
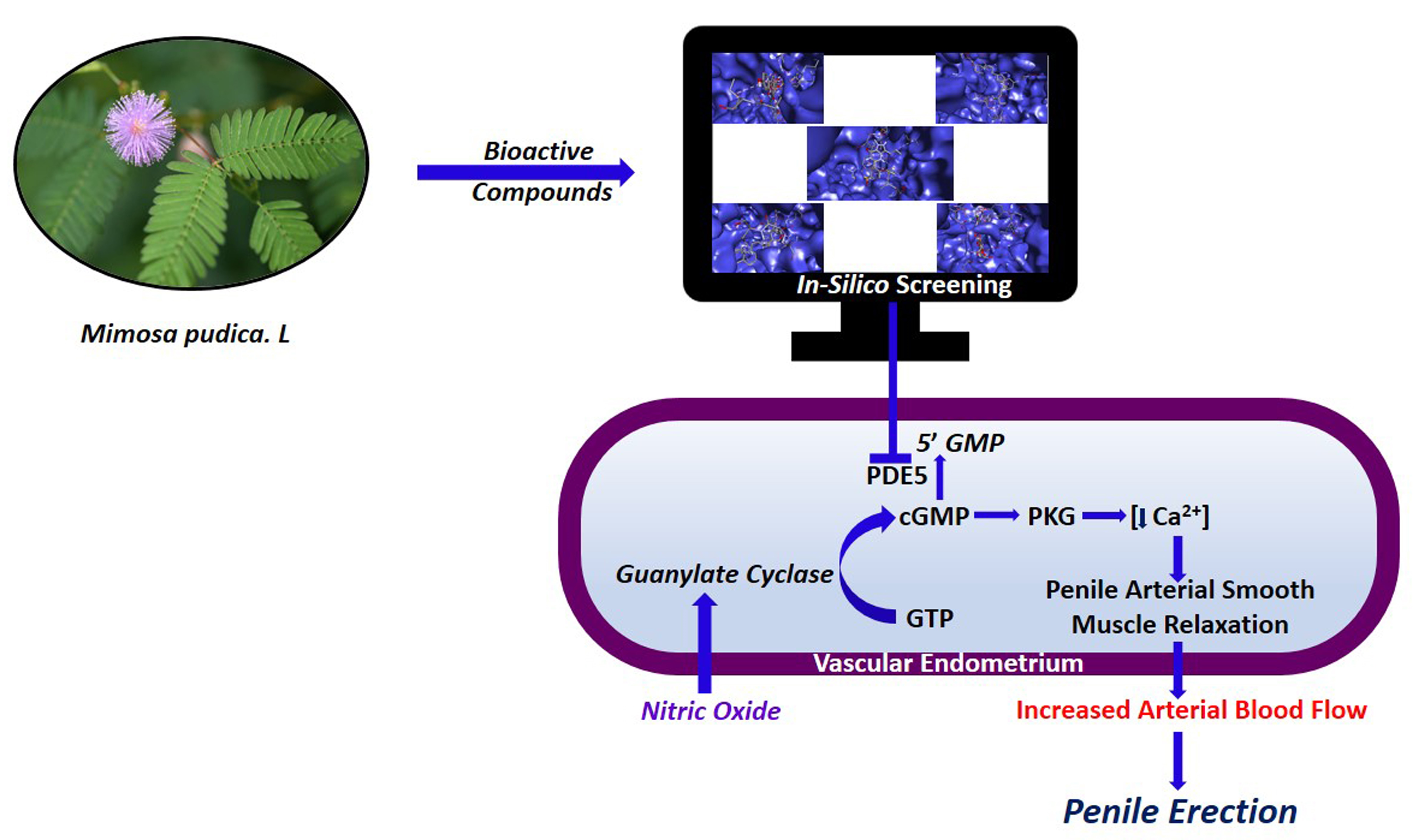
Aphrodisiac Performance of Bioactive Compounds from Mimosa pudica Linn.: In Silico Molecular Docking and Dynamics Simulation Approach


 , , , and
, , , and
Abstract
:
1. Introduction
2. Experimental Section
2.1. Bioactive Compounds Identification
2.2. Graph Theoretical Network Investigation
2.3. Protein Preparation
2.4. Active Binding Site Prediction
2.5. Molecular Docking
2.6. Pharmacokinetics and Physicochemical Properties Prediction
2.7. Molecular Dynamics (MD) Simulation Studies
2.8. Molecular Mechanics Poisson–Boltzmann Surface Area (MM/PBSA) Calculation
2.9. Density Functional Theory (DFT)
3. Result
3.1. Selection of Bioactive Compounds and Preparation
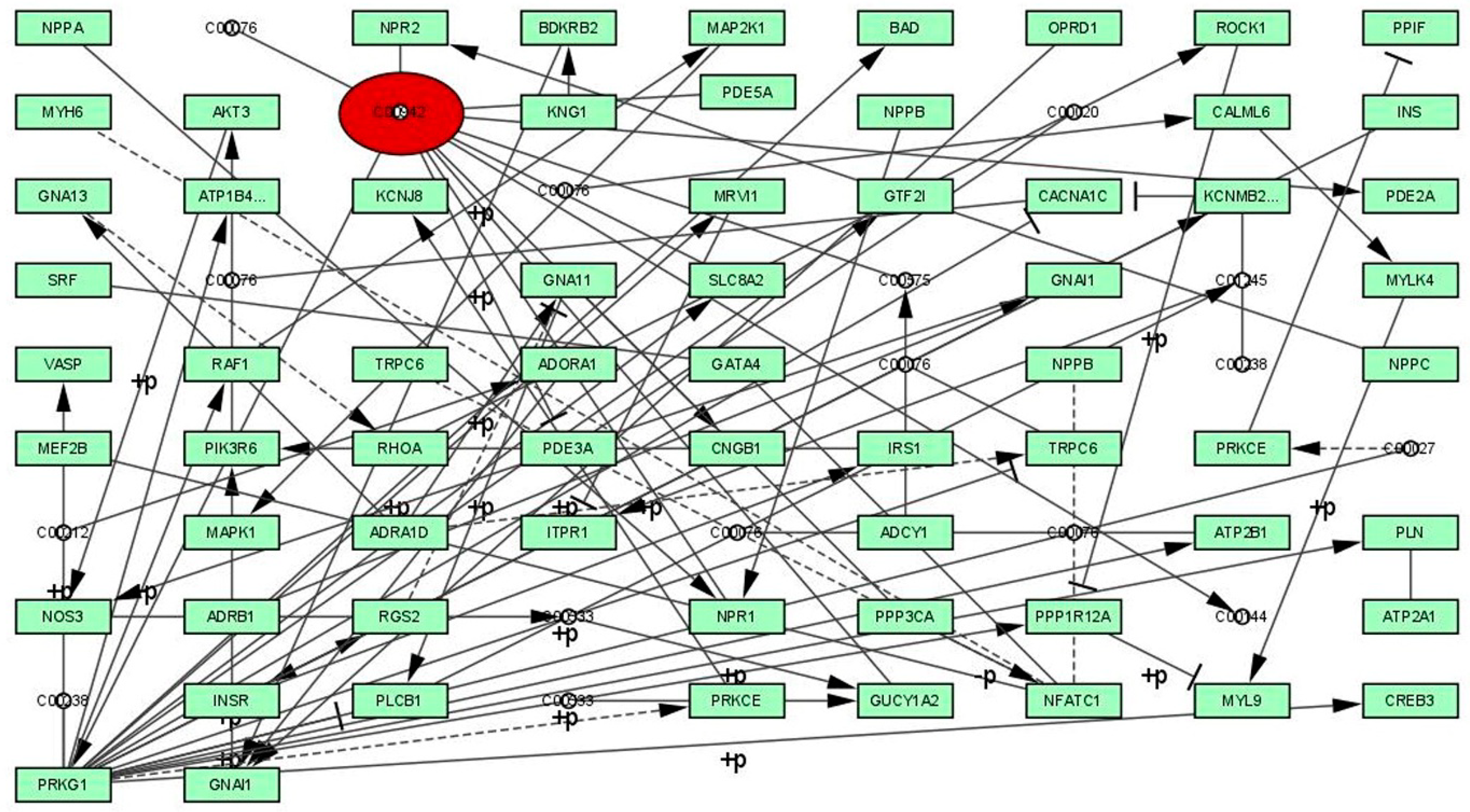
3.2. Graph Theoretical Network Analysis
3.3. Active Binding Site Identification
3.4. Molecular Docking
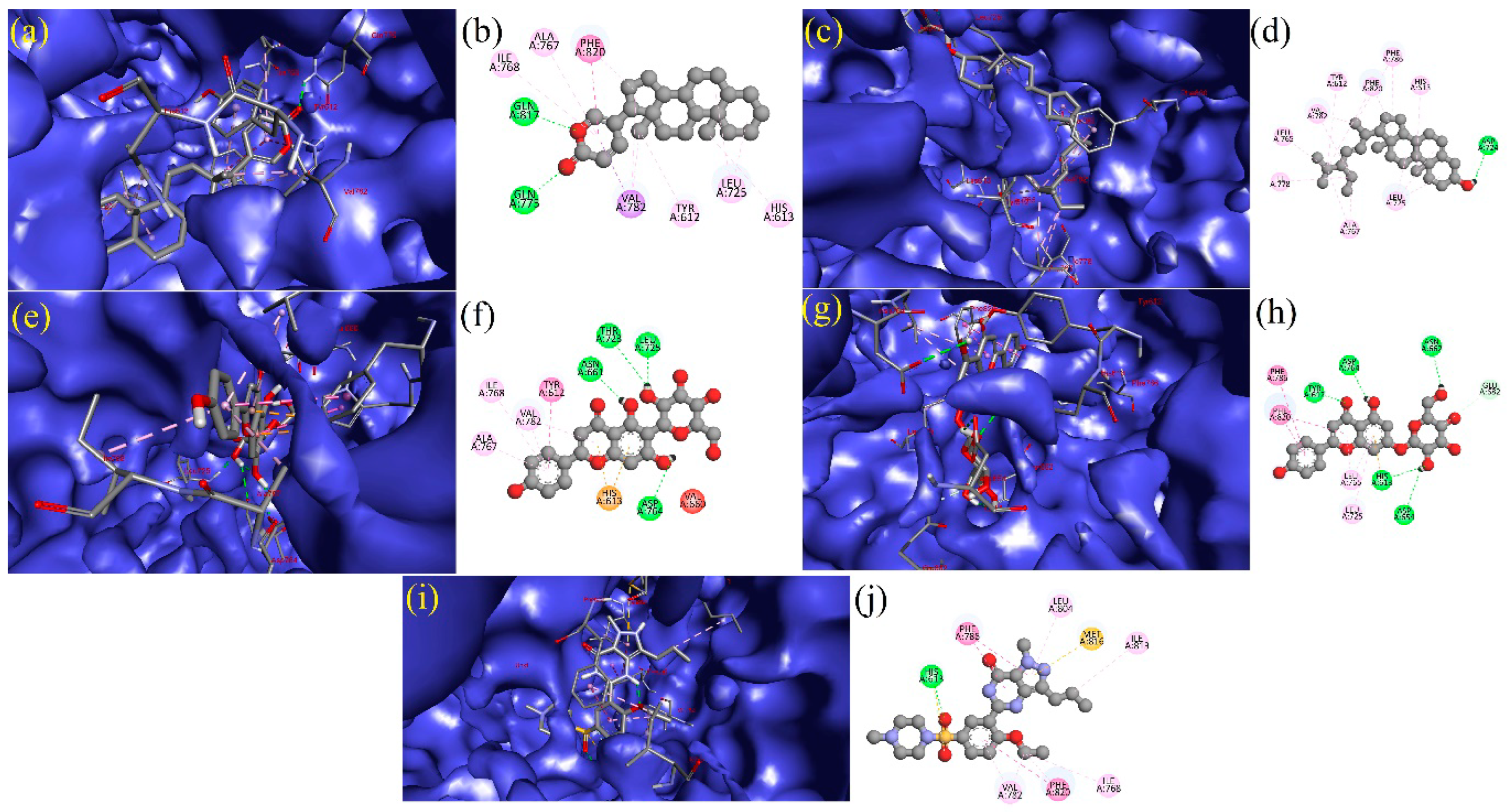
3.5. Interpretation of Receptor–Ligand Interactions
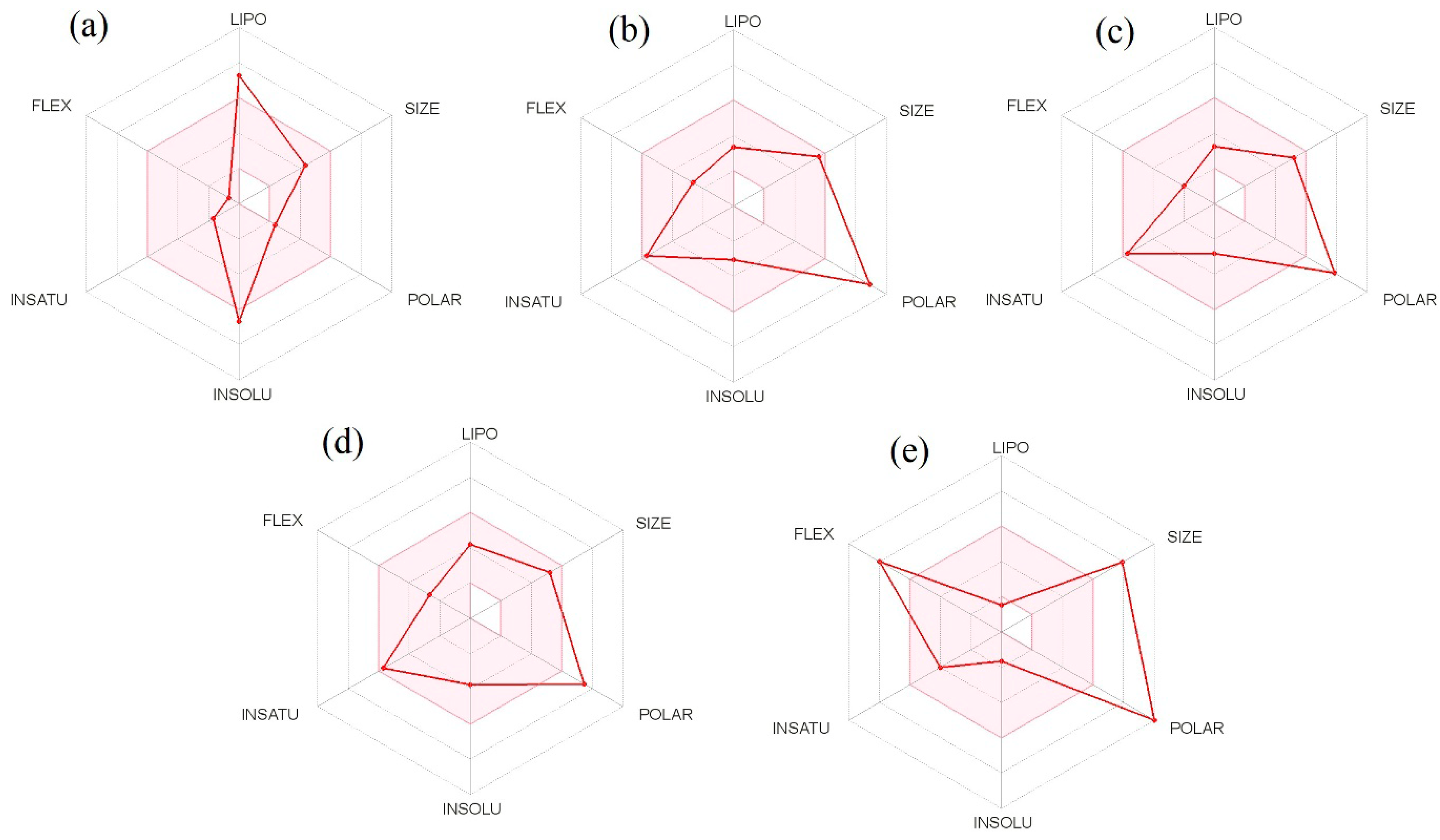
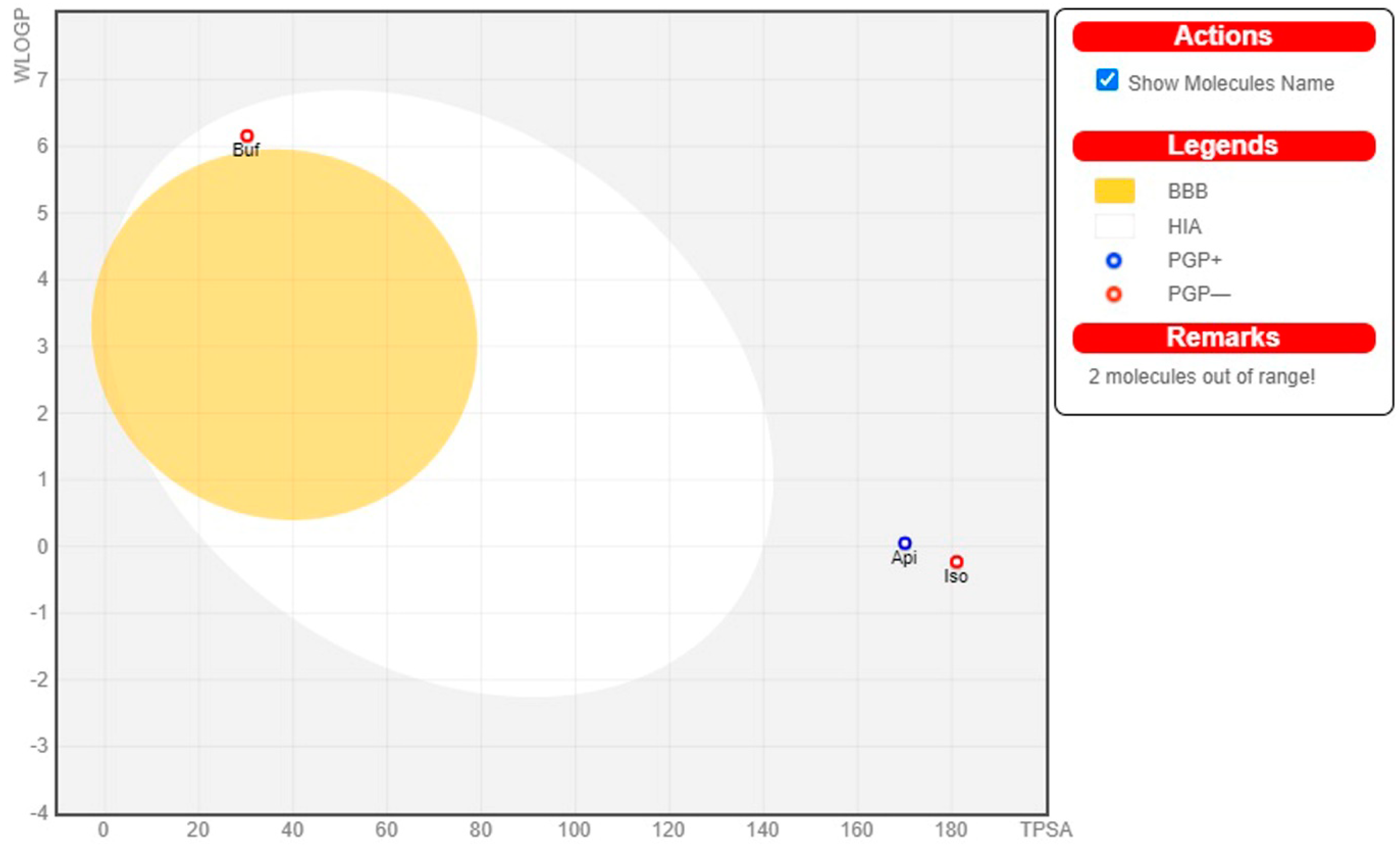
3.6. Pharmacokinetics and Physicochemical Properties of Bioactive Compounds
3.7. Analysis of Toxicity
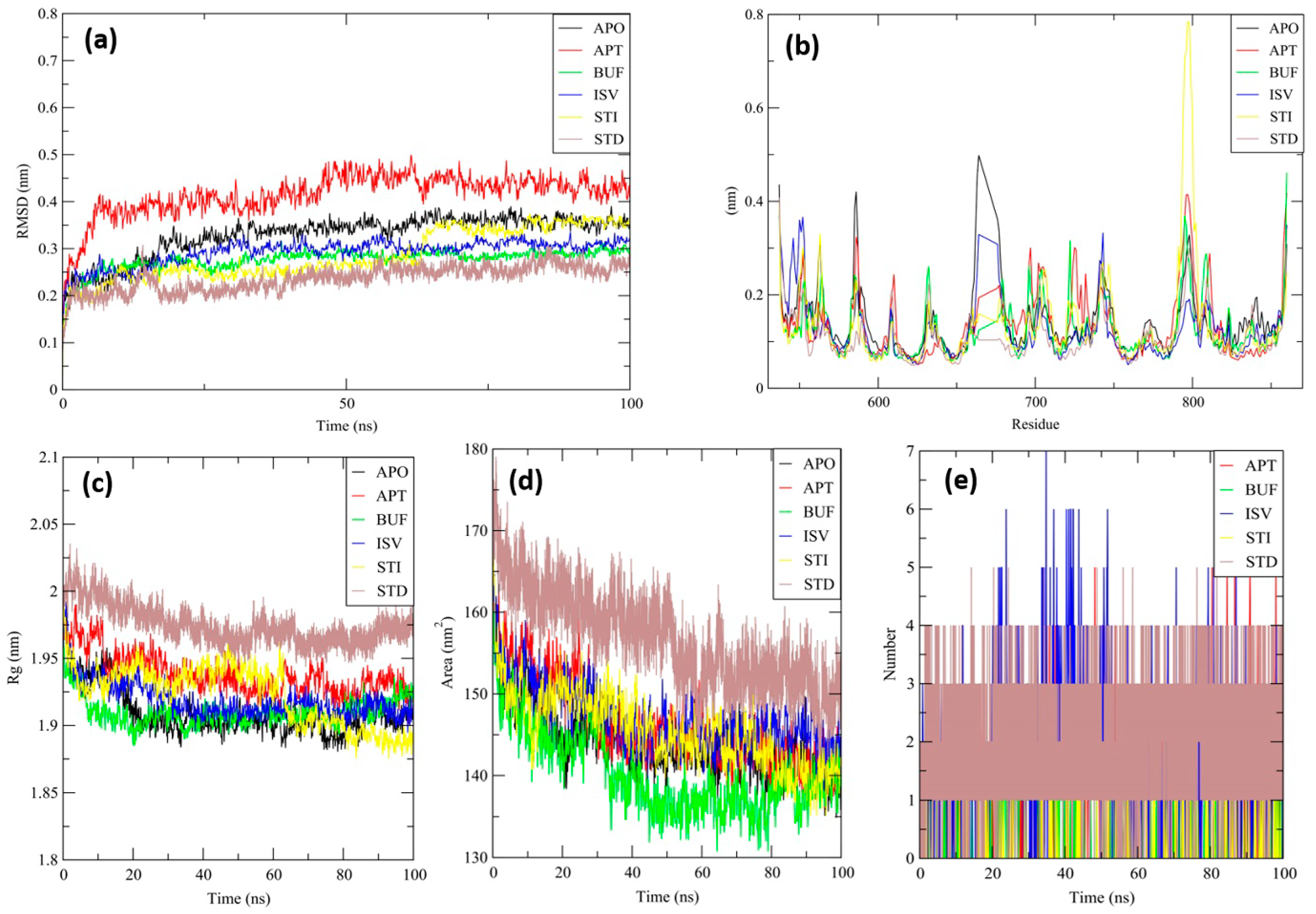
3.8. Molecular Dynamics (MD) Simulation
3.9. MM/PBSA—Binding Free Energy Analysis
3.10. Density Functional Theory
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Gott, M.; Hinchliff, S. How important is sex in later life? The views of older people. Soc. Sci. Med. 2003, 56, 1617–1628. [Google Scholar] [CrossRef]
- Lawrance, K.A.; Byers, E.S. Sexual satisfaction in long-term heterosexual relationships: The interpersonal exchange model of sexual satisfaction. Pers. Relatsh. 1995, 2, 267–285. [Google Scholar] [CrossRef]
- Monga, M.; Alexandrescu, B.; Katz, S.E.; Stein, M.; Ganiats, T. Impact of infertility on quality of life, marital adjustment, and sexual function. Urology 2004, 63, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Kubin, M.; Wagner, G.; Fugl-Meyer, A.R. Epidemiology of erectile dysfunction. Int. J. Impot. Res. 2003, 15, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, R.; Cummings, M. Erectile dysfunction: A weighty issue? Pract. Diabetes 2012, 29, 32–35. [Google Scholar] [CrossRef]
- Araujo, A.B.; Durante, R.; Feldman, H.A.; Goldstein, I.; McKinlay, J.B. The relationship between depressive symptoms and male erectile dysfunction: Cross-sectional results from the Massachusetts Male Aging Study. Psychosom. Med. 1998, 60, 458–465. [Google Scholar] [CrossRef]
- Yafi, F.A.; Jenkins, L.; Albersen, M.; Corona, G.; Isidori, A.M.; Goldfarb, S.; Maggi, M.; Nelson, C.J.; Parish, S.; Salonia, A. Erectile dysfunction. Nat. Rev. Dis. Primers 2016, 2, 16003. [Google Scholar] [CrossRef]
- Agarwal, A.; Nandipati, K.C.; Sharma, R.K.; Zippe, C.D.; Raina, R. Role of oxidative stress in pathophysiology of erectile dysfunction. J. Androl. 2005, 27, 335–347. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, C.; Ko, E.Y. Oxidative stress in the pathophysiology of male infertility. Andrologia 2021, 53, e13581. [Google Scholar] [CrossRef]
- Sikka, S.C. Relative impact of oxidative stress on male reproductive function. Curr. Med. Chem. 2001, 8, 851–862. [Google Scholar] [CrossRef]
- Jin, L.; Lagoda, G.; Leite, R.; Webb, R.C.; Burnett, A.L. NADPH oxidase activation: A mechanism of hypertension-associated erectile dysfunction. J. Sex. Med. 2008, 5, 544–551. [Google Scholar] [CrossRef] [PubMed]
- SIKKA, S.C.; Rajasekaran, M.; Hellstrom, W.J. Role of oxidative stress and antioxidants in male infertility. J. Androl. 1995, 16, 464–468. [Google Scholar] [PubMed]
- Kunwar, A.; Priyadarsini, K. Free radicals, oxidative stress and importance of antioxidants in human health. J. Med. Allied Sci. 2011, 1, 53–60. [Google Scholar]
- Agarwal, A.; Makker, K.; Sharma, R. Clinical relevance of oxidative stress in male factor infertility: An update. Am. J. Reprod. Immunol. 2008, 59, 2–11. [Google Scholar] [CrossRef]
- Burnett, A.L. Treatment strategies for erectile dysfunction. Ther. Strateg. 2007, 17, 247–260. [Google Scholar]
- Kalsi, J.; Muneer, A. Erectile dysfunction—An update of current practice and future strategies. J. Clin. Urol. 2013, 6, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Sehgal, V.N. Erectile Dysfunction/Impotence: An update of Drugs, Drug Delivery/Management, and Approaches or strategies. SKINmed 2019, 17, 256–259. [Google Scholar]
- Hellstrom, W.J.; Montague, D.K.; Moncada, I.; Carson, C.; Minhas, S.; Faria, G.; Krishnamurti, S. Implants, mechanical devices, and vascular surgery for erectile dysfunction. J. Sex. Med. 2010, 7, 501–523. [Google Scholar] [CrossRef]
- Kim, S.; Cho, M.C.; Cho, S.Y.; Chung, H.; Rajasekaran, M.R. Novel emerging therapies for erectile dysfunction. World J. Men’s Health 2021, 39, 48. [Google Scholar] [CrossRef] [Green Version]
- Citi, V.; Martelli, A.; Gorica, E.; Brogi, S.; Testai, L.; Calderone, V. Role of hydrogen sulfide in endothelial dysfunction: Pathophysiology and therapeutic approaches. J. Adv. Res. 2021, 27, 99–113. [Google Scholar] [CrossRef]
- Rahmatullah, M.; Ferdausi, D.; Mollik, M.A.H.; Azam, M.N.K.; Rahman, M.; Jahan, R. Ethnomedicinal survey of Bheramara area in Kushtia district, Bangladesh. Am. Eurasian J. Sustain. Agric. 2009, 3, 534–541. [Google Scholar]
- Singh, R.; Ali, A.; Jeyabalan, G.; Semwal, A. Current status of Indian medicinal plants with aphrodisiac potential. J. Acute Dis. 2013, 2, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, V.; Gurumani, V.; Kunjiappan, S.; Panneerselvam, T.; Somasundaram, B.; Kannan, S.; Chowdhury, A.; Saravanan, G.; Bhattacharjee, C. Optimization and analysis of microwave-assisted extraction of bioactive compounds from Mimosa pudica L. using RSM & ANFIS modeling. J. Food Meas. Charact. 2018, 12, 228–242. [Google Scholar]
- Subashini, T.; Srinivasan, T.P.; Kumar, K.S.; Ammari, A.A.; Amran, R.A.; Alhimaidi, A.R. Mimosa pudica alleviates streptozotocin-induced diabetes, glycemic stress and glutathione depletion in Wistar Albino Rats. J. King Saud Univ.-Sci. 2022, 34, 102037. [Google Scholar]
- Sarker, S. An Isolation, Characterization and In-vitro Evaluation Study of Cholinesterase Inhibitory and Antioxidant Activities of Mimosa pudica for the Treatment of Neurodegenerative Disorders; East West University: Dhaka, Bangladesh, 2017. [Google Scholar]
- Ahmad, H.; Sehgal, S.; Mishra, A.; Gupta, R. Mimosa pudica L. (Laajvanti): An overview. Pharmacogn. Rev. 2012, 6, 115. [Google Scholar]
- Meenatchisundaram, S.; Michael, A. Preliminary studies on antivenom activity of Mimosa pudica root extracts against russell’s viper and saw scaled viper venom by in vivo and in vitro methods. Pharmacologyonline 2009, 2, 372–374. [Google Scholar]
- Molina, M.; Contreras, C.; Tellez-Alcantara, P. Mimosa pudica may possess antidepressant actions in the rat. Phytomedicine 1999, 6, 319–323. [Google Scholar] [CrossRef]
- Bum, E.N.; Dawack, D.; Schmutz, M.; Rakotonirina, A.; Rakotonirina, S.; Portet, C.; Jeker, A.; Olpe, H.-R.; Herrling, P. Anticonvulsant activity of Mimosa pudica decoction. Fitoterapia 2004, 75, 309–314. [Google Scholar]
- Pande, M.; Pathak, A. Aphrodisiac activity of roots of Mimosa pudica Linn. ethanolic extract in mice. Int. J. Pharm. Sci. Nanotechnol. 2009, 2, 477–486. [Google Scholar] [CrossRef]
- Ambikabothy, J.; Ibrahim, H.; Ambu, S.; Chakravarthi, S.; Awang, K.; Vejayan, J. Efficacy evaluations of Mimosa pudica tannin isolate (MPT) for its anti-ophidian properties. J. Ethnopharmacol. 2011, 137, 257–262. [Google Scholar] [CrossRef]
- Zhang, J.; Yuan, K.; Zhou, W.-l.; Zhou, J.; Yang, P. Studies on the active components and antioxidant activities of the extracts of Mimosa pudica Linn. from southern China. Pharmacogn. Mag. 2011, 7, 35. [Google Scholar]
- Mohan, U.P.; Sriram, B.; Panneerselvam, T.; Devaraj, S.; MubarakAli, D.; Parasuraman, P.; Palanisamy, P.; Premanand, A.; Arunachalam, S.; Kunjiappan, S. Utilization of plant-derived Myricetin molecule coupled with ultrasound for the synthesis of gold nanoparticles against breast cancer. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020, 393, 1963–1976. [Google Scholar] [CrossRef]
- Mohanraj, K.; Karthikeyan, B.S.; Vivek-Ananth, R.; Chand, R.; Aparna, S.; Mangalapandi, P.; Samal, A. IMPPAT: A curated database of Indian medicinal plants, phytochemistry and therapeutics. Sci. Rep. 2018, 8, 4329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- RCSB, P. The Research Collaboratory for Structural Bioinformatics Protein Data Bank. 2014. Available online: http://www.rcsb.org/pdb (accessed on 1 May 2022).
- Biovia, D.S. Discovery studio visualizer, vision 17.2.0; Dassault Systèmes: San Diego, CA, USA, 2017.
- Kalimuthu, A.K.; Panneerselvam, T.; Pavadai, P.; Pandian, S.R.K.; Sundar, K.; Murugesan, S.; Ammunje, D.N.; Kumar, S.; Arunachalam, S.; Kunjiappan, S. Pharmacoinformatics-based investigation of bioactive compounds of Rasam (South Indian recipe) against human cancer. Sci. Rep. 2021, 11, 21488. [Google Scholar] [CrossRef]
- Rosy, J.C.; Ravinarayanan, H.; Gokila, P.; Navamuthumani, T.; Marimuthu, S.C.V.; Kunjiappan, S.; Sundar, K. In Silico Screening of Natural Metabolites as Inhibitors of Biosynthesis and Transport of Enterobactin. Biointerface Res. Appl. Chem. 2022, 13, 125. [Google Scholar]
- Ursu, O.; Rayan, A.; Goldblum, A.; Oprea, T.I. Understanding drug-likeness. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 760–781. [Google Scholar] [CrossRef]
- Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpí, J.L. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. AABC 2015, 8, 37. [Google Scholar]
- Hess, B.; van der Spoel, L.E.; Lindahl, E. GROMACS Groningen Machine for Chemical Simulations, User Manual Version 4.5.4; University of Groningen: Groningen, The Netherlands, 2011.
- Pandian, S.R.K.; Kunjiappan, S.; Pavadai, P.; Sundarapandian, V.; Chandramohan, V.; Sundar, K. Delivery of Ursolic Acid by Polyhydroxybutyrate Nanoparticles for Cancer Therapy: In silico and in vitro Studies. Drug Res. 2022, 72, 72–81. [Google Scholar]
- Tieleman, D.P.; Berendsen, H. Molecular dynamics simulations of a fully hydrated dipalmitoylphosphatidylcholine bilayer with different macroscopic boundary conditions and parameters. J. Chem. Phys. 1996, 105, 4871–4880. [Google Scholar] [CrossRef] [Green Version]
- Lounnas, V.; Lüdemann, S.K.; Wade, R.C. Towards molecular dynamics simulation of large proteins with a hydration shell at constant pressure. Biophys. Chem. 1999, 78, 157–182. [Google Scholar] [CrossRef]
- Rajagopal, G.; Nivetha, A.; Sundar, M.; Panneerselvam, T.; Murugesan, S.; Parasuraman, P.; Kumar, S.; Ilango, S.; Kunjiappan, S. Mixed phytochemicals mediated synthesis of copper nanoparticles for anticancer and larvicidal applications. Heliyon 2021, 7, e07360. [Google Scholar] [CrossRef] [PubMed]
- Pandian, S.R.K.; Pavadai, P.; Vellaisamy, S.; Ravishankar, V.; Palanisamy, P.; Sundar, L.M.; Chandramohan, V.; Sankaranarayanan, M.; Panneerselvam, T.; Kunjiappan, S. Formulation and evaluation of rutin-loaded solid lipid nanoparticles for the treatment of brain tumor. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2021, 394, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Theivendren, P.; Kunjiappan, S.; Govindraj, S.; Chandrasekarn, J.; Pavadai, P.; Saraswathy, G.R.; Murugan, I. Graph theoretical analysis, insilico modeling and formulation of pyrimidine nanoparticles as p38α MAP kinases inhibitors: A quantitative proteomics approach. Drug Res. 2019, 69, 100–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Lateef, H.M.A.; Khalaf, M.M.; Shehata, M.R.; Abu-Dief, A.M. Fabrication, DFT Calculation, and Molecular Docking of Two Fe (III) Imine Chelates as Anti-COVID-19 and Pharmaceutical Drug Candidate. Int. J. Mol. Sci. 2022, 23, 3994. [Google Scholar] [CrossRef] [PubMed]
- Tüzün, B.; Kaya, C. Investigation of DNA–RNA molecules for the efficiency and activity of corrosion inhibition by DFT and molecular docking. J. Bio-Tribo-Corros. 2018, 4, 69. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. Revision E.01; Gaussian: Wallinford, CT, USA, 2013. [Google Scholar]
- Ruiz-Garcia, A.; Bermejo, M.; Moss, A.; Casabo, V.G. Pharmacokinetics in drug discovery. J. Pharm. Sci. 2008, 97, 654–690. [Google Scholar] [CrossRef]
- Widodo, H. Plants used as aphrodisiacs by the Dayak ethnic groups in central Kalimantan, Indonesia. Biodiversitas: J. Biol. Divers. 2019, 20, 1859–1865. [Google Scholar]
- Zoete, V.; Grosdidier, A.; Michielin, O. Docking, virtual high throughput screening and in silico fragment-based drug design. J. Cell. Mol. Med. 2009, 13, 238–248. [Google Scholar] [CrossRef] [Green Version]
- Fujii, J.; Iuchi, Y.; Matsuki, S.; Ishii, T. Cooperative function of antioxidant and redox systems against oxidative stress in male reproductive tissues. Asian J. Androl. 2003, 5, 231–242. [Google Scholar] [PubMed]
- Wood, K.C.; Hsu, L.L.; Gladwin, M.T. Sickle cell disease vasculopathy: A state of nitric oxide resistance. Free Radic. Biol. Med. 2008, 44, 1506–1528. [Google Scholar] [CrossRef] [PubMed]
- Ostfeld, R.J.; Allen, K.E.; Aspry, K.; Brandt, E.J.; Spitz, A.; Liberman, J.; Belardo, D.; O’Keefe, J.H.; Aggarwal, M.; Miller, M. Vasculogenic Erectile Dysfunction: The Impact of Diet and Lifestyle. Am. J. Med. 2021, 134, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Masuku, N.P.; Unuofin, J.O.; Lebelo, S.L. Promising role of medicinal plants in the regulation and management of male erectile dysfunction. Biomed. Pharmacother. 2020, 130, 110555. [Google Scholar] [CrossRef] [PubMed]
- Baskararaj, S.; Theivendren, P.; Palanisamy, P.; Kannan, S.; Pavadai, P.; Arunachalam, S.; Sankaranarayanan, M.; Mohan, U.P.; Ramasamy, L.; Kunjiappan, S. Optimization of bioactive compounds extraction assisted by microwave parameters from Kappaphycus alvarezii using RSM and ANFIS modeling. J. Food Meas. Charact. 2019, 13, 2773–2789. [Google Scholar] [CrossRef]
- Hossain, S.; Sarkar, B.; Prottoy, M.N.I.; Araf, Y.; Taniya, M.A.; Ullah, M.A. Thrombolytic activity, drug likeness property and ADME/T analysis of isolated phytochemicals from ginger (Zingiber officinale) using in silico approaches. Mod. Res. Inflamm. 2019, 8, 29–43. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. [Google Scholar] [CrossRef] [Green Version]
- Ghose, A.K.; Herbertz, T.; Hudkins, R.L.; Dorsey, B.D.; Mallamo, J.P. Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. ACS Chem. Neurosci. 2012, 3, 50–68. [Google Scholar] [CrossRef] [Green Version]
- Epriliati, I.; Ginjom, I.R. Bioavailability of Phytochemicals. Phytochemicals—A Global Perspective of their Role in Nutrition and Health; Rao, V., Ed.; InTech: Rijeka, Croatia, 2012; pp. 401–428. [Google Scholar]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef]
- Truzzi, F.; Tibaldi, C.; Zhang, Y.; Dinelli, G.; D′ Amen, E. An overview on dietary polyphenols and their biopharmaceutical classification system (BCS). Int. J. Mol. Sci. 2021, 22, 5514. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, M.; Sharma, P.; Singh, R. Identification of potential CYP51 inhibiting anti-Aspergillus phytochemicals using molecular docking and ADME/T studies. Chem. Biol. Lett. 2021, 8, 18–21. [Google Scholar]
- Roy, S.; Samant, L.R.; Chowdhary, A. In silico pharmacokinetics analysis and ADMET of phytochemicals of Datura metel Linn. and Cynodon dactylon Linn. J. Chem. Pharm. Res 2015, 7, 385–388. [Google Scholar]
- Brown, A.C. An overview of herb and dietary supplement efficacy, safety and government regulations in the United States with suggested improvements. Part 1 of 5 series. Food Chem. Toxicol. 2017, 107, 449–471. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, A.; Amir, A.; Hussain, W.; Ghaffar, A.; Rasool, N. In silico computations of selective phytochemicals as potential inhibitors against major biological targets of diabetes mellitus. Curr. Comput.-Aided Drug Des. 2019, 15, 401–408. [Google Scholar] [CrossRef]
- Rizwan, K.; Majeed, I.; Bilal, M.; Rasheed, T.; Shakeel, A.; Iqbal, S. Phytochemistry and Diverse Pharmacology of Genus Mimosa: A Review. Biomolecules 2022, 12, 83. [Google Scholar] [CrossRef]
- Hafsa, A.; Sakshi, S.; Anurag, M.; Rajiv, G. Mimosa pudica L. Laajvanti): An overview. Pharm. Rev. 2012, 6, 115–124. [Google Scholar]
- Islam, A.; Kabir, M.S.H.; Dash, R.; Emran, T.B.; Uddin, M.Z.; Nesa, K.; Uddin, M.M.N.; Ahsan, M.T. Virtual screening for potential COX-inhibiting constituents from Mimosa pudica. J. Appl. Pharm. Sci. 2015, 5, 71–75. [Google Scholar] [CrossRef] [Green Version]
- Ueda, M.; Yamamura, S. Leaf-opening substance of Mimosa pudica L.; chemical studies on the other leaf movement of mimosa. Tetrahedron Lett. 1999, 40, 353–356. [Google Scholar] [CrossRef]
- Bourbouloux, A.; Fleurat-Lessard, P.; Roblin, G. Effects of jasmonic acid on motor cell physiology in Mimosa pudica L. and Cassia fasciculata Michx. Plant Cell Physiol. 1994, 35, 389–396. [Google Scholar]
- Muhammad, G.; Hussain, M.A.; Jantan, I.; Bukhari, S.N.A. Mimosa pudica L., a high-value medicinal plant as a source of bioactives for pharmaceuticals. Compr. Rev. Food Sci. Food Saf. 2016, 15, 303–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasnuva, S.; Qamar, U.; Ghafoor, K.; Sahena, F.; Jahurul, M.; Rukshana, A.; Juliana, M.; Al-Juhaimi, F.Y.; Jalifah, L.; Jalal, K. α-glucosidase inhibitors isolated from Mimosa pudica L. Nat. Prod. Res. 2019, 33, 1495–1499. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Betweenness | Closeness | Degree | Eccentricity | Eigen Vector | Radiality | Stress |
|---|---|---|---|---|---|---|---|
| PRKG1 | 4355.8 | 0.004524887 | 20 | 0.111111111 | 0.664578142 | 10.96341 | 6142 |
| C00942 | 3193.75 | 0.004166667 | 10 | 0.125 | 0.241942799 | 10.73171 | 4126 |
| NFATC1 | 805.6666667 | 0.002304147 | 6 | 0.076923077 | 0.005206962 | 8.365854 | 1684 |
| GNA11 | 1051.166667 | 0.003194888 | 4 | 0.090909091 | 0.084715899 | 9.841463 | 2202 |
| GNAI1 | 718.5 | 0.001883239 | 4 | 0.076923077 | 0.0000377 | 7.378049 | 856 |
| PPP1R12A | 752.0666667 | 0.003571429 | 3 | 0.1 | 0.1554146 | 10.2439 | 964 |
| NPR1 | 247.75 | 0.003267974 | 3 | 0.111111111 | 0.056256814 | 9.926829 | 350 |
| GUCY1A2 | 1814 | 0.003663004 | 3 | 0.125 | 0.056401272 | 10.32927 | 2366 |
| KCNMB2 | 110 | 0.003448276 | 3 | 0.1 | 0.189210899 | 10.12195 | 176 |
| ADRA1D | 185.8333333 | 0.003144654 | 3 | 0.090909091 | 0.054578549 | 9.780488 | 228 |
| PLCB1 | 673 | 0.003717472 | 3 | 0.1 | 0.16797582 | 10.37805 | 1314 |
| NOS3 | 1596.5 | 0.002777778 | 3 | 0.1 | 0.002934907 | 9.268293 | 2020 |
| PIK3R6 | 1192.5 | 0.002150538 | 3 | 0.083333333 | 0.000154 | 7.987805 | 1476 |
| TRPC6 | 311.1 | 0.003597122 | 3 | 0.1 | 0.159374597 | 10.46341 | 400 |
| CACNA1C | 86.2 | 0.003436426 | 3 | 0.1 | 0.189210899 | 10.10976 | 176 |
| MYL9 | 373.4 | 0.002915452 | 2 | 0.090909091 | 0.034521634 | 9.47561 | 516 |
| ITPR1 | 2 | 0.002873563 | 2 | 0.090909091 | 0.040978905 | 9.414634 | 2 |
| NPR2 | 148 | 0.003367003 | 2 | 0.111111111 | 0.053617712 | 10.03659 | 176 |
| MRVI1 | 125 | 0.003472222 | 2 | 0.1 | 0.149354275 | 10.14634 | 136 |
| RGS2 | 510 | 0.003703704 | 2 | 0.1 | 0.158612643 | 10.36585 | 1134 |
| RHOA | 19.03333333 | 0.002557545 | 2 | 0.083333333 | 0.010336014 | 8.890244 | 32 |
| ROCK1 | 130.5 | 0.002915452 | 2 | 0.090909091 | 0.03508655 | 9.47561 | 142 |
| C01245 | 21 | 0.00295858 | 2 | 0.090909091 | 0.044232116 | 9.536585 | 24 |
| PPP3CA | 909.8333333 | 0.002688172 | 2 | 0.083333333 | 0.019035121 | 9.121951 | 1918 |
| GATA4 | 148 | 0.002012072 | 2 | 0.071428571 | 0.001153931 | 7.597561 | 286 |
| PLN | 148 | 0.003484321 | 2 | 0.1 | 0.147279271 | 10.15854 | 176 |
| RAF1 | 292 | 0.003558719 | 2 | 0.1 | 0.147604105 | 10.23171 | 348 |
| MAP2K1 | 148 | 0.002832861 | 2 | 0.090909091 | 0.032711014 | 9.353659 | 176 |
| SLC8A2 | 86.2 | 0.003460208 | 2 | 0.1 | 0.147279271 | 10.13415 | 176 |
| ATP2B1 | 86.2 | 0.003436426 | 2 | 0.1 | 0.147279271 | 10.10976 | 176 |
| AKT3 | 1271 | 0.002415459 | 2 | 0.090909091 | 0.000654 | 8.609756 | 1580 |
| C00533 | 1663 | 0.003095975 | 2 | 0.111111111 | 0.012560447 | 9.914634 | 2100 |
| C00575 | 148 | 0.003389831 | 2 | 0.111111111 | 0.053617712 | 10.06098 | 176 |
| PDE3A | 148 | 0.003355705 | 2 | 0.111111111 | 0.053617712 | 10.02439 | 176 |
| PRKCE | 148.5 | 0.002793296 | 2 | 0.090909091 | 0.032711014 | 9.487805 | 176 |
| PRKCE | 148.5 | 0.003367003 | 2 | 0.1 | 0.147279271 | 10.23171 | 176 |
| MYLK4 | 231.8 | 0.002439024 | 2 | 0.083333333 | 0.007667322 | 8.658537 | 348 |
| CALML6 | 86.2 | 0.002079002 | 2 | 0.076923077 | 0.00169918 | 7.792683 | 176 |
| IRS1 | 292 | 0.001872659 | 2 | 0.076923077 | 0.0000341 | 7.146341 | 348 |
| INSR | 148 | 0.001666667 | 2 | 0.071428571 | 0.00000756 | 6.341463 | 176 |
| GNAI1 | 148.5 | 0.002309469 | 2 | 0.090909091 | 0.00065 | 8.573171 | 176 |
| BDKRB2 | 148 | 0.001683502 | 2 | 0.071428571 | 0.00000837 | 6.414634 | 176 |
| ADORA1 | 148 | 0.001666667 | 2 | 0.071428571 | 0.00000837 | 6.341463 | 176 |
| GNA13 | 32.53333333 | 0.002617801 | 2 | 0.083333333 | 0.013741295 | 9 | 50 |
| C00027 | 255 | 0.003472222 | 2 | 0.1 | 0.147604105 | 10.14634 | 348 |
| S. No | Compound ID | Bioactive Molecule | Binding Affinity |
|---|---|---|---|
| 1. | 94477 | Mimosinamine | −5.50 |
| 2. | 125409 | Beta-D-xylopyranose | −5.70 |
| 3. | 190359 | Mimosinic Acid | −6.00 |
| 4. | 370 | Gallic Acid | −6.20 |
| 5. | 951 | Dl- Norepinephrine | −6.30 |
| 6. | 164619 | D-Pinitol | −6.40 |
| 7. | 3862 | Mimosine | −6.60 |
| 8. | 1153 | DL-tyrosine | −6.60 |
| 9. | 71684438 | Octadecadienoicacid | −6.80 |
| 10. | 94715 | D-Glucopyranuronate | −6.80 |
| 11. | 5280441 | Vitexin | −7.00 |
| 12. | 440473 | L-Mimosine | −7.10 |
| 13. | 5281166 | Jasmonic Acid | −7.10 |
| 14. | 5375199 | Abscisic acid | −7.30 |
| 15. | 100927206 | Mimopudine | −7.80 |
| 16. | 5280489 | Beta-Carotene | −8.60 |
| 17. | 5281679 | Methylquercetin | −9.30 |
| 18. | 64971 | Betulinic Acid | −9.70 |
| 19. | 222284 | Beta-sitosterol | −10.30 |
| 20. | 5490064 | Avicularin | −10.50 |
| 21. | 5281675 | Orientin | −10.60 |
| 22. | 114776 | Isoorientin | −10.70 |
| 23. | 70698280 | Cassiaoccidentalin B | −10.80 |
| 24. | 5280804 | Isoquercitrin | −10.90 |
| 25. | 162350 | Isovitexin | −11.20 |
| 26. | 5280704 | Apigetrin | −11.20 |
| 27. | 5280794 | Stigmasterol | −11.40 |
| 28. | 46173848 | Bufadienolide | −12.30 |
| Standard drug | |||
| 29. | 135398744 | Sildenafil | −9.8 |
| Parameters | Bufadienolide (CID:46173848) | Stigmasterol (CID:5280794) | Apigetrin (CID:5280704) | Isovitexin (CID:162350) | Sildenafil (CID: 135398744) |
|---|---|---|---|---|---|
| Formula | C24H34O2 | C21H20O12 | C21H20O10 | C21H20O10 | C22H30N6O4S |
| MW (g mol−1) | 354.53 | 464.38 | 432.38 | 432.38 | 474.58 |
| Num. heavy atoms | 26 | 33 | 31 | 31 | 33 |
| Num. arom. heavy atoms | 6 | 16 | 16 | 16 | 15 |
| Fraction Csp3 | 0.79 | 0.29 | 0.29 | 0.29 | 0.50 |
| Num. rotatable bonds | 1 | 4 | 4 | 3 | 7 |
| Num. H-bond acceptors | 2 | 12 | 10 | 10 | 8 |
| Num. H-bond donors | 0 | 8 | 6 | 7 | 1 |
| Molar Refractivity | 107.49 | 110.6 | 106.11 | 106.61 | 134.56 |
| TPSA (Å2) | 30.21 | 210.51 | 170.5 | 181.5 | 121.80 |
| Solubility class | Poorly Soluble | Soluble | Soluble | Soluble | Soluble |
| GI absorption | High | Low | Low | Low | High |
| BBB permeation | NO | NO | NO | NO | NO |
| Violation of Lipinski’s rule of five | 1 | 2 | 1 | 1 | 1 |
| Violation of Veber rule | 0 | 0 | 0 | 0 | 0 |
| Bioavailability Score | 0.55 | 0.17 | 0.55 | 0.55 | 0.55 |
| Synthetic accessibility | 5.60 | 5.32 | 5.12 | 4.99 | 3.95 |
| Compound | AMES Toxicity | Max. Tolerated Dose (Human) | hERG Inhibition | LD50 | Hepatotoxicity | Carcinogenicity | Cytotoxicity | Skin Sensitization | T. pyriformis Toxicity | Minnow Toxicity |
|---|---|---|---|---|---|---|---|---|---|---|
| Bufadienlide | NO | −0.087 | No | 2.638 | No | No | No | NO | 0.417 | −1.896 |
| Stigmasterol | NO | 0.508 | NO | 2.624 | NO | No | No | NO | 0.285 | 2.706 |
| Apigetrin | NO | 0.698 | NO | 2.442 | NO | No | No | NO | 0.285 | 1.131 |
| Isovitexin | YES | 1.036 | NO | 3.034 | NO | No | No | NO | 0.285 | 2.803 |
| Sildenafil | NO | 0.209 | NO | 2.655 | YES | No | No | NO | 0.286 | 0.889 |
| S. No | Protein–ligand Complex | Average Backbone RMSD (nm) | Average Backbone RMSF (nm) |
|---|---|---|---|
| 1 | APO | 0.328935 ± 0.044482 | 0.140706 ± 0.068559 |
| 2 | API | 0.415632 ± 0.044550 | 0.135675 ± 0.072969 |
| 3 | BUF | 0.276698 ± 0.022447 | 0.126093 ± 0.060376 |
| 4 | ISV | 0.292700 ± 0.026475 | 0.121282 ± 0.060832 |
| 5 | STI | 0.285493 ± 0.052889 | 0.134828 ± 0.102423 |
| 6 | STD | 0.236710 ± 0.027702 | 0.111075 ± 0.052376 |
| S. No | Protein–ligand Complex | ΔGbind (kJ mol−1) |
|---|---|---|
| 1 | API | −212.407 ± 17.541 |
| 2 | BUF | −233.376 ± 14.471 |
| 3 | ISV | −211.953 ± 10.191 |
| 4 | STI | −210.678 ± 12.676 |
| 5 | STD | −164.117 ± 16.451 |
| Compound | HOMO | LUMO | Energy Gap | ||
|---|---|---|---|---|---|
| BUF | −9.27337 |  | −5.57126 |  | 3.702111 |
| STI | −9.84236 |  | −4.7873 |  | 5.055062 |
| API | −8.94139 |  | −5.55303 |  | 3.388364 |
| ISO | −8.93432 |  | −5.55249 |  | 3.381833 |
| STD | −8.35254 |  | −5.51112 |  | 2.841414 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palanichamy, C.; Pavadai, P.; Panneerselvam, T.; Arunachalam, S.; Babkiewicz, E.; Ram Kumar Pandian, S.; Shanmugampillai Jeyarajaguru, K.; Nayak Ammunje, D.; Kannan, S.; Chandrasekaran, J.; et al. Aphrodisiac Performance of Bioactive Compounds from Mimosa pudica Linn.: In Silico Molecular Docking and Dynamics Simulation Approach. Molecules 2022, 27, 3799. https://doi.org/10.3390/molecules27123799
Palanichamy C, Pavadai P, Panneerselvam T, Arunachalam S, Babkiewicz E, Ram Kumar Pandian S, Shanmugampillai Jeyarajaguru K, Nayak Ammunje D, Kannan S, Chandrasekaran J, et al. Aphrodisiac Performance of Bioactive Compounds from Mimosa pudica Linn.: In Silico Molecular Docking and Dynamics Simulation Approach. Molecules. 2022; 27(12):3799. https://doi.org/10.3390/molecules27123799
Chicago/Turabian StylePalanichamy, Chandrasekar, Parasuraman Pavadai, Theivendren Panneerselvam, Sankarganesh Arunachalam, Ewa Babkiewicz, Sureshbabu Ram Kumar Pandian, Kabilan Shanmugampillai Jeyarajaguru, Damodar Nayak Ammunje, Suthendran Kannan, Jaikanth Chandrasekaran, and et al. 2022. "Aphrodisiac Performance of Bioactive Compounds from Mimosa pudica Linn.: In Silico Molecular Docking and Dynamics Simulation Approach" Molecules 27, no. 12: 3799. https://doi.org/10.3390/molecules27123799