexes. Beilstein J. Org. Chem. 2014, 10, 2789–2799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guendouzi, O.; Guendouzi, A.; Ouici, H.B.; Brahim, H.; Boumediene, M.; Elkeurti, M. A quantum chemical study of encapsulation and stabilization of gallic acid in β-cyclodextrin as a drug delivery system. Can. J. Chem. 2020, 98, 204–214. [Google Scholar] [CrossRef]
- Yang, L.; Li, D.; Guo, B.; Wei, D. Theoretical Study on the Inclusion Interaction of β-Cyclodextrin with Gabapentin and Its Stability. J. Struct. Chem. 2019, 60, 564–574. [Google Scholar] [CrossRef]
- Asztemborska, M.; Ceborska, M.; Pietrzak, M. Complexation of tropane alkaloids by cyclodextrins. Carbohydr. Polym. 2020, 209, 74–81. [Google Scholar] [CrossRef]
- Abdel-Mottaleb, M.S.A.; Hamed, E.; Saif, M.; Hafez, H.S. Binding, and thermodynamics of β-cyclodextrin inclusion complexes with some coumarin laser dyes and coumarin-based enzyme substrates: A simulation study. J. Incl. Phenom. Macrocycl. Chem. 2018, 92, 319–327. [Google Scholar] [CrossRef]
- Diamantis, D.A.; Ramesova, S.; Chatzigiannis, C.M.; Degano, I.; Gerogianni, P.S.; Karadima, K.E.; Perikleous, S.; Rekkas, D.; Gerothanassis, I.P.; Galaris, D.; et al. Exploring the oxidation and iron binding profile of a cyclodextrin encapsulated quercetin complex unveiled a controlled complex dissociation through a chemical stimulus. Biochim. Biophys. Acta (BBA) Gen. Subj. 2018, 1862, 1913–1924. [Google Scholar] [CrossRef]
- Guendouzi, A.; Mekelleche, S.M.; Brahim, H.; Litim, K. Quantitative conformational stability host-guest complex of Carvacrol and Thymol with β-cyclodextrin: A theoretical investigation. J. Incl. Phenom. Macrocycl. Chem. 2017, 89, 143–155. [Google Scholar] [CrossRef]
- Abdelaali, M.; Fatiha, M.; Leila, N.; Nora, M.; Mouna, C.; Sakina, H.; Eddine, K.D. Computational approach in the study of the inclusion processes of Thymol with β-cyclodextrin. J. Mol. Liq. 2017, 242, 714–721. [Google Scholar] [CrossRef]
- Aree, T. β-Cyclodextrin Inclusion Complexes with Catechol-Containing Antioxidants Protocatechuic Aldehyde and Protocatechuic Acid—An Atomistic Perspective on Structural and Thermodynamic Stabilities. Molecules 2021, 26, 3574. [Google Scholar] [CrossRef] [PubMed]
- Aree, T.; Jongrungruangchok, S. Structure–antioxidant activity relationship of β-cyclodextrin inclusion complexes with olive tyrosol, hydroxytyrosol and oleuropein: Deep insights from X-ray analysis, DFT calculation and DPPH assay. Carbohydr. Polym. 2018, 199, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Zhuang, S.; Liu, W.; Lin, L.; Sun, L. Computational investigation on the chiral differentiation of D- and L-penicillamine by β-cyclodextrin. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 248, 119277. [Google Scholar] [CrossRef]
- Yang, S.; Wu, F.; Yu, F.; Gu, L.; Wang, H.; Liu, Y.; Chu, Y.; Wang, F.; Fang, X.; Ding, C.-F. Distinction of chiral penicillamine using metal-ion coupled cyclodextrin complex as chiral selector by trapped ion mobility-mass spectrometry and a structure investigation of the complexes. Anal. Chim. Acta 2021, 1184, 339017. [Google Scholar] [CrossRef]
- Știufiuc, G.F.; Toma, V.; Onaciu, A.; Chiș, V.; Lucaciu, C.M.; Știufiuc, R.I. Proving Nanoscale Chiral Interactions of Cyclodextrins and Propranolol Enantiomers by Means of SERS Measurements Performed on a Solid Plasmonic Substrate. Pharmaceutics 2021, 13, 1594. [Google Scholar] [CrossRef]
- Lee, S.-S.; Lee, J.-U.; Oh, J.H.; Park, S.; Hong, Y.; Min, B.K.; Lee, H.H.L.; Kim, H.I.; Kong, X.; Lee, S.; et al. Chiral differentiation of d- and l-isoleucine using permethylated β-cyclodextrin: Infrared multiple photon dissociation spectroscopy, ion-mobility mass spectrometry, and DFT calculations. Phys. Chem. Chem. Phys. 2018, 20, 30428–30436. [Google Scholar] [CrossRef]
- Lee, S.; Park, S.; Hong, Y.; Lee, J.U.; Kim, J.H.; Yoon, D.; Kong, X.; Lee, S.; Bin Oh, H. Chiral differentiation of d- and l-alanine by permethylated β-cyclodextrin: IRMPD spectroscopy and DFT methods. Phys. Chem. Chem. Phys. 2017, 19, 14729–14737. [Google Scholar] [CrossRef]
- Reyes-Reyes, M.L.; Roa-Morales, G.; Melgar-Fernández, R.; Reyes-Pérez, H.; Gómez-Oliván, L.M.; Gonzalez-Rivas, N.; Bautista-Renedo, J.; Balderas-Hernández, P. Chiral recognition of abacavir enantiomers by (2-hydroxy)propyl-β-cyclodextrin: UHPLC, NMR and DFT studies. J. Incl. Phenom. Macrocycl. Chem. 2015, 82, 373–382. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazurek, A.H.; Szeleszczuk, Ł. Current Status of Quantum Chemical Studies of Cyclodextrin Host–Guest Complexes. Molecules 2022, 27, 3874. https://doi.org/10.3390/molecules27123874
Mazurek AH, Szeleszczuk Ł. Current Status of Quantum Chemical Studies of Cyclodextrin Host–Guest Complexes. Molecules. 2022; 27(12):3874. https://doi.org/10.3390/molecules27123874
Chicago/Turabian StyleMazurek, Anna Helena, and Łukasz Szeleszczuk. 2022. "Current Status of Quantum Chemical Studies of Cyclodextrin Host–Guest Complexes" Molecules 27, no. 12: 3874. https://doi.org/10.3390/molecules27123874