
Unlocking the Secrets of Cancer Stem Cells with γ-Secretase Inhibitors: A Novel Anticancer Strategy

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. The Role of the Notch Signaling Pathway in Cancer
3. The Role of GSIs in Notch Signaling Pathway Inhibition
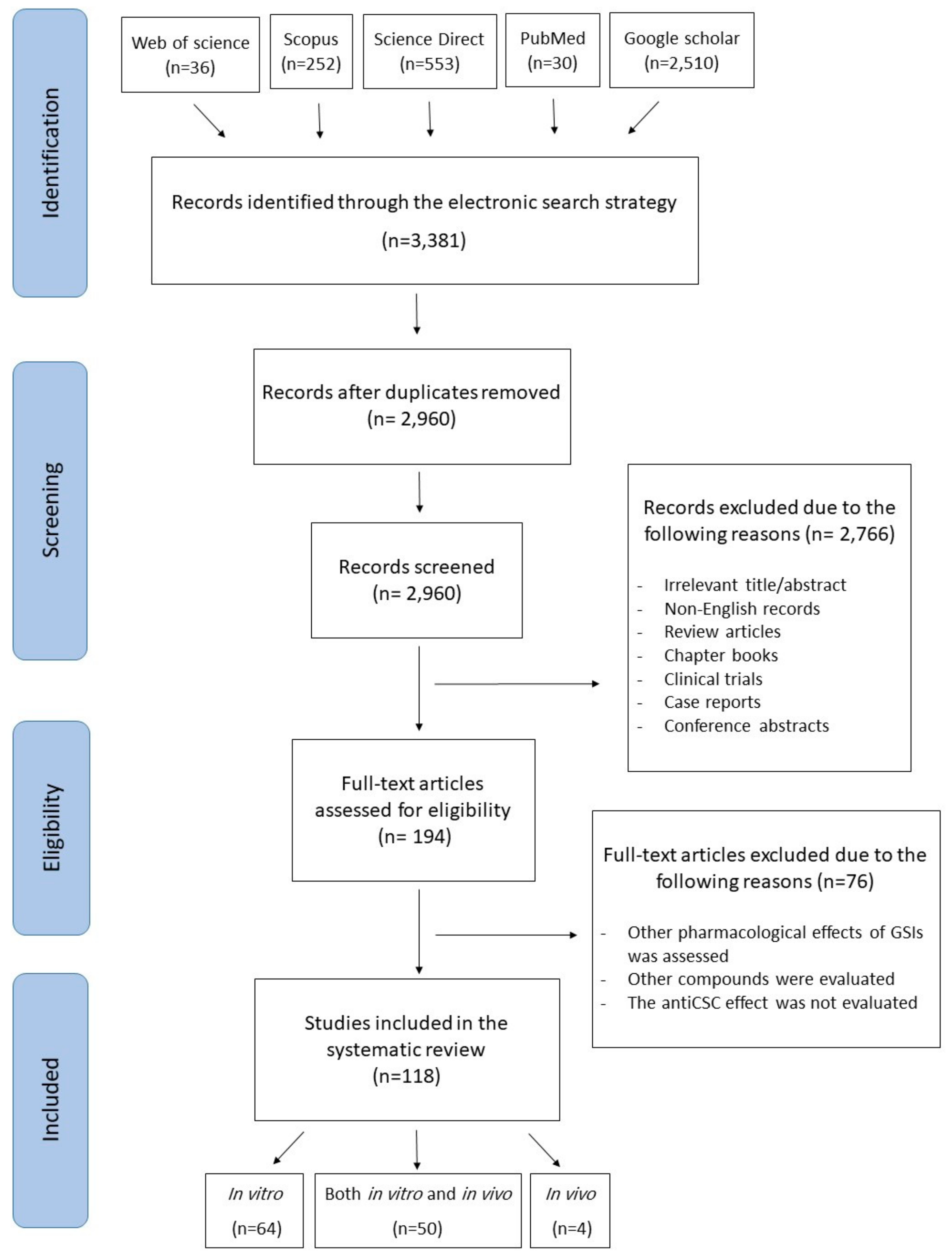
4. Methodology for the Literature Search and Study Selection
5. Anticancer Activities of GSIs against CSCs
5.1. Adenoid Cystic Carcinoma
5.2. Blood Cancer
5.3. Brain Cancer
5.4. Breast Cancer
5.5. Colorectal Carcinoma
5.6. Gastric Cancer
5.7. Head and Neck Cancer
5.8. Liver Cancer and Cholangiocarcinoma
5.9. Lung Cancer
5.10. Melanoma
5.11. Osteosarcoma
5.12. Ovarian Cancer
5.13. Pancreatic Carcinoma
5.14. Prostate Cancer
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 1–18. [Google Scholar]
- Lathia, J.D.; Liu, H. Overview of cancer stem cells and stemness for community oncologists. Target. Oncol. 2017, 12, 387–399. [Google Scholar] [CrossRef] [Green Version]
- Akbarzadeh, M.; Maroufi, N.F.; Tazehkand, A.P.; Akbarzadeh, M.; Bastani, S.; Safdari, R.; Farzane, A.; Fattahi, A.; Nejabati, H.R.; Nouri, M. Current approaches in identification and isolation of cancer stem cells. J. Cell. Physiol. 2019, 234, 14759–14772. [Google Scholar] [CrossRef] [PubMed]
- Kemper, K.; Sprick, M.R.; de Bree, M.; Scopelliti, A.; Vermeulen, L.; Hoek, M.; Zeilstra, J.; Pals, S.T.; Mehmet, H.; Stassi, G. The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation. Cancer Res. 2010, 70, 719–729. [Google Scholar] [CrossRef] [Green Version]
- Weiswald, L.-B.; Bellet, D.; Dangles-Marie, V. Spherical cancer models in tumor biology. Neoplasia 2015, 17, 1–15. [Google Scholar]
- Liu, J.; Sato, C.; Cerletti, M.; Wagers, A. Notch signaling in the regulation of stem cell self-renewal and differentiation. Curr. Top. Dev. Biol. 2010, 92, 367–409. [Google Scholar] [PubMed]
- Venkatesh, V.; Nataraj, R.; Thangaraj, G.S.; Karthikeyan, M.; Gnanasekaran, A.; Kaginelli, S.B.; Kuppanna, G.; Kallappa, C.G.; Basalingappa, K.M. Targeting Notch signalling pathway of cancer stem cells. Stem Cell Investig. 2018, 5. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, P.; Osipo, C.; Foreman, K.; Golde, T.; Osborne, B.; Miele, L. Rational targeting of Notch signaling in cancer. Oncogene 2008, 27, 5124–5131. [Google Scholar] [PubMed] [Green Version]
- Curry, C.L.; Reed, L.L.; Golde, T.E.; Miele, L.; Nickoloff, B.J.; Foreman, K.E. Gamma secretase inhibitor blocks Notch activation and induces apoptosis in Kaposi’s sarcoma tumor cells. Oncogene 2005, 24, 6333–6344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, E.R.; Lendahl, U. Therapeutic modulation of Notch signalling—are we there yet? Nat. Rev. Drug Discov. 2014, 13, 357–378. [Google Scholar] [PubMed]
- Orza, A.; Casciano, D.; Biris, A. Nanomaterials for targeted drug delivery to cancer stem cells. Drug Metab. Rev. 2014, 46, 191–206. [Google Scholar] [CrossRef]
- Nie, P.; Vartak, A.; Li, Y.M. γ-Secretase inhibitors and modulators: Mechanistic insights into the function and regulation of γ-Secretase. Semin. Cell Dev. Biol. 2020, 105, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Pine, S.R. Rethinking Gamma-secretase Inhibitors for Treatment of Non–small-Cell Lung Cancer: Is notch the target? Clin. Cancer Res. 2018, 68, 7–30. [Google Scholar] [CrossRef] [Green Version]
- Golde, T.E.; Koo, E.H.; Felsenstein, K.M.; Osborne, B.A.; Miele, L. γ-Secretase inhibitors and modulators. Biochim. Biophys. Acta—Biomater. 2013, 1828, 2898–2907. [Google Scholar] [CrossRef] [Green Version]
- Allenspach, E.J.; Maillard, I.; Aster, J.C.; Pear, W.S. Notch signaling in cancer. Cancer Biol. Ther. 2002, 1, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Ellisen, L.W.; Bird, J.; West, D.C.; Soreng, A.L.; Reynolds, T.C.; Smith, S.D.; Sklar, J. TAN-1, the human homolog of the Drosophila Notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 1991, 66, 649–661. [Google Scholar] [CrossRef]
- Fabbri, G.; Rasi, S.; Rossi, D.; Trifonov, V.; Khiabanian, H.; Ma, J.; Grunn, A.; Fangazio, M.; Capello, D.; Monti, S.; et al. Analysis of the chronic lymphocytic leukemia coding genome: Role of NOTCH1 mutational activation. J. Exp. Med. 2011, 208, 1389–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houde, C.; Li, Y.; Song, L.; Barton, K.; Zhang, Q.; Godwin, J.; Nand, S.; Toor, A.; Alkan, S.; Smadja, N.V.; et al. Overexpression of the NOTCH ligand JAG2 in malignant plasma cells from multiple myeloma patients and cell lines. Blood 2004, 104, 3697–3704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Liu, M.; Gonzalez-Perez, R.R. Role of Notch and its oncogenic signaling crosstalk in breast cancer. Biochim. Biophys. Acta—Rev. Cancer 2011, 1815, 197–213. [Google Scholar]
- Song, L.L.; Peng, Y.; Yun, J.; Rizzo, P.; Chaturvedi, V.; Weijzen, S.; Kast, W.M.; Stone, P.J.B.; Santos, L.; Loredo, A.; et al. Notch-1 associates with IKKα and regulates IKK activity in cervical cancer cells. Oncogene 2008, 27, 5833–5844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miele, L.; Miao, H.; Nickoloff, B.J. NOTCH signaling as a novel cancer therapeutic target. Curr. Cancer Drug Targets 2006, 6, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Gu, K.; Li, Q.; Lin, H.; Zhu, J.; Mo, J.; He, S.; Lu, X.; Jiang, X.; Sun, H. Gamma secretase inhibitors: A patent review (2013-2015). Expert Opin. Ther. Pat. 2017, 27, 851–866. [Google Scholar] [CrossRef]
- Hyde, L.A.; McHugh, N.A.; Chen, J.; Zhang, Q.; Manfra, D.; Nomeir, A.A.; Josien, H.; Bara, T.; Clader, J.W.; Zhang, L. Studies to investigate the in vivo therapeutic window of the γ-secretase inhibitor N2-[(2S)-2-(3, 5-difluorophenyl)-2-hydroxyethanoyl]-N1-[(7S)-5-methyl-6-oxo-6, 7-dihydro-5H-dibenzo [b, d] azepin-7-yl]-L-alaninamide (LY411, 575) in the CRND8 mouse. J. Pharmacol. Exp. Ther. 2006, 319, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.-B.S.; Zhang, H.; Damelin, M.; Geles, K.G.; Grindley, J.C.; Dirks, P.B. Tumour-initiating cells: Challenges and opportunities for anticancer drug discovery. Nat. Rev. Drug Discov. 2009, 8, 806–823. [Google Scholar] [CrossRef] [PubMed]
- Kolb, E.A.; Gorlick, R.; Keir, S.T.; Maris, J.M.; Lock, R.; Carol, H.; Kurmasheva, R.T.; Reynolds, C.P.; Kang, M.H.; Wu, J. Initial testing (stage 1) by the pediatric preclinical testing program of RO4929097, a γ-secretase inhibitor targeting notch signaling. Pediatr. Blood Cancer 2012, 58, 815–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.C.; Pavlicek, A.; Zhang, Q.; Lira, M.E.; Painter, C.L.; Yan, Z.; Zheng, X.; Lee, N.V.; Ozeck, M.; Qiu, M. Biomarker and pharmacologic evaluation of the γ-secretase inhibitor PF-03084014 in breast cancer models. Clin. Cancer Res. 2012, 18, 5008–5019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyoshi, T.; Nakamura, M.; Yanai, K.; Nagai, S.; Wada, J.; Koga, K.; Nakashima, H.; Sato, N.; Tanaka, M.; Katano, M. γ-Secretase inhibitors enhance taxane-induced mitotic arrest and apoptosis in colon cancer cells. Gastroenterology 2008, 134, 131–144. [Google Scholar] [CrossRef]
- Aleksic, T.; Feller, S.M. Gamma-secretase inhibition combined with platinum compounds enhances cell death in a large subset of colorectal cancer cells. Cell Commun. Signal. 2008, 6, 8. [Google Scholar] [CrossRef] [Green Version]
- Pellegrinet, L.; Rodilla, V.; Liu, Z.; Chen, S.; Koch, U.; Espinosa, L.; Kaestner, K.H.; Kopan, R.; Lewis, J.; Radtke, F. Dll1-and dll4-mediated notch signaling are required for homeostasis of intestinal stem cells. Gastroenterology 2011, 140, 1230–1240. [Google Scholar] [CrossRef] [Green Version]
- Efferson, C.L.; Winkelmann, C.T.; Ware, C.; Sullivan, T.; Giampaoli, S.; Tammam, J.; Patel, S.; Mesiti, G.; Reilly, J.F.; Gibson, R.E. Downregulation of Notch pathway by a γ-secretase inhibitor attenuates AKT/mammalian target of rapamycin signaling and glucose uptake in an ERBB2 transgenic breast cancer model. Cancer Res. 2010, 70, 2476–2484. [Google Scholar] [CrossRef] [Green Version]
- Schott, A.F.; Landis, M.D.; Dontu, G.; Griffith, K.A.; Layman, R.M.; Krop, I.; Paskett, L.A.; Wong, H.; Dobrolecki, L.E.; Lewis, M.T.; et al. Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clin. Cancer Res. 2013, 19, 1512–1524. [Google Scholar] [CrossRef] [Green Version]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; Group, P. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panaccione, A.; Chang, M.T.; Carbone, B.E.; Guo, Y.; Moskaluk, C.A.; Virk, R.K.; Chiriboga, L.; Prasad, M.L.; Judson, B.; Mehra, S.; et al. NOTCH1 and SOX10 are essential for proliferation and radiation resistance of cancer stem-like cells in adenoid cystic carcinoma. Clin. Cancer Res. 2016, 22, 2083–2095. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, Y.; Malek, S.N.; Zheng, P.; Liu, Y. Targeting HIF1α eliminates cancer stem cells in hematological malignancies. Cell Stem Cell 2011, 8, 399–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikram, M.; Lim, Y.; Baek, S.Y.; Jin, S.; Jeong, Y.H.; Kwak, J.Y.; Yoon, S. Co-targeting of Tiam1/Rac1 and Notch ameliorates chemoresistance against doxorubicin in a biomimetic 3D lymphoma model. Oncotarget 2018, 9, 2058–2075. [Google Scholar] [CrossRef] [Green Version]
- Gal, H.; Amariglio, N.; Trakhtenbrot, L.; Jacob-Hirsh, J.; Margalit, O.; Avigdor, A.; Nagler, A.; Tavor, S.; Ein-Dor, L.; Lapidot, T.; et al. Gene expression profiles of AML derived stem cells; similarity to hematopoietic stem cells. Leukemia 2006, 20, 2147–2154. [Google Scholar] [CrossRef] [PubMed]
- Tatarek, J.; Cullion, K.; Ashworth, T.; Gerstein, R.; Aster, J.C.; Kelliher, M.A. Notch1 inhibition targets the leukemia-initiating cells in a Tal1/Lmo2 mouse model of T-ALL. Blood 2011, 118, 1579–1590. [Google Scholar] [CrossRef] [PubMed]
- Okuhashi, Y.; Itoh, M.; Nara, N.; Tohda, S. Effects of combination of notch inhibitor plus hedgehog inhibitor or Wnt inhibitor on growth of leukemia cells. Anticancer Res. 2011, 31, 893–906. [Google Scholar]
- Okuhashi, Y.; Itoh, M.; Arai, A.; Nara, N.; Tohda, S. γ-secretase inhibitors induce erythroid differentiation in erythroid leukemia cell lines. Anticancer Res. 2010, 30, 4071–4074. [Google Scholar]
- Issa, M.E.; Berndt, S.; Carpentier, G.; Pezzuto, J.M.; Cuendet, M. Bruceantin inhibits multiple myeloma cancer stem cell proliferation. Cancer Biol. Ther. 2016, 17, 966–975. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.Y.; Fu, L.A.; Li, S.Z.; Chen, Y.; Li, J.C.; Han, J.; Liang, L.; Li, L.; Ji, C.C.; Zheng, M.H.; et al. Hif-1α and Hif-2α differentially regulate Notch signaling through competitive interaction with the intracellular domain of Notch receptors in glioma stem cells. Cancer Lett. 2014, 314, 67–76. [Google Scholar] [CrossRef]
- Li, Y.; He, Z.C.; Zhang, X.N.; Liu, Q.; Chen, C.; Zhu, Z.; Chen, Q.; Shi, Y.; Yao, X.H.; Cui, Y.H.; et al. Stanniocalcin-1 augments stem-like traits of glioblastoma cells through binding and activating NOTCH1. Cancer Lett. 2018, 416, 66–74. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Q.R.; Xie, W.F.; Wang, M. De DAPT suppresses the proliferation of human glioma cell line SHG-44. Asian Pac. J. Trop. Med. 2014, 7, 552–556. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–835. [Google Scholar] [CrossRef]
- Kristoffersen, K.; Villingshøj, M.; Poulsen, H.S.; Stockhausen, M.T. Level of Notch activation determines the effect on growth and stem cell-like features in glioblastoma multiforme neurosphere cultures. Cancer Biol. Ther. 2013, 14, 625–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pistollato, F.; Rampazzo, E.; Persano, L.; Abbadi, S.; Frasson, C.; Denaro, L.; D’Avella, D.; Panchision, D.M.; Puppa, A.D.; Scienza, R.; et al. Interaction of hypoxia-inducible factor-1α and Notch signaling regulates medulloblastoma precursor proliferation and fate. Stem Cells 2010, 28, 1918–1929. [Google Scholar] [CrossRef] [Green Version]
- Kristoffersen, K.; Nedergaard, M.K.; Villingshøj, M.; Borup, R.; Broholm, H.; Kjær, A.; Poulsen, H.S.; Stockhausen, M.T. Inhibition of Notch signaling alters the phenotype of orthotopic tumors formed from glioblastoma multiforme neurosphere cells but does not hamper intracranial tumor growth regardless of endogene Notch pathway signature. Cancer Biol. Ther. 2014, 15, 862–877. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Chen, H.; Zhang, J.; Chen, Y.; Wang, M.; Ma, J.; Hong, L.; Liu, N.; Fan, Q.; Lu, X.; et al. Increased Notch signaling enhances radioresistance of malignant stromal cells induced by glioma stem/progenitor cells. PLoS ONE 2015, 10, e0142594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gal, H.; Makovitzki, A.; Amariglio, N.; Rechavi, G.; Ram, Z.; Givol, D. A rapid assay for drug sensitivity of glioblastoma stem cells. Biochem. Biophys. Res. Commun. 2007, 12, 1205–1219. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Chen, H.; Liang, Q. Endothelial Cells Promote Formation of Medulloblastoma Stem-Like Cells via Notch Pathway Activation. J. Mol. Neurosci. 2017, 63, 152–158. [Google Scholar] [CrossRef]
- Kanabur, P.; Guo, S.; Simonds, G.R.; Kelly, D.F.; Gourdie, R.G.; Verbridge, S.S.; Sheng, Z. Patient-derived glioblastoma stem cells respond differentially to targeted therapies. Oncotarget 2016, 7, 86406–86419. [Google Scholar] [CrossRef]
- Floyd, D.H.; Kefas, B.; Seleverstov, O.; Mykhaylyk, O.; Dominguez, C.; Comeau, L.; Plank, C.; Purow, B. Alpha-secretase inhibition reduces human glioblastoma stem cell growth in vitro and in vivo by inhibiting Notch. Neuro. Oncol. 2012, 14, 1215–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, N.I.; Guilhamon, P.; Desai, K.; McAdam, R.F.; Langille, E.; O’Connor, M.; Lan, X.; Whetstone, H.; Coutinho, F.J.; Vanner, R.J.; et al. ASCL1 Reorganizes Chromatin to Direct Neuronal Fate and Suppress Tumorigenicity of Glioblastoma Stem Cells. Cell Stem Cell 2017, 21, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Hirai, N.; Aoki, K.; Suzuki, R.; Fujita, S.; Nakayama, H.; Hayashi, M.; Ito, K.; Sakurai, T.; Iwabuchi, S. The oncogene addiction switch from NOTCH to PI3K requires simultaneous targeting of NOTCH and PI3K pathway inhibition in glioblastoma. Cancers (Basel) 2019, 11, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wakeman, T.P.; Lathia, J.D.; Hjelmeland, A.B.; Wang, X.F.; White, R.R.; Rich, J.N.; Sullenger, B.A. Notch promotes radioresistance of glioma stem cells. Stem Cells 2010, 28, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Khaki, L.; Zhu, T.S.; Soules, M.E.; Talsma, C.E.; Gul, N.; Koh, C.; Zhang, J.; Li, Y.M.; Maciaczyk, J.; et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 2010, 28, 5–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Matsui, W.; Khaki, L.; Stearns, D.; Chun, J.; Li, Y.M.; Eberhart, C.G. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 2006, 66, 7445–7452. [Google Scholar] [CrossRef] [Green Version]
- Dai, L.; He, J.; Liu, Y.; Byun, J.; Vivekanandan, A.; Pennathur, S.; Fan, X.; Lubman, D.M. Dose-dependent proteomic analysis of glioblastoma cancer stem cells upon treatment with γ-secretase inhibitor. Proteomics 2011, 11, 4529–4540. [Google Scholar] [CrossRef] [Green Version]
- Charles, N.; Ozawa, T.; Squatrito, M.; Bleau, A.M.; Brennan, C.W.; Hambardzumyan, D.; Holland, E.C. Perivascular Nitric Oxide Activates Notch Signaling and Promotes Stem-like Character in PDGF-Induced Glioma Cells. Cell Stem Cell 2010, 6, 141–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornara, O.; Bartek, J.; Rahbar, A.; Odeberg, J.; Khan, Z.; Peredo, I.; Hamerlik, P.; Bartek, J.; Stragliotto, G.; Landázuri, N.; et al. Cytomegalovirus infection induces a stem cell phenotype in human primary glioblastoma cells: Prognostic significance and biological impact. Cell Death Differ. 2016, 23, 261–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, S.; Nakada, M.; Yamada, D.; Nakano, I.; Todo, T.; Ino, Y.; Hoshii, T.; Tadokoro, Y.; Ohta, K.; Ali, M.A.E.; et al. Strong therapeutic potential of γ-secretase inhibitor MRK003 for CD44-high and CD133-low glioblastoma initiating cells. J. Neurooncol. 2015, 121, 239–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natsumeda, M.; Maitani, K.; Liu, Y.; Miyahara, H.; Kaur, H.; Chu, Q.; Zhang, H.D.; Kahlert, U.; Eberhart, C.G. Targeting Notch Signaling and Autophagy Increases Cytotoxicity in Glioblastoma Neurospheres. Brain Pathol. 2016, 26, 713–723. [Google Scholar] [CrossRef]
- Lin, J.; Zhang, X.M.; Yang, J.C.; Ye, Y.B.; Luo, S.Q. γ-secretase inhibitor-I enhances radiosensitivity of glioblastoma cell lines by depleting CD133+ tumor cells. Arch. Med. Res. 2010, 41, 519–529. [Google Scholar] [CrossRef]
- Ulasov, I.V.; Nandi, S.; Dey, M.; Sonabend, A.M.; Lesniak, M.S. Inhibition of sonic hedgehog and notch pathways enhances sensitivity of cd133+ glioma stem cells to temozolomide therapy. Mol. Med. 2011, 17, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Pietras, A.; Katz, A.M.; Ekström, E.J.; Wee, B.; Halliday, J.J.; Pitter, K.L.; Werbeck, J.L.; Amankulor, N.M.; Huse, J.T.; Holland, E.C. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell 2014, 14, 357–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Cheng, Z.; Liu, J.; Torre-Healy, L.; Lathia, J.D.; Nakano, I.; Guo, Y.; Thompson, R.C.; Freeman, M.L.; Wang, J. Inhibition of Farnesyltransferase Potentiates NOTCH-Targeted Therapy against Glioblastoma Stem Cells. Stem Cell Rep. 2017, 9, 1948–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.Y.; Zheng, M.H.; Cheng, G.; Li, L.; Liang, L.; Gao, F.; Wei, Y.N.; Fu, L.A.; Han, H. Notch signaling contributes to the maintenance of both normal neural stem cells and patient-derived glioma stem cells. BMC Cancer 2011, 11, 82. [Google Scholar] [CrossRef] [Green Version]
- Dai, L.; Liu, Y.; He, J.; Flack, C.G.; Talsma, C.E.; Crowley, J.G.; Muraszko, K.M.; Fan, X.; Lubman, D.M. Differential profiling studies of N-linked glycoproteins in glioblastoma cancer stem cells upon treatment with γ-secretase inhibitor. Proteomics 2011, 11, 4021–4028. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Ding, C.; Wang, F.; Deng, W.; Yu, M.; Meng, Q.; Sun, P. Effects of NOTCH1 signaling inhibitor γ-secretase inhibitor II on growth of cancer stem cells. Oncol. Lett. 2018, 16, 6095–6099. [Google Scholar] [CrossRef]
- Hsu, E.C.; Kulp, S.K.; Huang, H.L.; Tu, H.J.; Salunke, S.B.; Sullivan, N.J.; Sun, D.; Wicha, M.S.; Shapiro, C.L.; Chen, C.S. Function of integrin-linked kinase in modulating the stemness of il-6-abundant breast cancer cells by regulating γ-secretase-mediated notch1 activation in caveolae. Neoplasia (U. S.) 2015, 17, 497–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.H.; Kuo, K.T.; Bamodu, O.A.; Lin, Y.C.; Yang, R.B.; Yeh, C.T.; Chao, T.Y. Upregulated SCUBE2 expression in breast cancer stem cells enhances triple negative breast cancer aggression through modulation of notch signaling and epithelial-to-mesenchymal transition. Exp. Cell Res. 2018, 370, 444–453. [Google Scholar] [CrossRef]
- Cho, Y.; Lee, H.W.; Kang, H.G.; Kim, H.Y.; Kim, S.J.; Chun, K.H. Cleaved CD44 intracellular domain supports activation of stemness factors and promotes tumorigenesis of breast cancer. Oncotarget 2015, 6, 8709–8721. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Yang, H.; Li, X.; Han, L.; Xu, N.; Shi, A. Signaling pathway inhibitors target breast cancer stem cells in triple-negative breast cancer. Oncol. Rep. 2019, 41, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Roberts, A.L.; Dunphy, K.A.; Bigelow, C.; Yan, H.; Jerry, D.J. Repression of mammary stem/progenitor cells by p53 is mediated by notch and separable from apoptotic activity. Stem Cells 2011, 29, 119–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGowan, P.M.; Simedrea, C.; Ribot, E.J.; Foster, P.J.; Palmieri, D.; Steeg, P.S.; Allan, A.L.; Chambers, A.F. Notch1 inhibition alters the CD44 hi/CD24 lo population and reduces the formation of brain metastases from breast cancer. Mol. Cancer Res. 2011, 9, 834–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, F.; Okuda, H.; Watabe, M.; Kobayashi, A.; Pai, S.K.; Liu, W.; Pandey, P.R.; Fukuda, K.; Hirota, S.; Sugai, T.; et al. Hypoxia-induced Jagged2 promotes breast cancer metastasis and self-renewal of cancer stem-like cells. Oncogene 2011, 30, 4075–4086. [Google Scholar] [CrossRef] [Green Version]
- Boelens, M.C.; Wu, T.J.; Nabet, B.Y.; Xu, B.; Qiu, Y.; Yoon, T.; Azzam, D.J.; Twyman-Saint Victor, C.; Wiemann, B.Z.; Ishwaran, H.; et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell 2014, 159, 499–513. [Google Scholar] [CrossRef] [Green Version]
- Farnie, G.; Willan, P.M.; Clarke, R.B.; Bundred, N.J. Combined Inhibition of ErbB1/2 and Notch Receptors Effectively Targets Breast Ductal Carcinoma In Situ (DCIS) Stem/Progenitor Cell Activity Regardless of ErbB2 Status. PLoS ONE 2013, 8, e56840. [Google Scholar] [CrossRef] [Green Version]
- Harrison, H.; Simões, B.M.; Rogerson, L.; Howell, S.J.; Landberg, G.; Clarke, R.B. Oestrogen increases the activity of oestrogen receptor negative breast cancer stem cells through paracrine EGFR and Notch signalling. Breast Cancer Res. 2013, 15, R21. [Google Scholar] [CrossRef] [PubMed]
- Buckley, N.E.; Nic An Tsaoir, C.B.; Blayney, J.K.; Oram, L.C.; Crawford, N.T.; D’Costa, Z.C.; Quinn, J.E.; Kennedy, R.D.; Harkin, D.P.; Mullan, P.B. BRCA1 is a key regulator of breast differentiation through activation of Notch signalling with implications for anti-endocrine treatment of breast cancers. Nucleic Acids Res. 2013, 41, 8601–8614. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Vasudevan, S.; Sengupta, S. 6-shogaol inhibits breast cancer cells and stem cell-like spheroids by modulation of notch signaling pathway and induction of autophagic cell death. PLoS ONE 2015, e0137614. [Google Scholar] [CrossRef]
- Mamaeva, V.; Niemi, R.; Beck, M.; Özliseli, E.; Desai, D.; Landor, S.; Gronroos, T.; Kronqvist, P.; Pettersen, I.K.N.; McCormack, E. Inhibiting notch activity in breast cancer stem cells by glucose functionalized nanoparticles carrying γ-secretase inhibitors. Mol. Ther. 2016, 24, 926–936. [Google Scholar] [CrossRef]
- Phillips, T.M.; Kim, K.; Vlashi, E.; McBride, W.H.; Pajonk, F. Effects of recombinant erythropoietin on breast cancer-initiating cells. Neoplasia 2007, 9, 1122–1129. [Google Scholar] [CrossRef] [Green Version]
- Lagadec, C.; Vlashi, E.; Alhiyari, Y.; Phillips, T.M.; Bochkur Dratver, M.; Pajonk, F. Radiation-induced notch signaling in breast cancer stem cells. Int. J. Radiat. Oncol. Biol. Phys. 2013, 87, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Shang, L.; Brooks, M.D.; Jiagge, E.; Zhu, Y.; Buschhaus, J.M.; Conley, S.; Fath, M.A.; Davis, A.; Gheordunescu, E.; et al. Targeting Breast Cancer Stem Cell State Equilibrium through Modulation of Redox Signaling. Cell Metab. 2018, 28, 69–86. [Google Scholar] [CrossRef] [Green Version]
- Shah, D.; Wyatt, D.; Baker, A.T.; Simms, P.; Peiffer, D.S.; Fernandez, M.; Rakha, E.; Green, A.; Filipovic, A.; Miele, L.; et al. Inhibition of her2 increases jagged1-dependent breast cancer stem cells: Role for membrane jagged1. Clin. Cancer Res. 2018, 24, 4566–4578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angelo, R.C.; Ouzounova, M.; Davis, A.; Choi, D.; Tchuenkam, S.M.; Kim, G.; Luther, T.; Quraishi, A.A.; Senbabaoglu, Y.; Conley, S.J.; et al. Notch reporter activity in breast cancer cell lines identifies a subset of cells with stem cell activity. Mol. Cancer Ther. 2015, 14, 779–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondratyev, M.; Kreso, A.; Hallett, R.M.; Girgis-Gabardo, A.; Barcelon, M.E.; Ilieva, D.; Ware, C.; Majumder, P.K.; Hassell, J.A. Gamma-secretase inhibitors target tumor-initiating cells in a mouse model of ERBB2 breast cancer. Oncogene 2012, 31, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Xing, F.; Kobayashi, A.; Okuda, H.; Watabe, M.; Pai, S.K.; Pandey, P.R.; Hirota, S.; Wilber, A.; Mo, Y.Y.; Moore, B.E.; et al. Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating Notch signalling in brain. EMBO Mol. Med. 2013, 5, 384–396. [Google Scholar] [CrossRef] [PubMed]
- McClements, L.; Yakkundi, A.; Papaspyropoulos, A.; Harrison, H.; Ablett, M.P.; Jithesh, P.V.; McKeen, H.D.; Bennett, R.; Donley, C.; Kissenpfennig, A.; et al. Targeting treatment-resistant breast cancer stem cells with FKBPL and Its peptide derivative, AD-01, via the CD44 pathway. Clin. Cancer Res. 2013, 19, 3881–3893. [Google Scholar] [CrossRef] [Green Version]
- Harrison, H.; Farnie, G.; Howell, S.J.; Rock, R.E.; Stylianou, S.; Brennan, K.R.; Bundred, N.J.; Clarke, R.B. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010, 70, 709–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grudzien, P.; Lo, S.; Albain, K.S.; Robinson, P.; Rajan, P.; Strack, P.R.; Golde, T.E.; Miele, L.; Foreman, K.E. Inhibition of notch signaling reduces the stem-like population of breast cancer cells and prevents mammosphere formation. Anticancer Res. 2010, 30, 3853–3867. [Google Scholar]
- Wang, D.; Xu, J.; Liu, B.; He, X.; Zhou, L.; Hu, X.; Qiao, F.; Zhang, A.; Xu, X.; Zhang, H.; et al. IL6 blockade potentiates the anti-tumor effects of γ-secretase inhibitors in Notch3-expressing breast cancer. Cell Death Differ. 2018, 25, 330–339. [Google Scholar] [CrossRef]
- Azzam, D.J.; Zhao, D.; Sun, J.; Minn, A.J.; Ranganathan, P.; Drews-Elger, K.; Han, X.; Picon-Ruiz, M.; Gilbert, C.A.; Wander, S.A.; et al. Triple negative breast cancer initiating cell subsets differ in functional and molecular characteristics and in γ-secretase inhibitor drug responses. EMBO Mol. Med. 2013, 5, 1502–1522. [Google Scholar] [CrossRef]
- Debeb, B.G.; Cohen, E.N.; Boley, K.; Freiter, E.M.; Li, L.; Robertson, F.M.; Reuben, J.M.; Cristofanilli, M.; Buchholz, T.A.; Woodward, W.A. Pre-clinical studies of Notch signaling inhibitor RO4929097 in inflammatory breast cancer cells. Breast Cancer Res. Treat. 2012, 134, 495–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Bommer, G.T.; Zhao, J.; Green, M.; Sands, E.; Zhai, Y.; Brown, K.; Burberry, A.; Cho, K.R.; Fearon, E.R. Mutant kras promotes hyperplasia and alters differentiation in the colon epithelium but does not expand the presumptive stem cell pool. Gastroenterology 2011, 141, 1003–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, P.; Wang, L.; Chen, K.Y.; Srinivasan, T.; Murthy, P.K.L.; Tung, K.L.; Varanko, A.K.; Chen, H.J.; Ai, Y.; King, S.; et al. A miR-34a-Numb Feedforward Loop Triggered by Inflammation Regulates Asymmetric Stem Cell Division in Intestine and Colon Cancer. Cell Stem Cell 2016, 18, 189–202. [Google Scholar] [CrossRef] [Green Version]
- Bu, P.; Chen, K.Y.; Chen, J.H.; Wang, L.; Walters, J.; Shin, Y.J.; Goerger, J.P.; Sun, J.; Witherspoon, M.; Rakhilin, N.; et al. A microRNA miR-34a-regulated bimodal switch targets notch in colon cancer stem cells. Cell Stem Cell 2013, 12, 602–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fender, A.W.; Nutter, J.M.; Fitzgerald, T.L.; Bertrand, F.E.; Sigounas, G. Notch-1 Promotes Stemness and Epithelial to Mesenchymal Transition in Colorectal Cancer. J. Cell. Biochem. 2015, 116, 2517–2527. [Google Scholar] [CrossRef]
- Moon, C.M.; Kwon, J.H.; Kim, J.S.; Oh, S.H.; Jin Lee, K.; Park, J.J.; Pil Hong, S.; Cheon, J.H.; Kim, T.I.; Kim, W.H. Nonsteroidal anti-inflammatory drugs suppress cancer stem cells via inhibiting PTGS2 (cyclooxygenase 2) and NOTCH/HES1 and activating PPARG in colorectal cancer. Int. J. Cancer 2014, 134, 519–529. [Google Scholar] [CrossRef]
- Lu, J.; Ye, X.; Fan, F.; Xia, L.; Bhattacharya, R.; Bellister, S.; Tozzi, F.; Sceusi, E.; Zhou, Y.; Tachibana, I.; et al. Endothelial Cells Promote the Colorectal Cancer Stem Cell Phenotype through a Soluble Form of Jagged-1. Cancer Cell 2013, 23, 171–185. [Google Scholar] [CrossRef] [Green Version]
- Arcaroli, J.J.; Powell, R.W.; Varella-Garcia, M.; McManus, M.; Tan, A.C.; Quackenbush, K.S.; Pitts, T.M.; Gao, D.; Spreafico, A.; Dasari, A.; et al. ALDH+ tumor-initiating cells exhibiting gain in NOTCH1 gene copy number have enhanced regrowth sensitivity to a γ-secretase inhibitor and irinotecan in colorectal cancer. Mol. Oncol. 2012, 6, 370–381. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Ariyama, H.; Stancikova, J.; Sakitani, K.; Asfaha, S.; Renz, B.W.; Dubeykovskaya, Z.A.; Shibata, W.; Wang, H.; Westphalen, C.B.; et al. Mist1 Expressing Gastric Stem Cells Maintain the Normal and Neoplastic Gastric Epithelium and Are Supported by a Perivascular Stem Cell Niche. Cancer Cell 2015, 28, 800–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barat, S.; Chen, X.; Bui, K.C.; Bozko, P.; Götze, J.; Christgen, M.; Krech, T.; Malek, N.P.; Plentz, R.R. Gamma-secretase inhibitor IX (GSI) impairs concomitant activation of notch and wnt-beta-catenin pathways in CD441 gastric cancer stem cells. Stem Cells Transl. Med. 2017, 6, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Li, L.C.; Wang, D.L.; Wu, Y.Z.; Nian, W.Q.; Wu, Z.J.; Li, Y.; Ma, H.W.; Shao, J.H. Gastric tumor-initiating CD44+ cells and epithelial-mesenchymal transition are inhibited by γ-secretase inhibitor DAPT. Oncol. Lett. 2015, 10, 3293–3299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, K.J.; Wu, D.C.; Cheng, K.H.; Chen, L.T.; Hung, W.C. RECK Inhibits Stemness Gene Expression and Tumorigenicity of Gastric Cancer Cells by Suppressing ADAM-Mediated Notch1 Activation. J. Cell. Physiol. 2014, 229, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Kato, R.; Iwamuro, M.; Shiraha, H.; Horiguchi, S.; Tanaka, E.; Matsumoto, K.; Ohyama, A.; Sawahara, H.; Nagahara, T.; Uchida, D.; et al. Dipeptide γ-secretase inhibitor treatment enhances the anti-tumor effects of cisplatin against gastric cancer by suppressing cancer stem cell properties. Oncol. Lett. 2018, 16, 5426–5432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.F.; Huang, C.H.; Liu, C.J.; Hung, J.J.; Hsu, C.C.; Teng, S.C.; Wu, K.J. Twist1 induces endothelial differentiation of tumour cells through the Jagged1-KLF4 axis. Nat. Commun. 2014, 5, 4697. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Da Silva, T.G.; Jin, K.; Han, X.; Ranganathan, P.; Zhu, X.; Sanchez-Mejias, A.; Bai, F.; Li, B.; Fei, D.L.; et al. Notch signaling drives stemness and tumorigenicity of esophageal adenocarcinoma. Cancer Res. 2014, 74, 6364–6374. [Google Scholar] [CrossRef] [Green Version]
- Mendelson, J.; Song, S.; Li, Y.; Maru, D.M.; Mishra, B.; Davila, M.; Hofstetter, W.L.; Mishra, L. Dysfunctional transforming growth factor-β signaling with constitutively active notch signaling in Barrett’s esophageal adenocarcinoma. Cancer 2011, 117, 3691–3702. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.L.; Zhang, L.; Huang, C.F.; Ma, S.R.; Bu, L.L.; Liu, J.F.; Yu, G.T.; Liu, B.; Gutkind, J.S.; Kulkarni, A.B.; et al. NOTCH1 inhibition enhances the efficacy of conventional chemotherapeutic agents by targeting head neck cancer stem cell. Sci. Rep. 2016, 6, 24704. [Google Scholar] [CrossRef] [Green Version]
- Upadhyay, P.; Nair, S.; Kaur, E.; Aich, J.; Dani, P.; Sethunath, V.; Gardi, N.; Chandrani, P.; Godbole, M.; Sonawane, K.; et al. Notch pathway activation is essential for maintenance of stem-like cells in early tongue cancer. Oncotarget 2016, 7, 50437–50449. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, K.; Honda, M.; Yamashita, T.; Okada, H.; Shirasaki, T.; Nishikawa, M.; Nio, K.; Arai, K.; Sakai, Y.; Yamashita, T.; et al. Jagged1 DNA Copy Number Variation Is Associated with Poor Outcome in Liver Cancer. Am. J. Pathol. 2016, 24, 3145–3154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.X.; Xu, A.; Zhang, C.C.; Olson, P.; Chen, L.; Lee, T.K.; Cheung, T.T.; Lo, C.M.; Wang, X.Q. Notch inhibitor PF-03084014 inhibits hepatocellular carcinoma growth and metastasis via suppression of cancer stemness due to reduced activation of Notch1–Stat3. Mol. Cancer Ther. 2017, 16, 1531–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; Zhou, Y.; Zhai, B.; Liao, J.; Xu, W.; Zhang, R.; Li, J.; Zhang, Y.; Chen, L.; Qian, H.; et al. Sphere-forming cell subpopulations with cancer stem cell properties in human hepatoma cell lines. BMC Gastroenterol. 2011, 11, 71. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Xia, W.; Chen, L.; Wu, C.X.; Zhang, C.C.; Olson, P.; Wang, X.Q. Synergistic antitumor effect of a γ-secretase inhibitor PF-03084014 and sorafenib in hepatocellular carcinoma. Oncotarget 2018, 9, 34996–35007. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.; Song, K.; Han, C.; Zhang, J.; Lu, L.; Chen, W.; Wu, T. Epigenetic Silencing of miRNA-34a in Human Cholangiocarcinoma via EZH2 and DNA Methylation: Impact on Regulation of Notch Pathway. Am. J. Pathol. 2017. [Google Scholar] [CrossRef] [Green Version]
- Aoki, S.; Mizuma, M.; Takahashi, Y.; Haji, Y.; Okada, R.; Abe, T.; Karasawa, H.; Tamai, K.; Okada, T.; Morikawa, T.; et al. Aberrant activation of Notch signaling in extrahepatic cholangiocarcinoma: Clinicopathological features and therapeutic potential for cancer stem cell-like properties. BMC Cancer 2016, 16, 854. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; de la Cruz, C.C.; Sayles, L.C.; Alleyne-Chin, C.; Vaka, D.; Knaak, T.D.; Bigos, M.; Xu, Y.; Hoang, C.D.; Shrager, J.B.; et al. A Rare Population of CD24+ITGB4+Notchhi Cells Drives Tumor Propagation in NSCLC and Requires Notch3 for Self-Renewal. Cancer Cell 2013, 24, 59–74. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.P.; Yang, C.J.; Huang, M.S.; Yeh, C.T.; Wu, A.T.H.; Lee, Y.C.; Lai, T.C.; Lee, C.H.; Hsiao, Y.W.; Lu, J.; et al. Cisplatin selects for multidrug-resistant CD133+ cells in lung adenocarcinoma by activating notch signaling. Cancer Res. 2013, 73, 406–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H.; Lu, W.; Zhang, Y.; Liu, H.; Wang, Z.; Shen, Y. Specific inhibition of Notch1 signaling suppresses properties of lung cancer stem cells. J. Cancer Res. Ther. 2019, 15, 1547–1552. [Google Scholar] [CrossRef]
- Sullivan, J.P.; Spinola, M.; Dodge, M.; Raso, M.G.; Behrens, C.; Gao, B.; Schuster, K.; Shao, C.; Larsen, J.E.; Sullivan, L.A.; et al. Aldehyde dehydrogenase activity selects for lung adenocarcinoma stem cells dependent on notch signaling. Cancer Res. 2010, 70, 9937–9948. [Google Scholar] [CrossRef] [Green Version]
- Mao, Z.; Liu, J.; Mao, Z.; Huang, J.; Xie, S.; Liu, T. Blocking the NOTCH pathway can inhibit the growth of CD133-positive A549 cells and sensitize to chemotherapy. Biochem. Biophys. Res. Commun. 2014, 444, 670–675. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, W.; Guo, H.; Zhang, Y.; He, Y.; Lee, S.H.; Song, X.; Li, X.; Guo, Y.; Zhao, Y.; et al. NOTCH1 signaling regulates self-renewal and platinum chemoresistance of cancer stem-like cells in human non-small cell lung cancer. Cancer Res. 2017, 77, 3082–3091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, K.A.; Wang, L.; Korkaya, H.; Chen, G.; Maillard, I.; Beer, D.G.; Kalemkerian, G.P.; Wicha, M.S. Notch pathway activity identifies cells with cancer stem cell-like properties and correlates with worse survival in lung adenocarcinoma. Clin. Cancer Res. 2013, 19, 1972–1980. [Google Scholar] [CrossRef] [Green Version]
- Arasada, R.R.; Amann, J.M.; Rahman, M.A.; Huppert, S.S.; Carbone, D.P. EGFR blockade enriches for lung cancer stem-like cells through Notch3-dependent signaling. Cancer Res. 2014, 74, 5572–5584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, S.A.; Justilien, V.; Jamieson, L.; Murray, N.R.; Fields, A.P. Protein Kinase Cι Drives a NOTCH3-dependent Stem-like Phenotype in Mutant KRAS Lung Adenocarcinoma. Cancer Cell 2016, 29, 367–378. [Google Scholar] [CrossRef] [Green Version]
- Patenaude, A.; Woerher, S.; Umlandt, P.; Wong, F.; Ibrahim, R.; Kyle, A.; Unger, S.; Fuller, M.; Parker, J.; Minchinton, A.; et al. A novel population of local pericyte precursor cells in tumor stroma that require Notch signaling for differentiation. Microvasc. Res. 2015, 101, 38–47. [Google Scholar] [CrossRef]
- Kumar, D.; Kumar, S.; Gorain, M.; Tomar, D.; Patil, H.S.; Radharani, N.N.V.; Kumar, T.V.S.; Patil, T.V.; Thulasiram, H.V.; Kundu, G.C. Notch1-MAPK Signaling Axis Regulates CD133+ Cancer Stem Cell-Mediated Melanoma Growth and Angiogenesis. J. Investig. Dermatol. 2016, 136, 2462–2474. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, N.; Almeida, A.; Partyka, K.A.; Lu, Y.; Schwan, J.V.; Lambert, K.; Rogers, M.; Robinson, W.A.; Robinson, S.E.; Applegate, A.J.; et al. Combining a GSI and BCL-2 inhibitor to overcome melanoma’s resistance to current treatments. Oncotarget 2016, 7, 84594–84607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.; Sun, B.; Zhu, D.; Zhao, X.; Sun, R.; Zhang, Y.; Zhang, D.; Dong, X.; Gu, Q.; Li, Y.; et al. Notch4+ cancer stem-like cells promote the metastatic and invasive ability of melanoma. Cancer Sci. 2016, 107, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Fan, Z.; Fang, S.; Yang, J.; Gao, T.; Simões, B.M.; Eyre, R.; Guo, W.; Clarke, R.B. Cisplatin selects for stem-like cells in osteosarcoma by activating notch signaling. Oncotarget 2016, 7, 33055–33068. [Google Scholar] [CrossRef] [PubMed]
- Dai, G.; Deng, S.; Guo, W.; Yu, L.; Yang, J.; Zhou, S.; Gao, T. Notch pathway inhibition using DAPT, a γ-secretase inhibitor (GSI), enhances the antitumor effect of cisplatin in resistant osteosarcoma. Mol. Carcinog. 2019, 58, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Vathipadiekal, V.; Saxena, D.; Mok, S.C.; Hauschka, P.V.; Ozbun, L.; Birrer, M.J. Identification of a potential ovarian cancer stem cell gene expression profile from advanced stage papillary serous ovarian cancer. PLoS ONE 2012, 7, e29079. [Google Scholar] [CrossRef] [Green Version]
- McAuliffe, S.M.; Morgan, S.L.; Wyant, G.A.; Tran, L.T.; Muto, K.W.; Chen, Y.S.; Chin, K.T.; Partridge, J.C.; Poole, B.B.; Cheng, K.H.; et al. Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy. Proc. Natl. Acad. Sci. USA 2012, 109, E2939–E2948. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.Y.; Zhang, X.L.; Du, P.; Zheng, J.H. γ-Secretase inhibitor, DAPT inhibits self-renewal and stemness maintenance of ovarian cancer stem-like cells in vitro. Chin. J. Cancer Res. 2011, 23, 140–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zhang, W.; Niu, C.; Lin, C.; Wu, X.; Jian, Y.; Li, Y.; Ye, L.; Dai, Y.; Ouyang, Y.; et al. Nuclear orphan receptor NR2F6 confers cisplatin resistance in epithelial ovarian cancer cells by activating the Notch3 signaling pathway. Int. J. Cancer 2019, 145, 1921–1934. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Jiang, B.; Xu, B.; Lu, W.; Guo, Q.; Xie, Q.; Zhang, B.; Dong, X.; Chen, D.; Wu, Y. Delta like ligand 4 induces impaired chemo-drug delivery and enhanced chemoresistance in pancreatic cancer. Cancer Lett. 2013, 330, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Palagani, V.; El Khatib, M.; Kossatz, U.; Bozko, P.; Müller, M.R.; Manns, M.P.; Krech, T.; Malek, N.P.; Plentz, R.R. Epithelial Mesenchymal Transition and Pancreatic Tumor Initiating CD44+/EpCAM+ Cells Are Inhibited by γ-Secretase Inhibitor IX. PLoS ONE 2012, 7, e46514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbuzariu, A.; Rampoldi, A.; Daley-Brown, D.S.; Candelaria, P.; Harmon, T.L.; Lipsey, C.C.; Beech, D.J.; Quarshie, A.; Ilies, G.O.; Gonzalez-Perez, R.R. Leptin-Notch signaling axis is involved in pancreatic cancer progression. Oncotarget 2017, 8, 7740–7752. [Google Scholar] [CrossRef] [Green Version]
- Capodanno, Y.; Buishand, F.O.; Pang, L.Y.; Kirpensteijn, J.; Mol, J.A.; Argyle, D.J. Notch pathway inhibition targets chemoresistant insulinoma cancer stem cells. Endocr. Relat. Cancer 2018, 25, 131–144. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Song, S.Y.; Park, J.Y. Notch pathway activation is associated with pancreatic cancer treatment failure. Pancreatology 2014, 14, 48–53. [Google Scholar] [CrossRef]
- Bailey, J.M.; Alsina, J.; Rasheed, Z.A.; McAllister, F.M.; Fu, Y.Y.; Plentz, R.; Zhang, H.; Pasricha, P.J.; Bardeesy, N.; Matsui, W.; et al. DCLK1 marks a morphologically distinct subpopulation of cells with stem cell properties in preinvasive pancreatic cancer. Gastroenterology 2014, 146, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abel, E.V.; Kim, E.J.; Wu, J.; Hynes, M.; Bednar, F.; Proctor, E.; Wang, L.; Dziubinski, M.L.; Simeone, D.M. The notch pathway is important in maintaining the cancer stem cell population in pancreatic cancer. PLoS ONE 2014, 9, e91983. [Google Scholar] [CrossRef] [PubMed]
- Mizuma, M.; Rasheed, Z.A.; Yabuuchi, S.; Omura, N.; Campbell, N.R.; De Wilde, R.F.; De Oliveira, E.; Zhang, Q.; Puig, O.; Matsui, W.; et al. The gamma secretase inhibitor MRK-003 attenuates pancreatic cancer growth in preclinical models. Mol. Cancer Ther. 2012, 11, 1999–2009. [Google Scholar] [CrossRef] [Green Version]
- Yabuuchi, S.; Pai, S.G.; Campbell, N.R.; De Wilde, R.F.; De Oliveira, E.; Korangath, P.; Streppel, M.M.; Rasheed, Z.A.; Hidalgo, M.; Maitra, A.; et al. Notch signaling pathway targeted therapy suppresses tumor progression and metastatic spread in pancreatic cancer. Cancer Lett. 2013, 335, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Ning, J.; Wakimoto, H.; Wu, S.; Wu, C.L.; Humphrey, M.R.; Rabkin, S.D.; Martuza, R.L. Oncolytic Herpes Simplex Virus and PI3K Inhibitor BKM120 Synergize to Promote Killing of Prostate Cancer Stem-like Cells. Mol. Ther.—Oncolytics 2019, 13, 58–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, F.L.F.; Marchionni, L.; Gupta, A.; Kummangal, B.A.; Schaeffer, E.M.; Ross, A.E.; Berman, D.M. HES6 promotes prostate cancer aggressiveness independently of Notch signalling. J. Cell. Mol. Med. 2015, 19, 1624–1636. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Dai, J.; Keller, J.M.; Mizokami, A.; Xia, S.; Keller, E.T. Notch pathway inhibition using PF-03084014, a γ-secretase inhibitor (GSI), enhances the antitumor effect of docetaxel in prostate cancer. Clin. Cancer Res. 2015, 21, 4619–4629. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
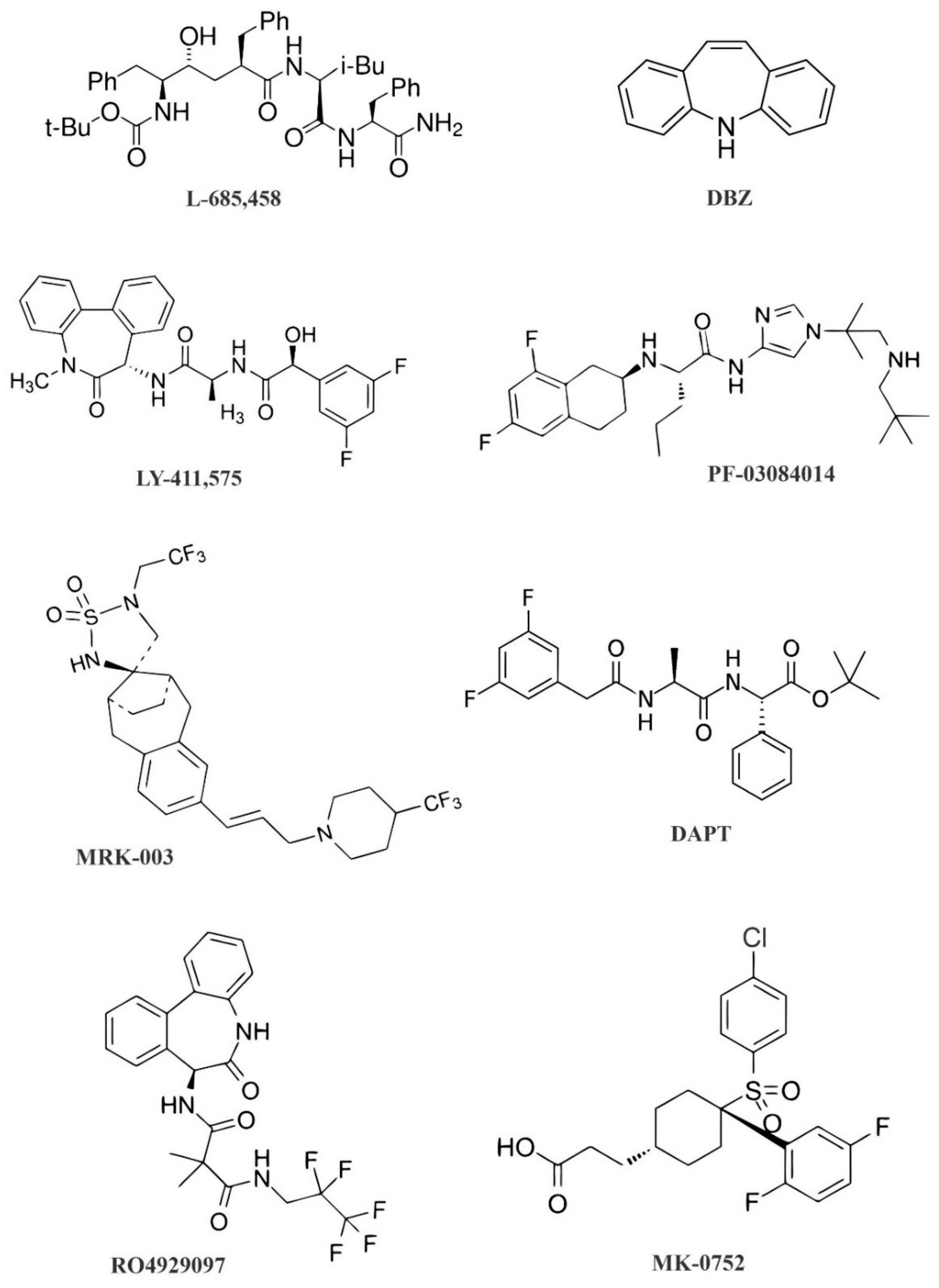{kind=link}
{kind=link}
| Cancer Type | Cell Type | Type and Properties of GSIs | Conc. | Exposure Time | Major Outcome | References |
|---|---|---|---|---|---|---|
| Adenoid cystic carcinoma | Accx11 cells | DAPT DAPT + radiation | 1–10 μM | 24–96 h | ↓SKP2 and N1ICD, ↓growth of ACC, ↓CD133+ cells, ↑p27Kip, ↑radio-sensitivity | Panaccione et al. [33] |
| Blood (Lymphoma) | Lymphoma cells | L-685,458 | 0.1–100 μM | 24 h | ↓CSCs, ↓colony formation | Wang et al. [34] |
| Blood (Lymphoma) | EL4 and A20 cells 3D cell culture | DAPT DAPT + NSC23766 | 5 μM | 24 h | ↓CSCs, ↑sensitivity to doxorubicin | Ikram et al. [35] |
| Blood (Lymphoma) | Mono-nuclear cells (MNC) | DAPT | 8–16 μM | 14 days | ↓CSCs, ↓colony formation ability, ↓proliferation | Gal et al. [36] |
| Blood (leukemia) | Tal1/Lmo2 leukemic cells | MRK-003 | 48 h | ↓leukemia-initiating cells | Tatarek et al. [37] | |
| Blood (leukemia) | DND-41, KOPT-K1, Jurkat, NB4, HL60, and OCI/AML-3 cells | Compound E Cy + Qu + Compound E | 10 μM | 1–7 days | ↓growth, ↓clonogenicity, ↓NOTCH1, ↓CSCs | Okuhashi et al. [38] |
| Blood (leukemia) | AA and HEL cell lines | DAPT GSI-XII Compound E | 20 μM (DAPT) 5 μM (GSI-XII) 10 μM (Compound E) | 1–7 days | ↓cell growth, ↓colony formation ability, ↑differentiation, ↓CSCs, ↑hemoglobin | Okuhashi et al. [39] |
| Blood (Multiple myeloma) | Human MM-CSCs | RO4929097 RO4929097 + Bruceantin | 10 µM | 24 h | ↓CSCs, ↓viability, ↓migration, ↓proliferation, ↓angiogenesis | Issa et al. [40] |
| Brain (Glioblastoma) | GSCs | DAPT | 25 μM | 7 days | ↓proliferation, ↓tumor spheres, ↓CD133 and GLAST, ↓tumor propagation | Hu et al. [41] |
| Brain (Glioma) | U87-MG and LN-22 cell lines | DAPT | 1 μM | 24 h | ↓sphere-forming capacity, ↓N1ICD and SOX2 expression | Li et al. [42] |
| Brain (Glioma) | SHG-44 cell line | DAPT | 0.5–10 μmol/L | 1–5 days | ↓proliferation, ↓CSCs, ↓CSC of CD133+ | Liu et al. [43] |
| Brain (Glioblastoma) | GBM-derived CD105+ cells | DAPT | 5 μM | 48 h | ↓transition between CD133+/CD144− and double-positive, ↓CSCs | Wang et al. [44] |
| Brain (Glioblastoma) | Human-derived GBM xenograft cells | DAPT | 1–10 µM | 3–14 days | ↓cell growth, ↓stem cell-like features, ↓CSCs | Kristoffersen et al. [45] |
| Brain (medulloblastoma) | Medulloblastoma-derived cells | DAPT | 10 μM | 8–72 h | ↓HIF-1α and hes1, ↓nestin+ cells, ↓CSCs, ↑β-III-tubulin+ cells, ↑neuronal differentiation | Pistollato et al. [46] |
| Brain (Glioma) | 029 and 036 neurosphere cultures Human-derived GBM xenograft cells | DAPT | 10 µM | 24 h | ↓brain CSCs | Kristoffersen et al. [47] |
| Brain (Glioma) | ihBTC2, SU3, SU3-5R, and C6 cells | DAPT DAPT + radiation | 2 μm | 24 h–8 days | ↑radio-sensitivity, ↑apoptosis, ↓CSCs | Yuntian et al. [48] |
| Brain (Glioma) | A172 cell line | DAPT DAPT + Gleevec + amph1D peptide | 2/5–25 μm | 24–48 h | ↓CSCs, ↑cell death | Gal et al. [49] |
| Brain (medulloblastoma) | D341 cells | DAPT DAPT + HBMEC/ D341Med | 2 μM | 48 h | ↓Bmi-1, CDK6, c-MYC, and CCND1 expression, ↓CD133+ cells, ↓CSCs | Wang et al. [50] |
| Brain (Glioma) | LN229 and U251 cell lines and primary cells isolated from GBM10 xenograft PDX models | DAPT DAPT + imatinib | 20 μM | 1–2 weeks | ↓growth inhibition, ↓GSCs | Kanabur et al. [51] |
| Brain (Glioma) | U251MG, U87MG, T98G, A172, and U373MG Adherent GBM cell lines 0308 and 0822 GBM stem cell lines | DAPT GSI-loaded MLs DAPT + INCB3619 | 12/5–100 μM | 1–6 days | ↓HES1 and HEY1, ↓YKL-40/CHI3L1, ↓CSCs | Floyd et al. [52] |
| Brain (Glioblastoma) | GSCs | L-685,458 or DAPT | 5 μM (L-685,458) 10 μM (DAPT) | 3, 7, 14 days | ↓GSCs, ↓sphere-forming cells | Park et al. [53] |
| Brain (Glioma) | U251, U87, A172, and LN18 cells | DAPT BMS-708163 RO4929097 RO4929097 + BMS-708163 | 0.5–18 μM | 72 h | ↓CSCs | Saito et al. [54] |
| Brain (Glioma) | T3359, T3691, T4105, T4302, and T4597 cells | DAPT L685,458 | 2 μM (DAPT) 0.5 μM (L685,458) | 4 h–3 weeks | ↑apoptosis, ↓growth, ↓clonogenic survival of GSCs | Wang et al. [55] |
| Brain (Glioblastoma) | GBM neurospheres | GSI-18 MRK-003 | 2 μM (GSI-18) 2–10 μM (MRK-003) | 2–5 days | ↓CSCs, ↓proliferation, ↑cell death, ↓STAT3 and AKT phosphorylation | Fan et al. [56] |
| Brain (Medulloblastoma) | DAOY, PFSK, D283Med, and D425Med cell lines | GSI-18 | 2 μmol/L | 48 h | ↓Hes1, ↓mRNA levels, ↓clonogenicity, ↓CSCs | Fan et al. [57] |
| Brain (Glioblastoma) | GBM neurosphere cultures | GSI-18 | 2–50 μM | 48 h | ↓proliferation, ↑apoptosis, ↑differentiation, ↓CSCs | Dai et al. [58] |
| Brain (Glioma) | T98G cell lines, human tumor neurospheres, and PDGF-induced glioma primary cells | MRK-003 MRK-003 + GSNO | 3 μM | 2, 6 h | ↓side population cells | Charles et al. [59] |
| Brain (Glioblastoma) | Primary GBM cell lines | MRK003 | 2 μM | 7 days | ↓neurosphere formation, ↓CSCs | Fornara et al. [60] |
| Brain (Glioblastoma) | MD13, 30R, Me83, 1123M, 528P, 157NS, and 146NS cells | MRK003 | 1–10 μM | 2–7 days | ↓viability, ↓sphere-formation capacity, ↑apoptosis, ↓Akt pathway, ↓CSCs | Tanaka et al. [61] |
| Brain (Glioma) | HSR-GBM1 and JHH520 neurosphere lines | MRK003 MRK003 + Chloroquine | 0.5–5 μM | 48–72 h | ↓proliferation, ↓CSCs ↑autophagy, ↓cell growth, ↓colony formation ability | Natsumeda et al. [62] |
| Brain (Glioma) | U87 and U251 cell lines | GSI-I with radiaton | 1–5 μmol/L | 4–72 h | ↓CSCs, ↓CD133+ cells | Lin et al. [63] |
| Brain (Glioma) | U87MG cell line | GSI-I GSI-I + TMZ/cyclopamine | 5 µmol/L | 5 days | ↓CD133+ cells, ↓CSCs, ↑TMZ therapeutic effect | Ulasov et al. [64] |
| Brain (Glioma) | PIGPCs | MK-003 MK-003 + TPA | 10 μM | 24 h | ↓stem cell markers, ↓glioma primary cultures, ↓CSCs | Pietras et al. [65] |
| Brain (Glioma) | UAB-1005, 1051, 1027A, and 1079 cell lines | RO4929097 RO4929097 + Farnesyltransferase | 20–500 nM | 24–72 h | ↓AKT, ↓cell-cycle progression, ↑radio-sensitizing, ↓CSCs | Ma et al. [66] |
| Brain (Glioblastoma) | GSCs | MK0752 | 25 µM | 1–7 days | ↓self-renewal ability, ↓proliferation, ↓GSCs, ↓secondary neurosphere | Hu et al. [67] |
| Brain (Glioblastoma) | GBM neurosphere cultures | Compound E | - | - | ↓CSCs | Dai et al. [68] |
| Brain (Glioma) | U87 cells | GSI-II | 0.2 µg | 20 days | ↓CD133, ↓proliferation, ↓CSCs | Ding et al. [69] |
| Breast | MCF-7, MDA-MB-468, and MDA-MB-231 cells | DAPT | 10 μM | 12–and/or 24-h | ↓CSCs, ↓ILK-induced Notch activation | Hsu et al. [70] |
| Breast | Human TNBC, Hs578T, and MDA-MB-231 cell lines | DAPT | 1–5 μm | 5–12 days | ↓BCSCs, ↓sphere formation capacity, ↓self-renewal ability, ↓NICD, SCUBE2, HES1 and jagged 1 | Chen et al. [71] |
| Breast | ZR-75–1, MCF-7, T-47D, JIMT, SK-BR-3, and MDA-MB-231 cell lines | DAPT | 2 and 5 μM | 12–24 h | ↓mammosphere formation, ↓CD44ICD cleavage, ↓CSCs | Cho et al. [72] |
| Breast | HCC38 and HCC1806 stem cell lines | DAPT | 10–40 µM | 1–14 days | ↓BCSCs, ↓mammosphere-forming ability, ↓cell invasion, ↓cell proliferation, ↑cell death | Li et al. [73] |
| Breast | Trp53−/−mammospheres and TM40A-let7s-p53KD mammospheres | DAPT | 5 μM | 7 days | ↓mammary stem/progenitor cells | Tao et al. [74] |
| Breast | Athymic nude nu/nu mice bearing HCC1806 cells | DAPT | 5 μmol/L | 24–48 h | ↓CSCs, ↓percentage of CD44hi/CD24lo cells, ↓colony formation ability | McGowan et al. [75] |
| Breast | MCF7 and MDA231, MDA231LM, and MDA231BoM cell lines | DAPT | 10–20 μM | 24–72 h | ↓Akt, ↓CSCs | Xing et al. [76] |
| Breast | CAF61a, HCC1937, THP-1, MDA-MB-231 (1833), MDA-MB-436, MDA-MB-231, Hs578T, MDA-MB-157, SKBR3, T47D, MCF7, HCC70, MDA-MB-468 cells | DAPT RT + DAPT | 10 μM | 48 h | ↓mammosphere-forming ability, ↓CD44+CD24low+ TRCs, ↑TIC gene expression signature, ↓CSCs | Boelens et al. [77] |
| Breast | SUM225 and MCF10DCIS cell lines | DAPT DAPT + lapatinib/gefitinib | 0.1, 5, 10, 20 µM | 5, 7, 21 days | ↓acini size, ↓mammosphere formation, ↓ErbB1/2 | Farnie et al. [78] |
| Breast | T47D, MDA-MB-231, MCF7, BT474 cells | DAPT DAPT + Gefitinib | 10 μM | 24–48 h | ↓CSCs, ↓oestrogen effect | Harrison et al. [79] |
| Breast | MCF-7 and T47D cell lines | DAPT DAPT + DSL/ Tamoxifen | 0/5–1 μM | 24 h | ↓tumoursphere growth, ↓ER-α promoter activity, ↓CSCs, ↓tamoxifen sensitivity | Buckley et al. [80] |
| Breast | MCF-7 and MDA-MB-231 cells | DAPT DAPT + 6-shogaol | 25–50 μM | 1–7 days | ↓CSCs, ↓proliferation, ↓colony formation capacity, ↓number of spheroids | Ray et al. [81] |
| Breast | MDA-MB-231 and MCF7 cells | DAPT DAPT loaded glucose-functionalized MSNs | 1 μg | 24 h | ↓CSCs, ↓ALDH activity | Mamaeva et al. [82] |
| Breast | MCF-7, T47D, and MDA-MB-231 cell lines | GSI-XVII | 5 μM | 30 min | ↓self-renewal, ↓CSCs, ↓primary sphere formation | Phillips et al. [83] |
| Breast | SUM159, MCF-7, and T47D cells | GSI-XVII GSI-XVII + radiation | 5 μM | 1–4 days | ↓CSCs, ↓DLL3, Notch2, Jagged1, and DLL1 gene expression | Lagadec et al. [84] |
| Breast | HCC1937, HCC1806, MCF7, T47D, SUM149, and SUM159 cells | GSI-I | 1–10 μM | 24 h | ↓BCSCs | Luo et al. [85] |
| Breast | MDA-MB-453, HCC1954 and MCF-7 cells | MRK-003 MRK-003 + lapatinib/trastuzumab | 5 μM | 7 days | ↓CSCs, ↓mammosphere formation, ↓proliferation of bulk HER2+ HCC1954 cells | Shah et al. [86] |
| Breast | MCF7, MDA-MB 436, MDA-MB 231, ZR-75-1, ZR-75-30, and T47D cell lines | MRK-003 MRK-003 + docetaxel | 10 mmol/L | 1–3 weeks | ↓breast CSCs, ↓self-renewal | D’Angelo et al. [87] |
| Breast | Primary tumor cells Mammospheres | MRK-003 | 0.01–10 μM | 2–4 days | ↓proliferation, ↓self-renewal ability, ↑apoptosis, ↑differentiation, ↓CSCs | Kondratyev et al. [88] |
| Breast | MDA-MB231, MDA-MB231BrM, CN34, and CN34BrM cells | DAPT | 5–10 µM | 48–72 h | ↓CSCs, ↓HES5 | Xing et al. [89] |
| Breast | ZR-75, MCF-7, and MDA-231 cell lines | compound E DAPT DAPT +AD-01 | 0.025–1.25 μmol/L (compound E) 10 μmol/L (DAPT) | 72 h | ↓BCSCs, ↓MSFE | McClements et al. [90] |
| Breast | MCF7, MDA-MB-231, and BT474 cells | DAPT DBZ | 10 μmol/L | 3–7 days | ↓ESA+/CD44+/CD24low cells, N1-ICD, ↓HEY2 and HES1, ↓CSCs | Harrison et al. [91] |
| Breast | MCF7, T47D-A18, T47D-C42, BT474, and SKBR3 cells | MRK003 GSI-I LY-411,575 | GSI-I (0.5 μM), MRK003 (10–20 μM), LY-411, 575 (25–50 μM) | 24–48 h 12–28 days | ↓CSCs, ↓mammosphere formation | Grudzien et al. [92] |
| Breast | SUM149 and SUM159, MCF-7, MDA-MB-231, and HCC1954 cell lines | RO4929097 | 10 μM | 7 days | ↓BCSCs | Wang et al. [93] |
| Breast | MDA-MB-231-luc and MCF-10A cells | DAPT | 5–10 µM | 1–12 days | ↓CSCs, ↓N1-ICD, ↓Sox2, ↓sphere formation ability | Azzam et al. [94] |
| Breast | SUM149 and SUM190 cells | RO4929097 RO4929097 + radiation | 0.1 nM–10 μM | 24–14 days | ↓Hey1, HeyL, and Hes1, ↓colony-forming capacity, ↓CSCs, ↓TNF-α, IL-8, and IL-6, ↑sensitivity to ionizing radiation | G. Debeb et al. [87] |
| Cholangiocarcinoma | CCLP1, SG231, HUCCT1, and TFK1 cells | DAPT | 10–40 μM | 24–72 h 10–14 days | ↓ growth, ↓colony formation, ↓CSCs | Kwon et al. [117] |
| Cholangiocarcinoma | HuCCT1, TFK-1, and RBE cell lines | DAPT DAPT + GEM | 20–40 μM | 1–4 days | ↓CD24+CD44+ cells, ↓colony-forming capacity, ↓CSCs | Aoki et al. [118] |
| Colorectal | IEC-6/KRAS G12V cells | DAPT | 10–20 μmol/L | 24 h | ↓CSCs, ↓Hes1 | Feng et al. [96] |
| Colorectal | CCSC cells | DAPT | ↓CCSCs, ↓symmetric CCSC-CCSC division, ↓asymmetric division, ↑non-CCSCs | Bu et al. [98] | ||
| Colorectal | HCT-116 cell line | DAPT | 10 μM | 1–10 days | ↓CSCs, ↓Smad-3, Jagged-1, and CD44, ↓Slug | Fender et al. [99] |
| Colorectal | Caco-2 and SW620 cell lines | DAPT JLK6 | 2.5–10 µM (JLK6) 2.5–30 µM (DAPT) | 1 h–14 days | ↓colosphere formation, ↓CSCs | Moon et al. [100] |
| Colorectal | HCT116 cells | L-685,458 | ↓CSCs, ↓NICD upregulation, ↓Aldefluor-positive cell population | Lu et al. [101] | ||
| Gastric | Human gastric cancer cell lines, corpus gland cultures, and Mist1+ stem cells | DAPT | 25 μM | 10 days | ↓Mist1+ stem cell proliferation | Hayakawa et al. [103] |
| Gastric | MKN45 cell line | DAPT | 2.5–15 µM | 24–96 h | ↓CSCs, ↓proliferation, ↓CD44+ cells | Barat et al. [104] |
| Gastric | MKN-45 cells | DAPT | 10 µM | 72 h | ↓CSCs, ↓EMT markers, ↓Hes1, ↓proliferation | Li et al. [105] |
| Gastric | GI2, CS12, MKN45 cells | DAPT | 5 μM | 24h | ↓sphere-forming ability, ↓sphere size and number, ↓stemness genes and CD133 | Hong et al. [106] |
| Gastric | SC-6-JCK, SH-10-TC, MKN74, and MKN45 cells | DAPT + Cisplatin | 25–50 µM | 24–72 h | ↓CSCs, ↓cell viability, ↓CD44highLgr-5high population | Kato et al. [107] |
| Head and neck | SAS, OECM1, and FADU cells | DAPT DAPT + Cetuximab | 100 μM | 1–14 days | ↓KLF4+/CD44+ cells | Chen et al. [108] |
| Head and neck | OE33, OE19, FLO1, JH-EsoAd1 cells | DAPT DAPT + 5-FU | 1–10 μM | 1–7 days | ↓CSCs, ↓HES1 expression, ↓NICD, ↑apoptosis, ↓proliferation, ↑sensitivity to chemotherapeutic agents | Wang et al. [109] |
| Head and neck | FLO-1, SKGT-4, BE3, and OE33 cells | Compound E | 500 nM–5 μM | 72 h | ↓CSCs, ↓proliferation | Mendelson et al. [110] |
| Head and neck | CAL27 and FaDu cell lines | DAPT DAPT + chemotherapeutic agents | 5–10 μM | 1–14 days | ↓CSCs, ↓CSCs markers | Zhao et al. [111] |
| Head and neck | AW13516, NT8e, CAL27, DOK cells | Compound E | 5–10 μM | 20 h–21 days | ↓CSCs, ↓spheroid-forming ability, ↓survival, migration, and transformation of the HNSCC cells | Upadhyay et al. [112] |
| Liver | Huh7, Huh6, HepG2, Hep3B, PLC/PRF/5, SKHep1, HLE, and THLE-5b cells | L-685,485 DAPT | 10 μmol/L | 24–168 h | ↓cell growth EpCAM+ cells, ↓CSCs, ↓HES1 | Kawaguchi et al. [113] |
| Liver | Hep3B, Huh7, PLC, MHCC97L, and MHCC97H cells Liver cancer spheres | PF-03084014 | 0.25–2 μM | 1–14 days | ↓CSCs, ↓proliferation, ↓self-renewal ability | Wu et al. [114] |
| Liver | MHCC97H, PLC/PRF/5, and HepG2 cells | MRK003 | 10 μM | 1–7 days | ↓CSCs, ↓sphere formation ability | Cao et al. [115] |
| Liver | MHCC97H and MHCC97L cells | PF-03084014 PF-03084014 + sorafenib | 0.1–0.25 μM | 24 h | ↓CSCs, ↓self-renewal ability, ↓proliferation, ↓spheroid formation | Yang et al. [116] |
| Lung | NSCLC tumor-propagating Cells | DAPT | 100 μm | 1–2 weeks | ↓self-renewal, ↓ tumor propagation, ↓Hes1 and Hey1, ↓CSCs | Zheng et al. [119] |
| Lung | A549, H460, PC9, H1299, and H661 cells | DAPT DAPT + Cisplatin | 10 μmol/L | 30 min–48 h | ↓CD133+ cells, ↑sensitivity to doxorubicin and paclitaxel, ↓Hes-1 | Liu et al. [120] |
| Lung | A549 human lung adenocarcinoma cell line | DAPT | 25–75 μM | 48 h | ↓LCSCs, ↓CD44+/CD24− cells | Cai et al. [121] |
| Lung | NSCLC and SCLC cells | DAPT | 25 μmol/L | 3–14 days | ↓ALDH+ cancer cells, ↓proliferation, ↓clonogenicity, ↑cell-cycle arrest, ↓CSCs | Sullivan et al. [122] |
| Lung | A549 cell line | DAPT CDDP + DAPT | 2 μM | 48 h | ↑cell-cycle arrest, ↓CSCs, ↓proliferation of CD133+ and CD133− cells | Liu et al. [123] |
| Lung | LCSCs and NSCLC cells | RO4929097 | 1–10 µM | 24–48 h | ↓p-STAT3,↓self-renewal, ↓LCSCs, ↓HES1 | Zhang et al. [124] |
| Lung | H1299, H441, H460, H358, and A549 cells | MRK-003 and MRK-003 + Docetaxel | 5–20 μM | 24–48 h | ↓CSCs, ↓sphere formation ability, ↓self-renewal, ↓NICD2 | Hassan et al. [125] |
| Lung | HCC2429, HCC827, H358, and HCC4006 cells | PF-03084014 PF-03084014 + erlotinib | 1 μM | 7 days | ↓CSCs, ↓ALDH+ cells, ↓EGFR | Arasada et al. [126] |
| Lung | H23, A549, H358, H661, H1437, H1299, H1703, H520, and ChagoK1 cells | DBZ DBZ + ANF | 5 μM | 3–30 days | ↓oncosphere growth, ↓cell viability, ↓soft agar growth | Ali et al. [127] |
| Lung | LLC cells | DFPAA | - | 7 days | ↓NG2+ cells, ↓CSCs | Patenaude et al. [128] |
| Melanoma | B16F10 cells | DFPAA | - | 7 days | ↓NG2+ cells, ↓CSCs | Patenaude et al. [128] |
| Melanoma | B16F10 and B16F1, SK-MEL-28, A375, and SK-MEL-2 cell lines | DAPT and L-685,458 | 5–15 μM | 12 h–4 weeks | ↓CSCs, ↓CD133, ↓metastasis, ↓melanoma growth, ↓angiogenesis, ↓CD133-dependent MAPK signaling | Kumar et al. [129] |
| Melanoma | WM852c, SK-MEL 28, 1205Lu, HT144, A375, and 451Lu cells | GSI-I GSI-I + ABT-737 | 0.83 μM | 24–48 h | ↓primary sphere formation, ↓ALDH+ cells, ↓cell viability, ↑apoptosis of the non-MICs, ↓self-renewability | Mukherjee et al. [130] |
| Melanoma | B16F10, A375, A875, MUM-2C, and MUM-2B cell lines Tumorospheres and the multicellular tumor spheroid (MTS) model | DAPT | 10 μM | 24–72 h | ↓CSCs, ↑E-cadherin, ↓VE-cadherin and Twist1, ↓metastasis | Lin et al. [131] |
| Osteosarcoma | U2OS, 143B, and MG63 cell lines | DAPT RO4929097 | 20 μM | 24 h | ↓spheroid-forming ability, ↓CSCs | Yu et al. [132] |
| Osteosarcoma | 143B, U2OS, and MG-63 cell lines | DAPT DAPT + Cisplatin | 5–50 μM | 24–72 h | ↓OSCs, ↓proliferation, ↓motility, ↑apoptosis, ↑cell-cycle arrest, ↑platinum-sensitivity, ↓ERK and AKT | Dai et al. [133] |
| Ovarian | SKOV3, A224, OVCAR-3, and UCI-107 cell lines | DAPT | 10–20 μg | 8 days | ↓Colony-formation, ↓SP cells | Vathipadiekal et al. [134] |
| Ovarian | 4412, 4306, OVCAR5, PA-1, OVCAR3, IGROV1, A2780, SKOV3, and OV2008 cells | GSI-I GSI-I + platinum | 1–10 μM | 1–3 days | ↑tumor platinum-sensitivity, ↓CSCs, ↑cell-cycle arrest, apoptosis, and DNA-damage | McAuliffe et al. [135] |
| Ovarian | SKOV3 and HO8910 cell lines | DAPT | 1–20 μg/mL | 1–3 days | ↓self-renewal ability, ↓proliferation, ↓CSCs, ↓OCSCs-specific surface markers expression, ↓Sox2 and Oct4 | Jiang et al. [136] |
| Ovarian | OVCAR3 and A2780 cells | RO4929097 RO4929097 + CDDP | 10 μM 10 mg/kg | 24 h | ↓proliferation, ↑apoptosis, ↓CSCs | Li et al. [137] |
| Pancreatic | Bxpc-3 and Panc-1 cell lines | DAPT | 1 and 10 μM | 48 h | ↓CD133+, ↓proliferation, ↓CSCs, ↓chemo-resistance | Kang et al. [138] |
| Pancreatic | BxPC3, KP3 cells | DAPT | 2.5–10 µM | 48–96 h | ↑apoptosis, ↓CSCs, ↓EMT | Palagani et al. [139] |
| Pancreatic | Panc-1, BxPC-3, MiaPaCa-2, AsPC-1 cell lines | DAPT DAPT + leptin | 20 µM | 1–10 days | ↓PCSCs, ↓proliferation, ↓leptin-induced CD133+ and ALDH+ cells, ↓tumorsphere formation | Harbuzariu et al. [140] |
| Pancreatic | CM cell line | DAPT DAPT + 5-FU | 2–80 μg/mL | 24–48 h | ↓clonogenicity, ↑sensitivity to 5-FU, ↓CSC-enriched spheres | Capodanno et al. [141] |
| Pancreatic | BxPC3 and HPAC cells | DAPT Gem + DAPT | 20 μM | 72 h | ↓CSCs, ↓CD24+CD44+ cells, ↓pAKT, Hes1, and β-catenin expression, ↓invasion, ↓migration | Lee et al. [142] |
| Pancreatic | DCLKHI/AcTubHI cells | DAPTMRK-300 | DAPT (10–100 nM) MRK-300 (0.72–5 μM) | 3–4 days | ↓CSCs, ↓AcTubHI cells, ↓PanIN progression, ↓mPanIN epithelial cells expressing Dclk1 | Bailey et al. [143] |
| Pancreatic | Pancreatic cancer cells | MK-0752 GSI + gemcitabine | 8 μM | 24–72 h | ↓CSCs, ↓tumorsphere formation, ↑apoptosis | Abel et al. [144] |
| Pancreatic | Pa03C, Pa14C, Pa16C, and Pa29C cells | MRK-003 MRK-003 + GEM | 2–5 μM | 48 h | ↓CSCs, ↓NICD, ↓colony-forming capacity, ↑apoptosis | Mizuma et al. [145] |
| Prostate | DU145 and TRAMP-C2 cell lines and PCSCs | PF-03084014 | 0/01–100 μM | 6 days | ↓PCSCs | Wang et al. [147] |
| Prostate | VCaP and LnCaP96 cell lines | DAPT | 1 nM–400 μM | 48–96 h | ↓CSCs, ↓NICD1 | Carvalho et al. [148] |
| Prostate | Du145, PC3, Du145R, and PC3R cells | PF-03084014 PF-03084014 + docetaxel | 0.1–10 μM | 48 h | ↓CSCs, ↑apoptosis, ↓epithelial to mesenchymal transition, ↓(cyclin E; EGFR, PI3K/AKT, NF-κB, and NF-κB pathway; BCL-XL, BCL-2) | Cui et al. [149] |
| Cancer Type | Animal Model | Type and Properties of GSIs | Dose | Exposure time | Major Outcome | References |
|---|---|---|---|---|---|---|
| Adenoid cystic carcinoma | Athymic NCr-nu/nu mice bearing Accx11 cells | DAPT DAPT + radiation | 50 mg/kg | 35 days | ↓SKP2 and N1ICD, ↓growth of ACC, ↓CD133+ cells, ↑p27Kip, ↑radio-sensitivity | Panaccione et al. [33] |
| Blood (leukemia) | Tal1/Lmo2 transgenic mice bearing Tal1/Lmo2 leukemic cells | MRK-003 | 150 mg/kg | 1–3 weeks | ↓leukemia-initiating cells | Tatarek et al. [37] |
| Brain (Glioma) | Immunocompromised mice bearing GBM neurosphere cells | DAPT | 10 µM | 7 days | ↓brain CSCs | Kristoffersen et al. [47] |
| Brain (medulloblastoma) | BALB/c nude mice xenograft models | DAPT DAPT + HBMEC/D341Med | 40 days | ↓tumor size and volume | Wang et al. [50] | |
| Brain (Glioma) | Balb/c mice bearing GBM stem cells | DAPT GSI-loaded MLs DAPT + INCB3619 | 0.5 mg/mL | 3 weeks | ↑survival rate, ↓CSCs | Floyd et al. [52] |
| Brain (Glioma) | Nude (nu/nu) mice bearing GICT25 cells | DAPT BMS-708163 RO4929097 RO4929097 + BMS-708163 | 10 mg/kg (RO4929097) 20 mg/kg (BKM120) | 5 weeks | ↓tumor growth, ↑survival rate | Saito et al. [54] |
| Brain (Glioma) | Flank and Intracranial Xenograft tumors nude mice bearing GBM neurosphere cells | GSI-18 MRK-003 | 2 μM (GSI-18) 2–10 μM (MRK-003) | 6 weeks | ↓tumor growth, ↑survival rate | Fan et al. [56] |
| Brain (Medulloblastoma) | Athymic (nude) mice tumor xenografts | GSI-18 | 0/5 mg | 5 days | ↓clonogenicity, ↓tumor growth | Fan et al. [57] |
| Brain (Glioma) | Subcutaneous Xenografts Athymic Nude Mice bearing U251 cells | GSI-I with radiaton | 2.5 mmol/L | 2–4 weeks | ↓tumor growth | Lin et al. [63] |
| Brain (Glioma) | Immunocompromised mice bearing U87-MG cells | GSI-I GSI-I + TMZ/cyclopamine | − | − | ↓CD133+ cells, ↓CSCs, ↑TMZ therapeutic effect | Ulasov et al. [64] |
| Brain (Glioma) | T4302, T4105, and T4597 xenograft tumors Athymic nude mice bearing T4105 CD133+ cells | RO4929097 RO4929097 + Farnesyltransferase | 30 mg/kg | 5–20 days | ↑radio-sensitizing, ↓tumor growth | Ma et al. [66] |
| Breast | Athymic nude nu/nu mice bearing HCC1806 cells | DAPT | 10–40 µM | 7–21 days | ↓tumor formation | Li et al. [73] |
| Breast | Athymic nude nu/nu mice bearing 231-BR cells | DAPT | 8 mg/kg | 14 days | ↓micro- and macro-metastases | McGowan et al. [75] |
| Breast | Nude mice bearing MDA231BoM cells | DAPT | − | − | ↓invasion | Xing et al. [76] |
| Breast | Nude mice bearing MDA-MB-231 1833 cells | DAPT RT + DAPT | 10 mg/kg | 0–20 days | ↓tumor growth | Boelens et al. [77] |
| Breast | NSG mice bearing breast CSCs | DAPT DAPT + Gefitinib | ↓Oestrogen effect | Harrison et al. [79] | ||
| Breast | Chicken eggs | DAPT DAPT loaded glucose-functionalized MSNs | 5 μg/mL | 5 days | ↓number of cancer cells per mg/tissue | Mamaeva et al. [82] |
| Breast | NOD/SCID mice bearing Sum159 and MCF7 cells | MRK-003 MRK-003 + docetaxel | 75 mg/kg | 3–10 weeks | ↓breast CSCs, ↓self-renewal, ↓tumor initiation ability | D’Angelo et al. [87] |
| Breast | FVB/N mice bearing mammospheres | MRK-003 | 150 mg/kg | 2 weeks | ↓viability, ↓reduced tumor-resident TIC | Kondratyev et al. [88] |
| Breast | NOD/SCID mouse bearing MDA-MB231, MDA-MB231BrM, CN34, and CN34BrM cells | Compound E | 10 mg/kg | 4 weeks | ↓growth, ↓metastasis | Xing et al. [89] |
| Breast | Athymic nude mice bearing MCF7, MDA-MB-231, and BT474 cells | DBZ | 1 mg/mL | 18–28 days | ↓tumor size and volume, ↑mice latency | Harrison et al. [91] |
| Breast | Nude mice bearing SUM149 cells | MK-0752 | 25 μM and 25 mg/kg | 10 weeks | ↓tumor growth | Wang et al. [93] |
| Breast | SCID/Beige mice bearing human tumorgrafts | MK-0752 MK-0752 + Docetaxel | 100 mg/kg | 3–21 days | ↓primary and secondary MSFE, ↓ALDH+ and CD44+/CD24− subpopulations, ↓NICD, ↓Hes1, Hey1, Hes5, and myc, ↓tumor growth, ↓BCSCs | Schott et al. [31] |
| Breast | Balb/C nude mice bearing MDA-MB-231-luc cells | RO4920927 | 30 mg/kg/day | 2 weeks | ↓tumor growth | Azzam et al. [94] |
| Cholangiocarcinoma | (NOD/SCID) female mice bearing HuCCT1, TFK-1 and RBE cell lines | DAPT DAPT + GEM | 40 μM | 10 days | ↓mice tumorigenicity, ↓viability | Aoki et al. [118] |
| Colorectal | Lgr5-EGFP-creERT2 transgenic mouse bearing colorectal cancer cells | DAPT | − | − | ↓Lgr5-GFP+ ISC, ↓Ascl2 levels, ↓CSCs | Bu et al. [97] |
| Colorectal | athymic (nu+/nu+) mice bearing CRC cells | PF-03084014 PF-03084014 + irinotecan | 125 mg/kg | 28 days | ↓tumor recurrence, ↓tumor growth, ↓ALDH+ population, ↓CSCs | Arcaroli et al. [102] |
| Gastric | Mist1- CreERT2;R26-mTmG mice bearing gastric cancer cells | DBZ | 30 μmol/kg | 14 days | ↓corpus organoid growth | Hayakawa et al. [103] |
| Gastric | Nude mice bearing MKN45 cells | DAPT | 10 mg/kg/body weight | 1 week | ↓migration, ↓invasion | Barat et al. [104] |
| Gastric | Nude mice bearing MKN-45 cells | DAPT | 10 mg/kg/body weight | 5 weeks | ↓tumor growth, ↓invasion | Li et al. [105] |
| Head and neck | Balb/c Nude mice bearing, SAS, OECM1, and FADU cells | DAPT DAPT + Cetuximab | 100 mg/kg | 6 weeks | ↓viability, ↓tumor growth | Chen et al. [108] |
| Head and neck | Immunocompro-mised mice bearing esophageal adenocarcinoma cells | DAPT DAPT + 5-FU | 20 mg/kg | 2–10 weeks | ↓tumor growth↑sensitivity to chemotherapeutic agents | Wang et al. [109] |
| Head and neck | BALB/c nude mice bearing CAL27 or SCC9 cells | DAPT DAPT + chemotherapeutic agents | 10–20 mg/kg | 2 weeks | ↓tumor self-renewal capacity | Zhao et al. [111] |
| Liver | NOD-SCID mouse bearing Huh7 cells | L-685,485 DAPT | 5 mg/kg (L-685,458) 20 mg/kg (DAPT) | 2,6 weeks | ↓tumor growth | Kawaguchi et al. [113] |
| Liver | Nude or SCID-beige mice bearing MHCC97H and MHCC97L cells | PF-03084014 | 100 mg/kg | 4 weeks | ↓tumor metastasis | Wu et al. [114] |
| Liver | SCID mice bearing 97H spheroid-derived cancer cells | PF-03084014 PF-03084014 + sorafenib | 100 mg/kg/day | 2–4 weeks | ↓spheroid formation | Yang et al. [116] |
| Lung | Nude mice bearing H460 cells | DAPT DAPT + Cisplatin | 2 mg/kg | 4 days | ↑sensitivity to doxorubicin and paclitaxel | Liu et al. [120] |
| Lung | NSG mice bearing LCSCs and NSCLC cells | RO4929097 | 5 mg/kg | 10 days | ↑platinum sensitivity | Zhang et al. [124] |
| Lung | NOD/SCID mice bearing H1299 cells | MRK-003 and MRK-003 + Docetaxel | - | 3 weeks | ↓tumorigenicity | Hassan et al. [125] |
| Lung | Immunodeficient mice bearing A549 cells | DBZ DBZ + ANF | 200 μg/kg | 8 weeks | ↓tumor growth | Ali et al. [127] |
| Melanoma | NCRNU nude mice bearing HT144 and WM852c cells | GSI-I GSI-I + ABT-737 | 0.83 μM | 21 days | ↓tumor-initiating capacity | Mukherjee et al. [130] |
| Osteosarcoma | NOD/SCID mice bearing 143B cells | DAPT | 10 mg/kg/d | 2 weeks | ↓tumor recurrence | Yu et al. [132] |
| Osteosarcoma | BALB/c-nu/nu nude mice bearing 143B cells | DAPT DAPT + Cisplatin | 8–10 mg/kg/d | 5 weeks | ↑platinum-sensitivity, ↓metastasis, ↓tumor growth | Dai et al. [133] |
| Ovarian | Balb/C athymic mice bearing SKOV3-SP and MP cells | DAPT | 5 mg/mL | 8 weeks | ↓Colony-formation, ↓SP cells | Vathipadiekal et al. [134] |
| Ovarian | SCID mouse bearing PA-1/luc, OVCAR5/luc, and SKOV3/luc cells Tumor xenografts | GSI-I GSI-I + platinum | 5 mg/kg | 18 days | ↑tumor platinum-sensitivity | McAuliffe et al. [135] |
| Ovarian | BALB/c nude mice bearing OVCAR3 and A2780 cells | RO4929097 RO4929097 + CDDP | 10 mg/kg | 6 weeks | ↓tumor growth, ↓tumor volume | Li et al. [137] |
| Pancreatic | Nude mice bearing BxPC3, KP3 cells | DAPT | 10 mg/kg/body weight | 5 weeks | ↓tumorigenesis | Palagani et al. [139] |
| Pancreatic | Chicken eggs | DAPT DAPT + 5-FU | 2–80 μg/mL | 11 days | ↓tumourigenicity, ↑sensitivity to 5-FU | Capodanno et al. [141] |
| Pancreatic | KCPdx1, KPCPdx1, and KCiMist1 mice bearing pancreatic cancer cells | MRK-300 | 100 mg/kg | 11–13 weeks | ↓tumorigenesis, ↓Dclk1-expressing cells | Bailey et al. [143] |
| Pancreatic | NOD/SCID mice bearing pancreatic cancer cells | RO4929097 GSI + gemcitabine | 30 mg/kg | 5 days | ↓tumor growth | Abel et al. [144] |
| Pancreatic | athymic nude mice bearing Pa03C, Pa14C, Pa16C, and Pa29C cells | MRK-003 MRK-003 + GEM | 150 mg/kg | 3 weeks | ↓tumor cell proliferation, ↑intratumoral necrosis | Mizuma et al. [145] |
| Prostate | nu/nu athymic mice bearing Panc215, Panc266, Panc354, and Panc265 xenografts | PF-03084014 PF-03084014 + GEM | 150 mg/kg | 4 weeks | ↓NICD, ↓Hes-1 and Hey-1, ↓CSCs, ↓angiogenesis, ↓proliferation, ↓tumor growth, ↓metastasis, ↑apoptosis, ↓angiogenesis, ↑tumor regression | Yabuuchi et al. [146] |
| Prostate | (NOD/SCID) mice bearing Du145, Du145R, PC3, and PC3R cells | PF-03084014 PF-03084014 + docetaxel | 150 mg/kg | 4 weeks | ↓tumor growth | Cui et al. [149] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghanbari-Movahed, M.; Ghanbari-Movahed, Z.; Momtaz, S.; Kilpatrick, K.L.; Farzaei, M.H.; Bishayee, A. Unlocking the Secrets of Cancer Stem Cells with γ-Secretase Inhibitors: A Novel Anticancer Strategy. Molecules 2021, 26, 972. https://doi.org/10.3390/molecules26040972
Ghanbari-Movahed M, Ghanbari-Movahed Z, Momtaz S, Kilpatrick KL, Farzaei MH, Bishayee A. Unlocking the Secrets of Cancer Stem Cells with γ-Secretase Inhibitors: A Novel Anticancer Strategy. Molecules. 2021; 26(4):972. https://doi.org/10.3390/molecules26040972
Chicago/Turabian StyleGhanbari-Movahed, Maryam, Zahra Ghanbari-Movahed, Saeideh Momtaz, Kaitlyn L. Kilpatrick, Mohammad Hosein Farzaei, and Anupam Bishayee. 2021. "Unlocking the Secrets of Cancer Stem Cells with γ-Secretase Inhibitors: A Novel Anticancer Strategy" Molecules 26, no. 4: 972. https://doi.org/10.3390/molecules26040972