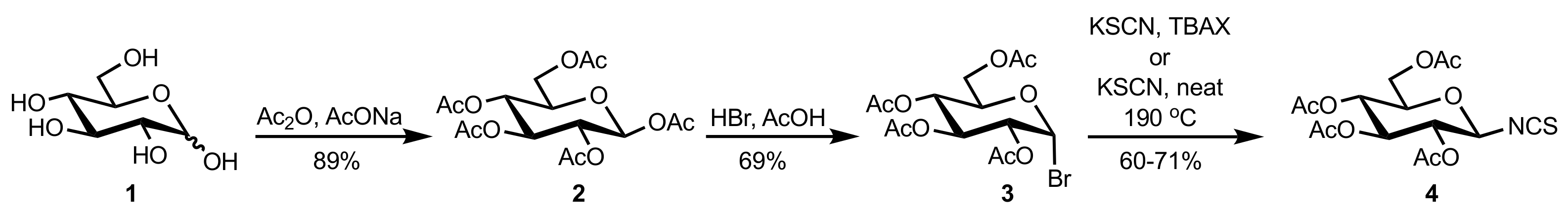
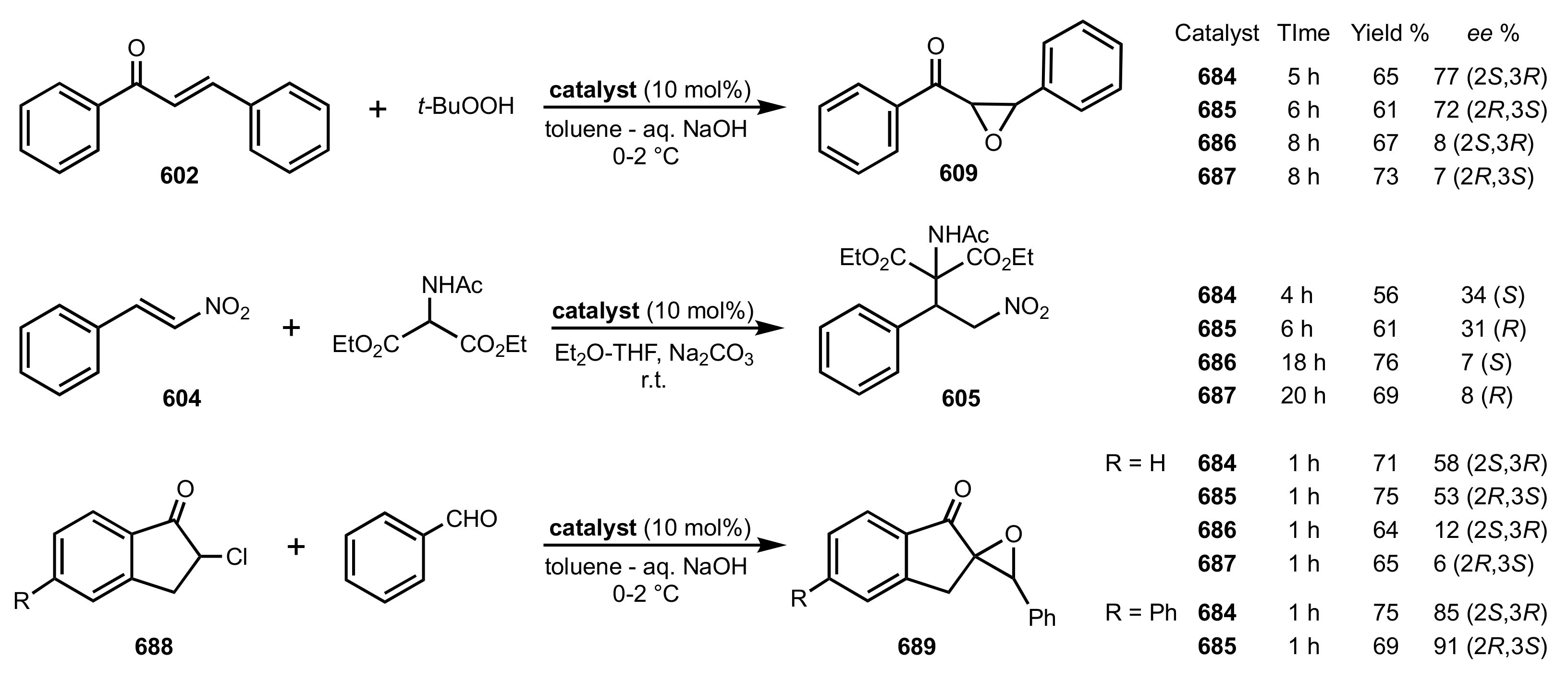
Scheme 1.
The preparation of d-glucopyranosyl isothiocyanate.
Scheme 1.
The preparation of d-glucopyranosyl isothiocyanate.
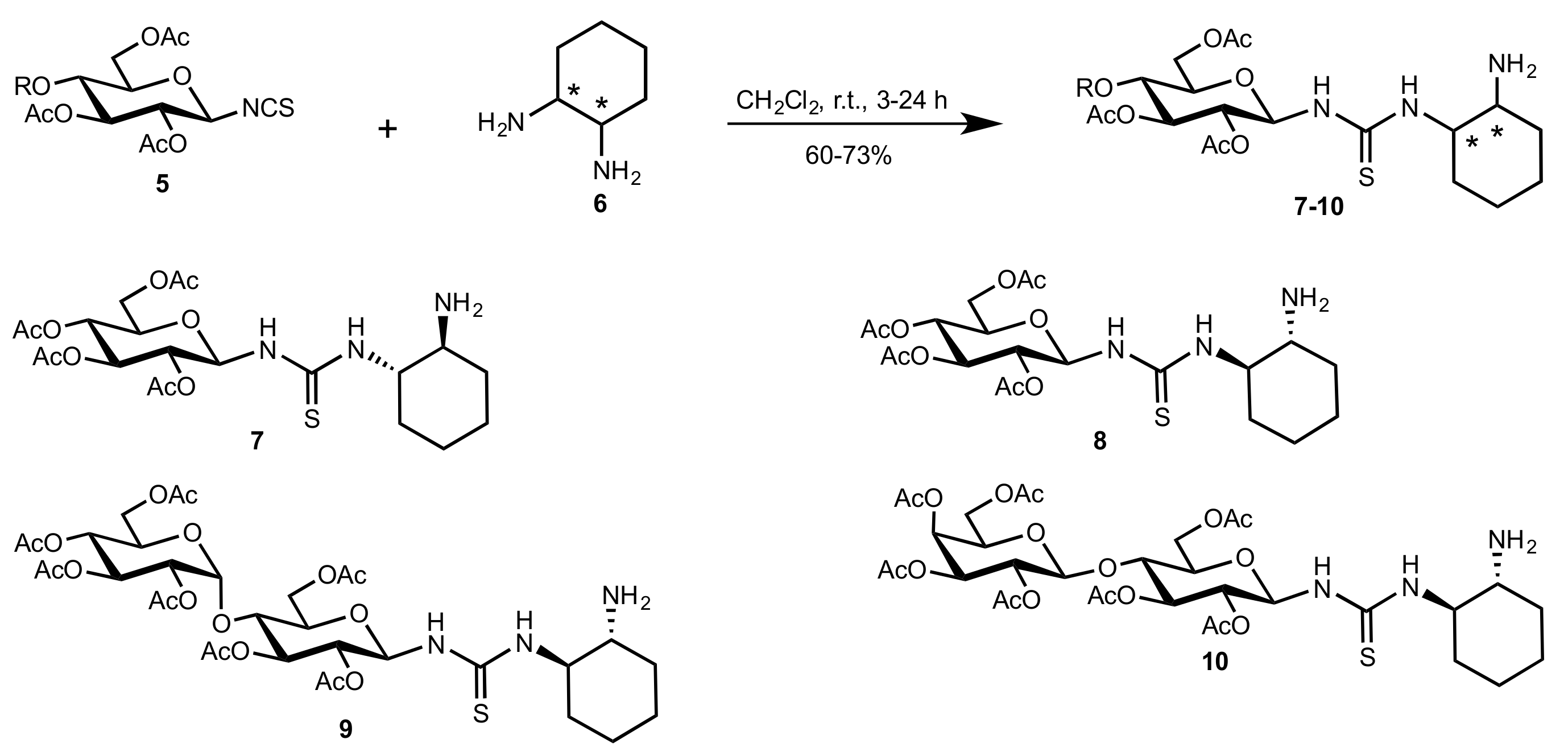
Scheme 2.
The synthesis of organocatalysts 7–10 (the asterisks indicate stereogenic carbon atoms).
Scheme 2.
The synthesis of organocatalysts 7–10 (the asterisks indicate stereogenic carbon atoms).
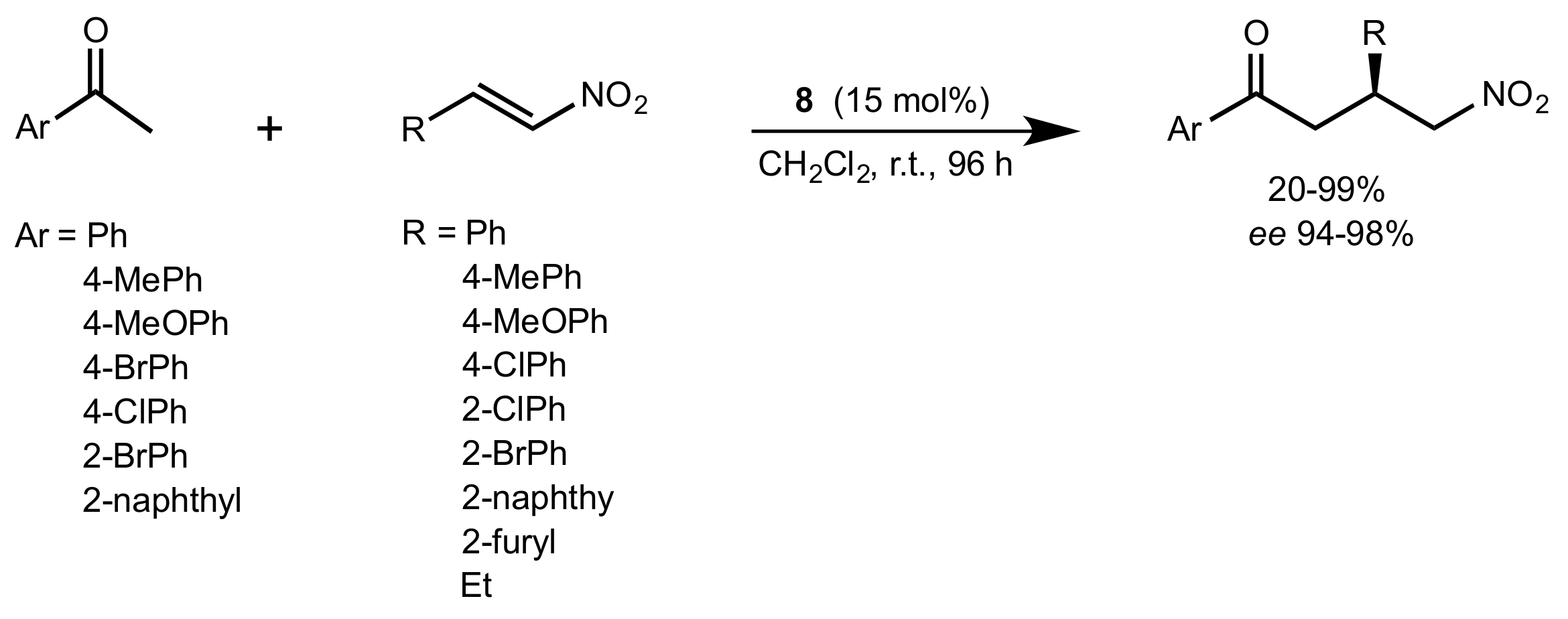
Scheme 3.
The Michael addition catalysed by 8.
Scheme 3.
The Michael addition catalysed by 8.
Figure 1.
Sugar thiourea organocatalysts employed by Wu and co-workers.
Figure 1.
Sugar thiourea organocatalysts employed by Wu and co-workers.
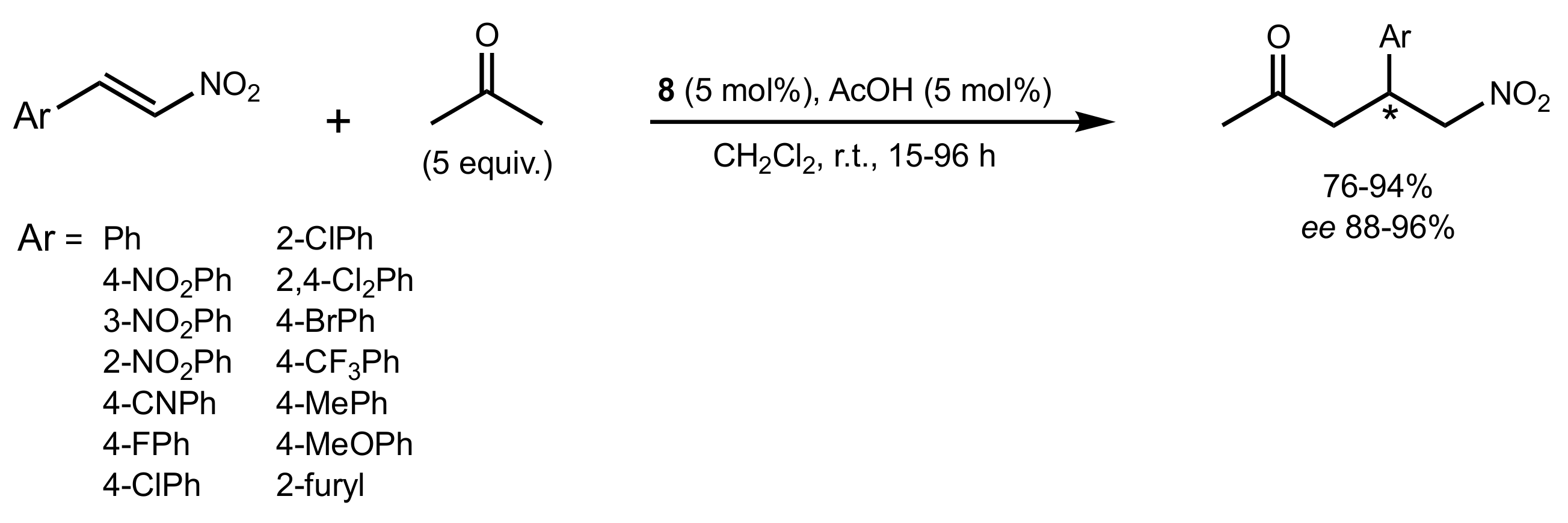
Scheme 4.
The Michael addition of acetone to nitrostyrenes catalysed by 8.
Scheme 4.
The Michael addition of acetone to nitrostyrenes catalysed by 8.
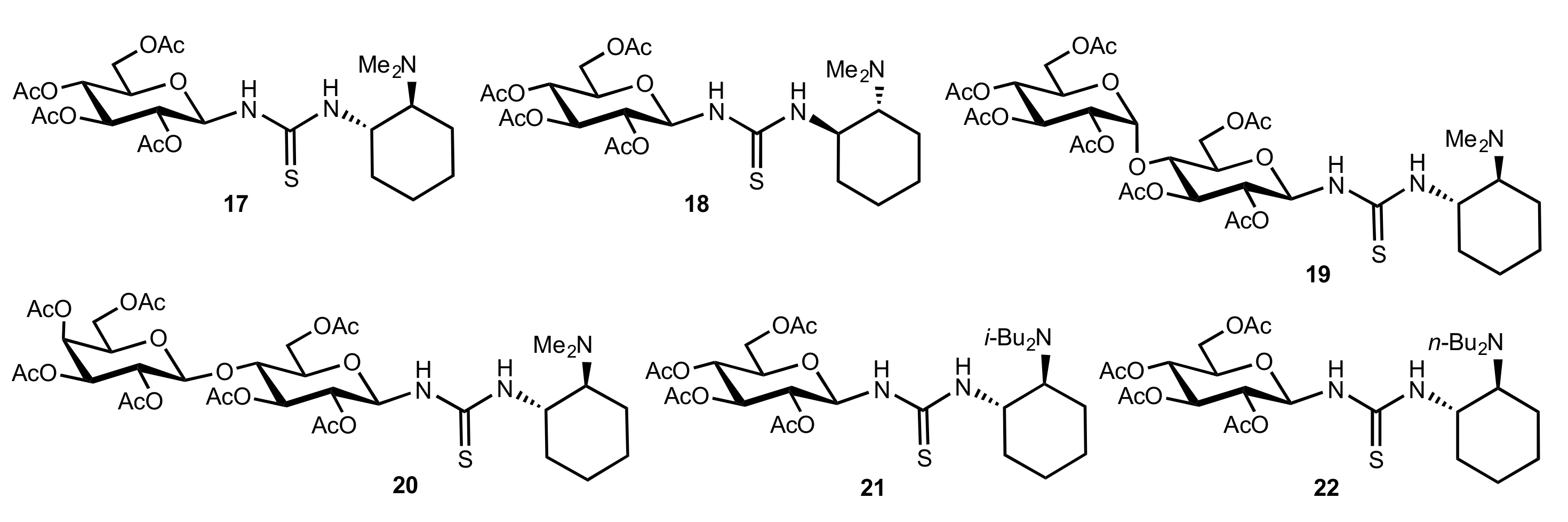
Figure 2.
The structure of organocatalysts 17–22.
Figure 2.
The structure of organocatalysts 17–22.
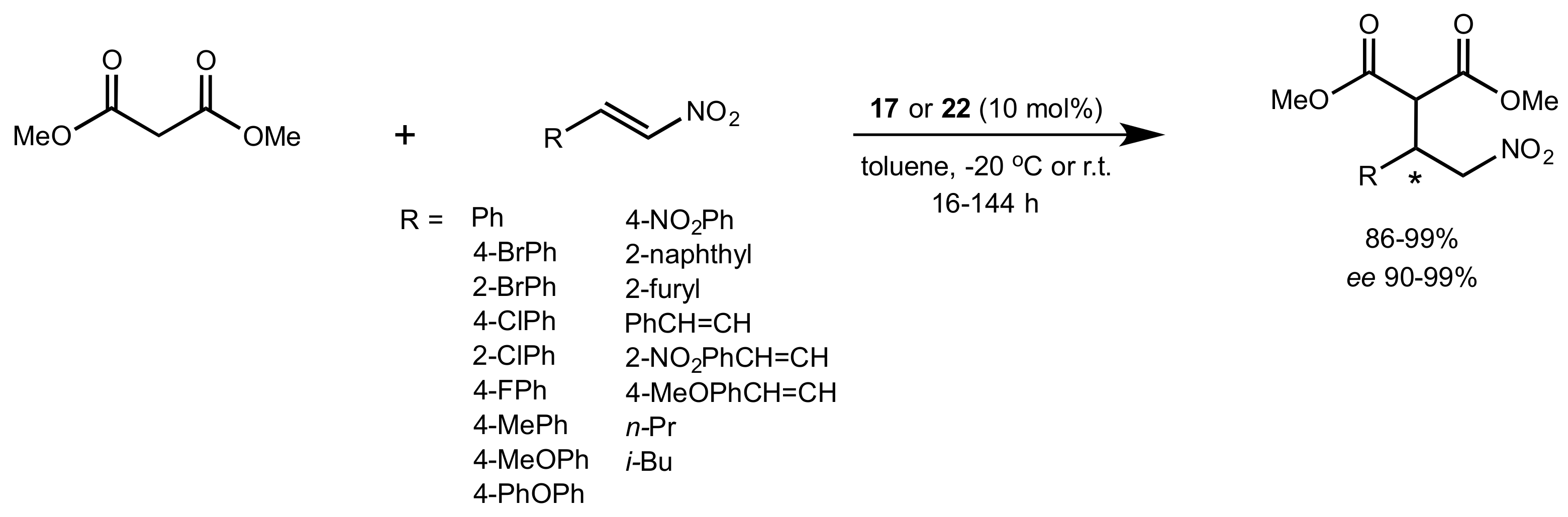
Scheme 5.
The Michael addition of methyl malonate to nitrostyrenes catalysed by 17 or 22.
Scheme 5.
The Michael addition of methyl malonate to nitrostyrenes catalysed by 17 or 22.
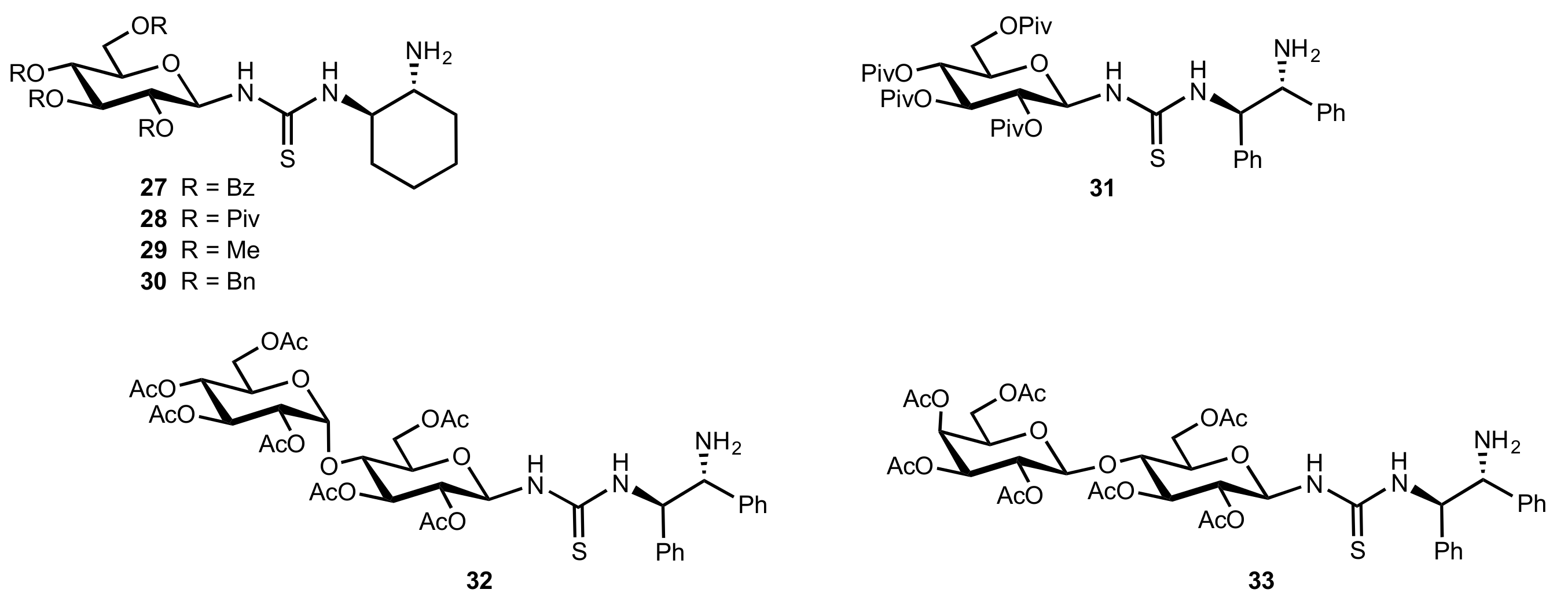
Figure 3.
Galactose-based organocatalyst 23.
Figure 3.
Galactose-based organocatalyst 23.
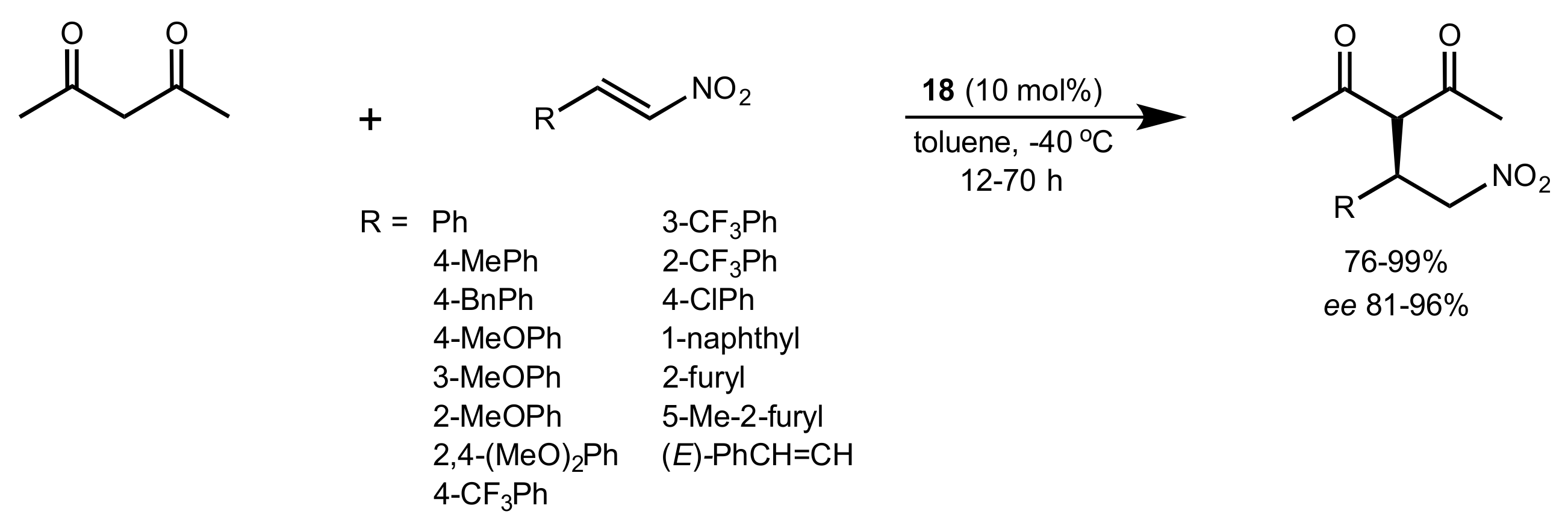
Scheme 6.
The Michael addition of acetylacetone to nitroolefins catalysed by 18.
Scheme 6.
The Michael addition of acetylacetone to nitroolefins catalysed by 18.
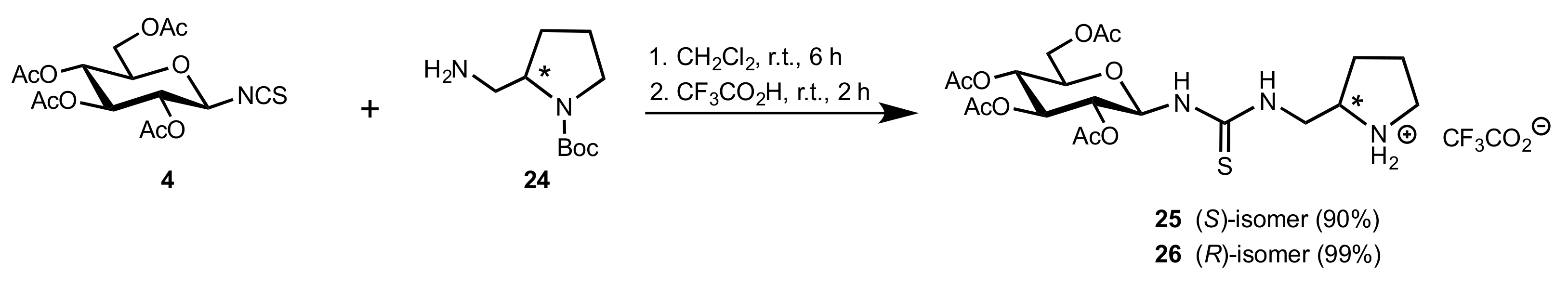
Scheme 7.
The synthesis of organocatalysts 25 and 26.
Scheme 7.
The synthesis of organocatalysts 25 and 26.
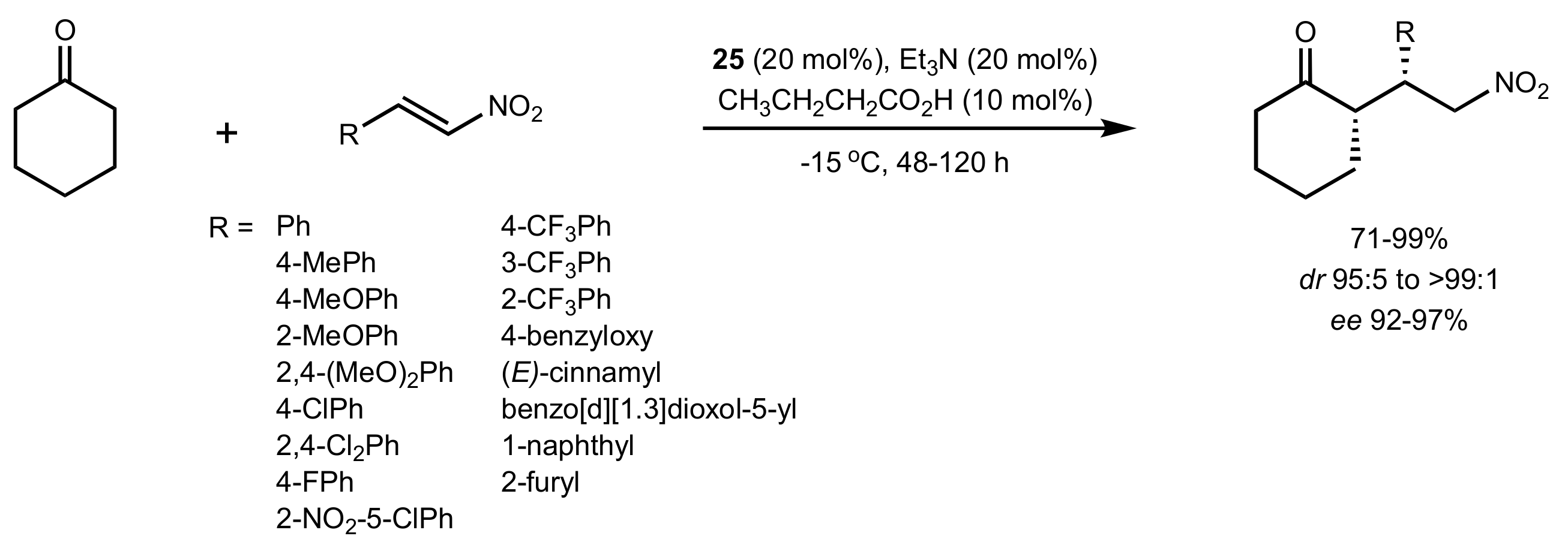
Scheme 8.
The Michael addition of cyclohexanone to nitroolefins catalysed by 25.
Scheme 8.
The Michael addition of cyclohexanone to nitroolefins catalysed by 25.
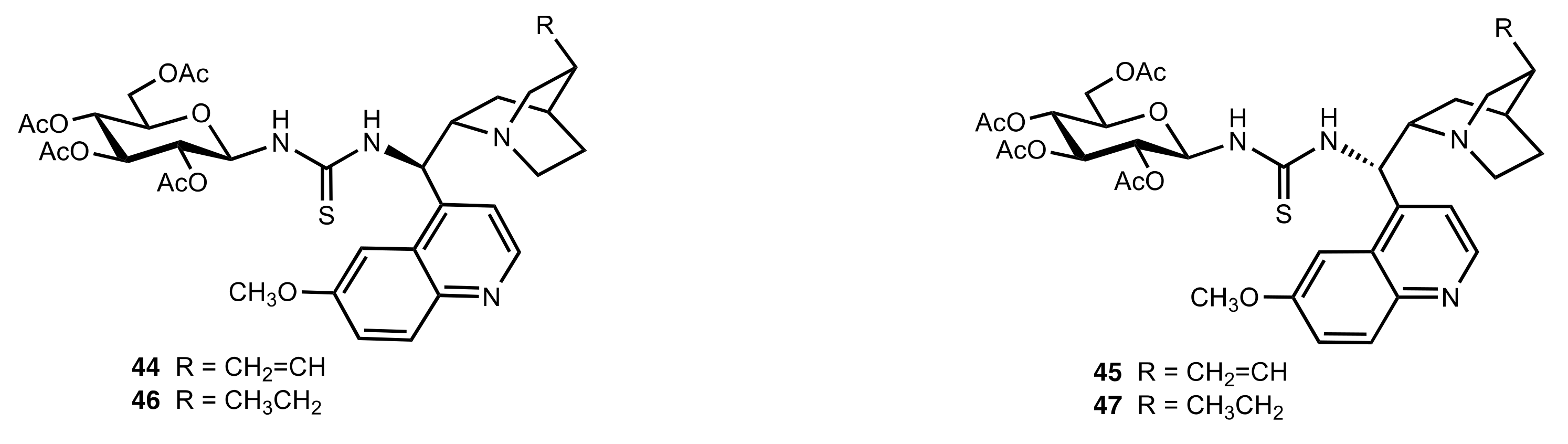
Figure 4.
Bifunctional organocatalysts used in the Michael addition of ketones to nitrodienes.
Figure 4.
Bifunctional organocatalysts used in the Michael addition of ketones to nitrodienes.
Scheme 9.
The Michael addition of aryl methyl ketones to nitrodienes catalysed by 28.
Scheme 9.
The Michael addition of aryl methyl ketones to nitrodienes catalysed by 28.
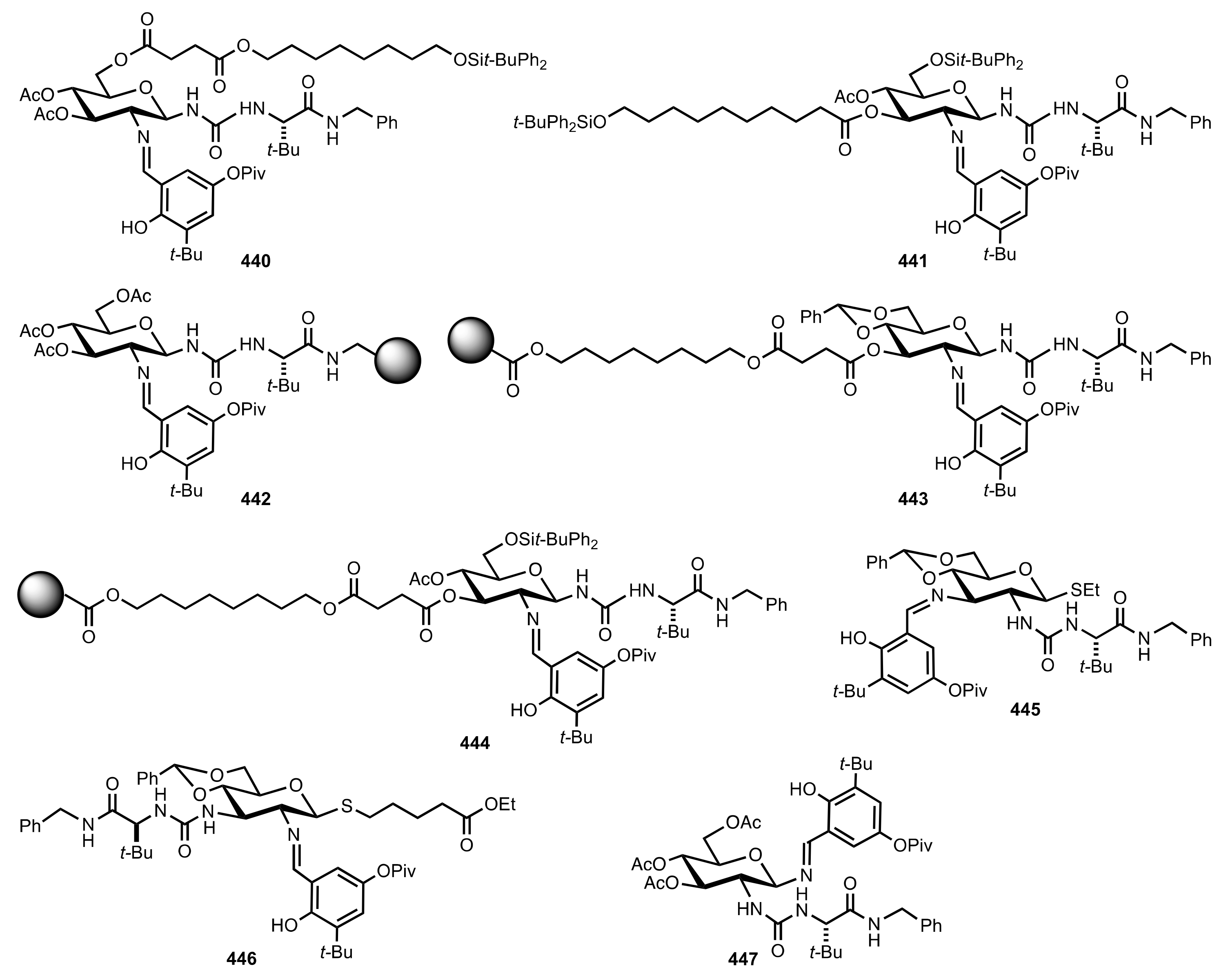
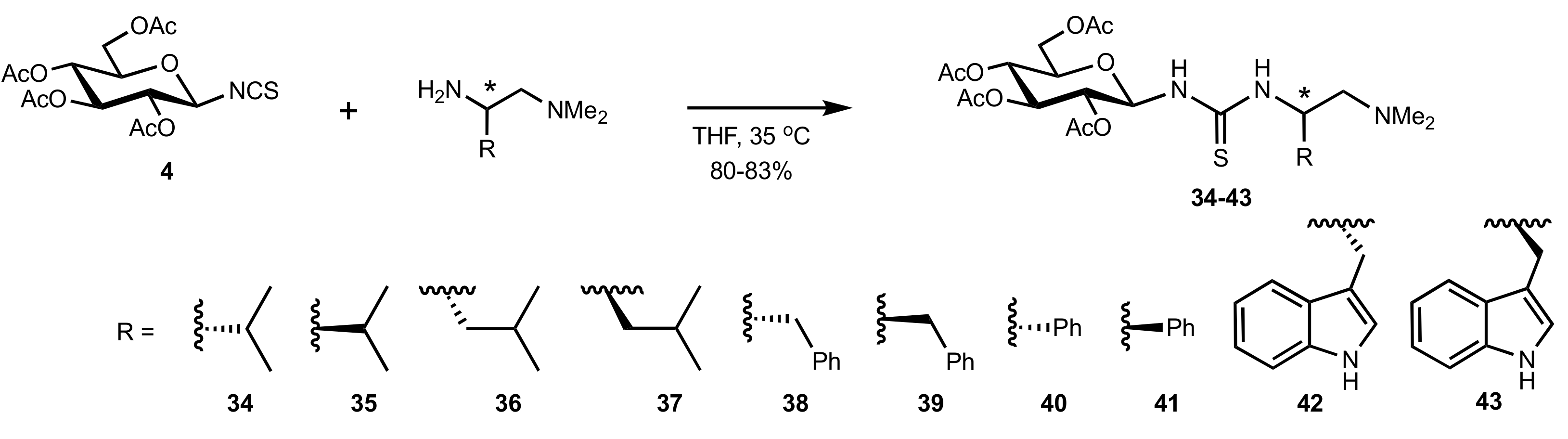
Scheme 10.
Preparation of catalysts containing carbohydrate and aminoacid moieties.
Scheme 10.
Preparation of catalysts containing carbohydrate and aminoacid moieties.
Scheme 11.
The Michael addition of acetylacetone to nitrostyrenes catalysed by 40 or 41.
Scheme 11.
The Michael addition of acetylacetone to nitrostyrenes catalysed by 40 or 41.
Figure 5.
Sugar thioureas containing quinine, quinidine, dihydroquinine and dihydroquinidine.
Figure 5.
Sugar thioureas containing quinine, quinidine, dihydroquinine and dihydroquinidine.
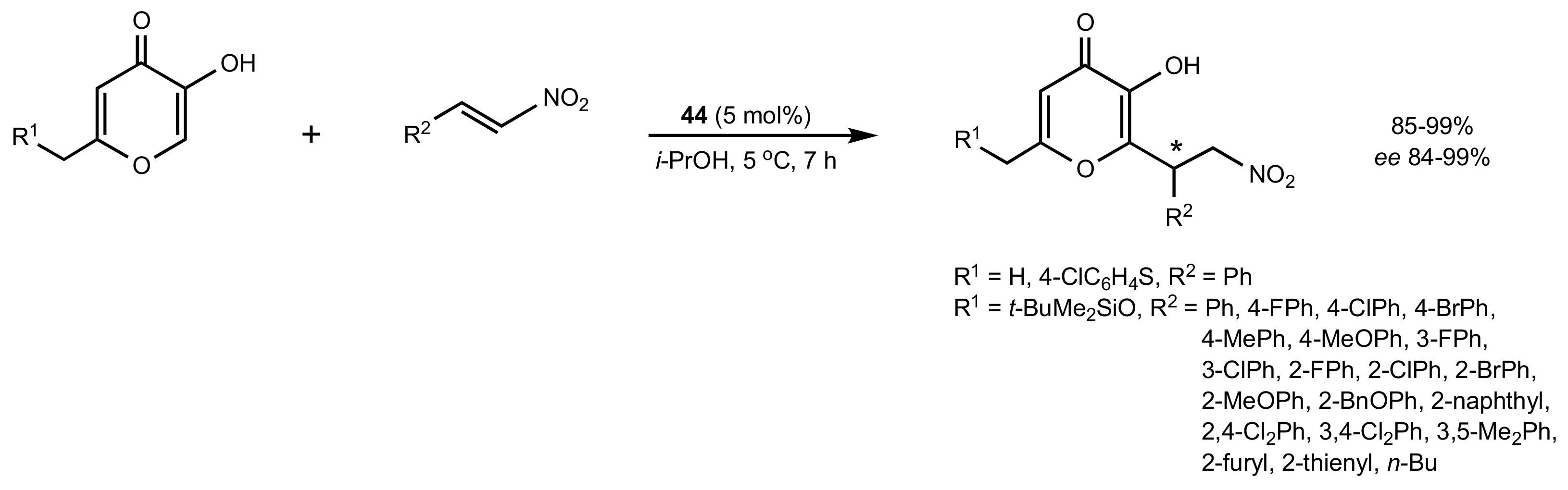
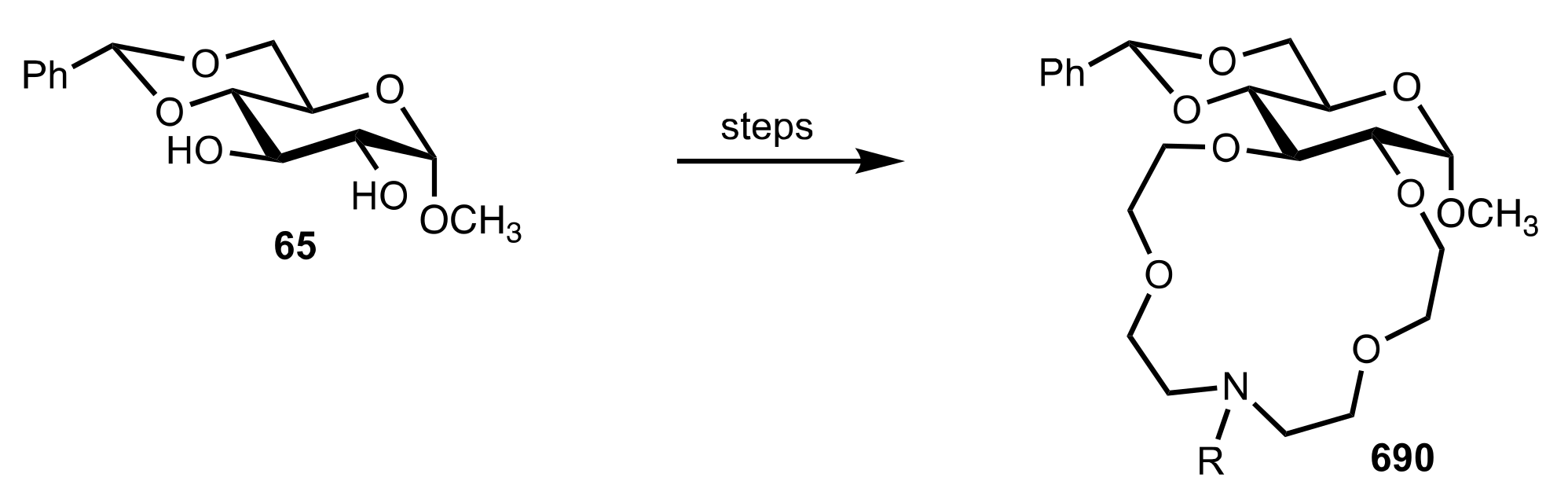
Scheme 12.
The Michael addition of kojic acid derivatives to nitroalkenes.
Scheme 12.
The Michael addition of kojic acid derivatives to nitroalkenes.
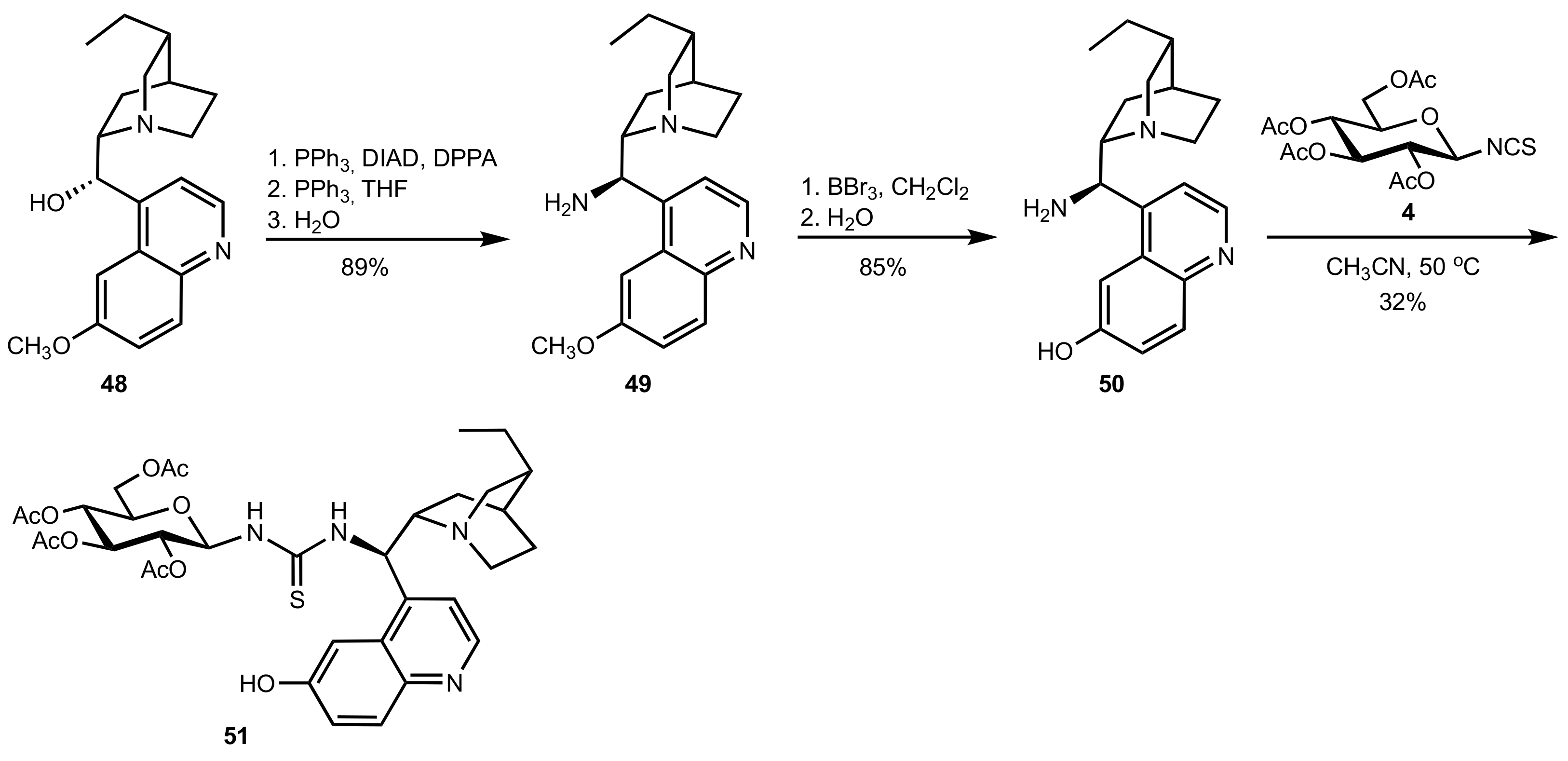
Scheme 13.
The synthesis of dihydroquinine-derived organocatalyst 51.
Scheme 13.
The synthesis of dihydroquinine-derived organocatalyst 51.
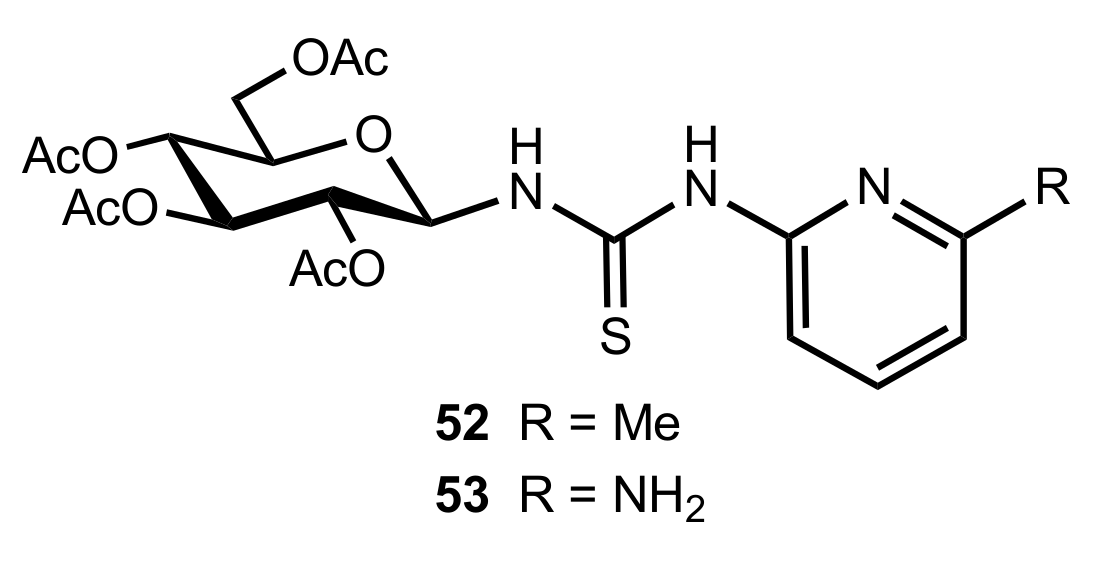
Figure 6.
The structure of organocatalysts 52 and 53.
Figure 6.
The structure of organocatalysts 52 and 53.
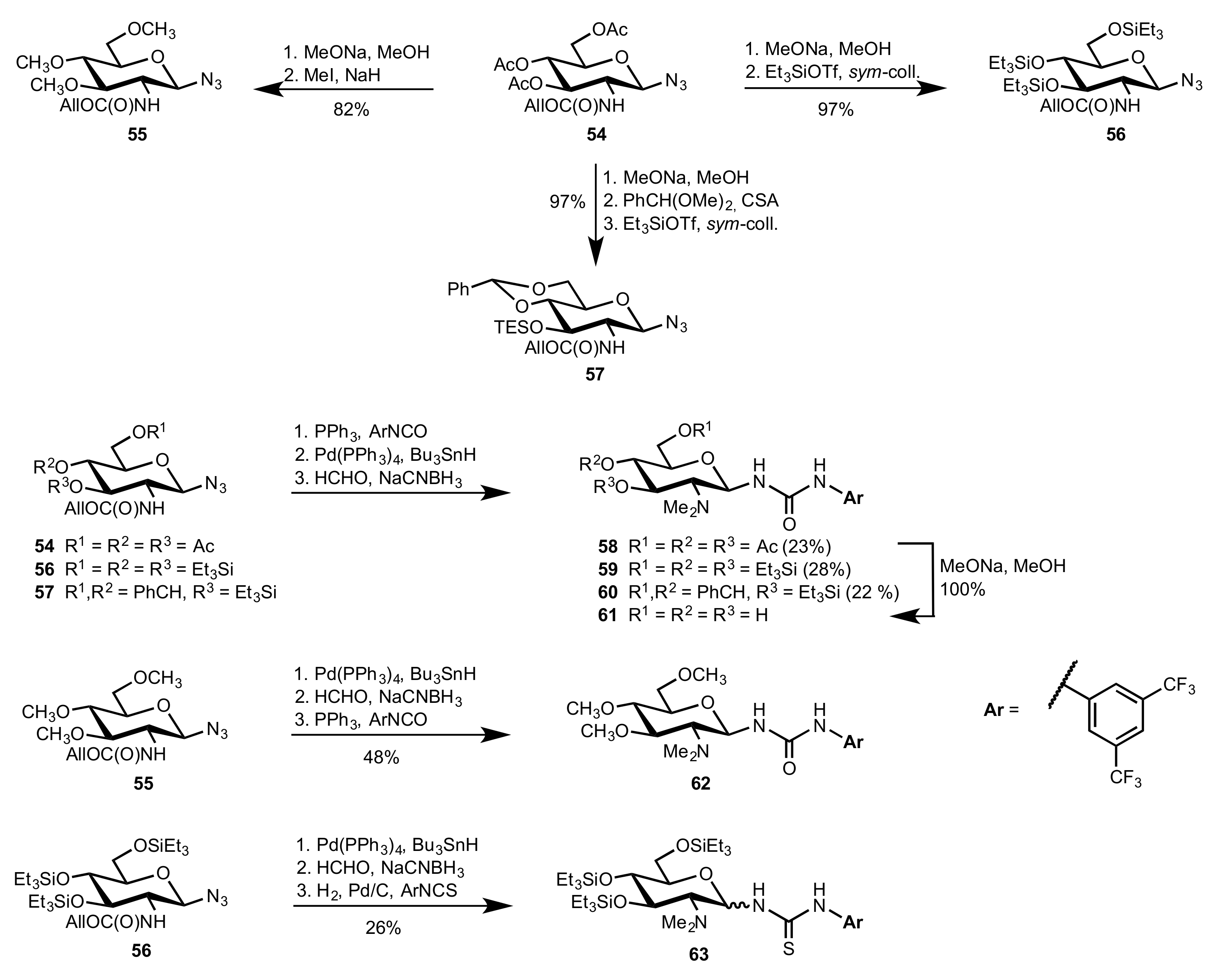
Scheme 14.
The synthesis of organocatalysts 58–63 (CSA = 10-camphorsulfonic acid; sym-coll. = sym-collidine, 2,4,6-trimethylpyridine).
Scheme 14.
The synthesis of organocatalysts 58–63 (CSA = 10-camphorsulfonic acid; sym-coll. = sym-collidine, 2,4,6-trimethylpyridine).
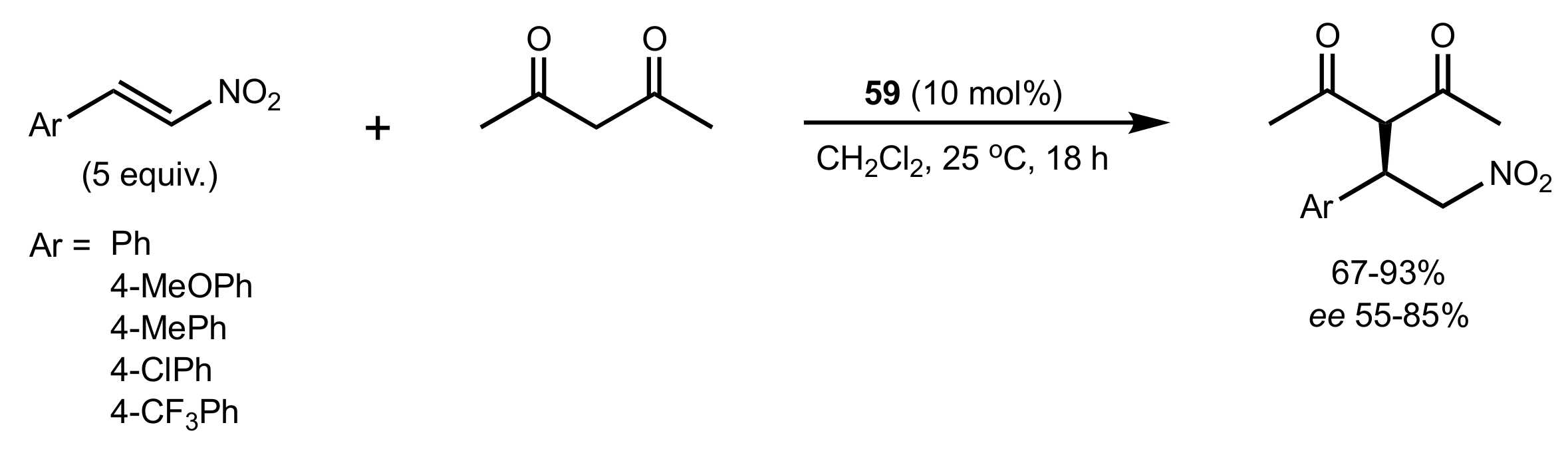
Scheme 15.
The Michael addition catalysed by 59.
Scheme 15.
The Michael addition catalysed by 59.
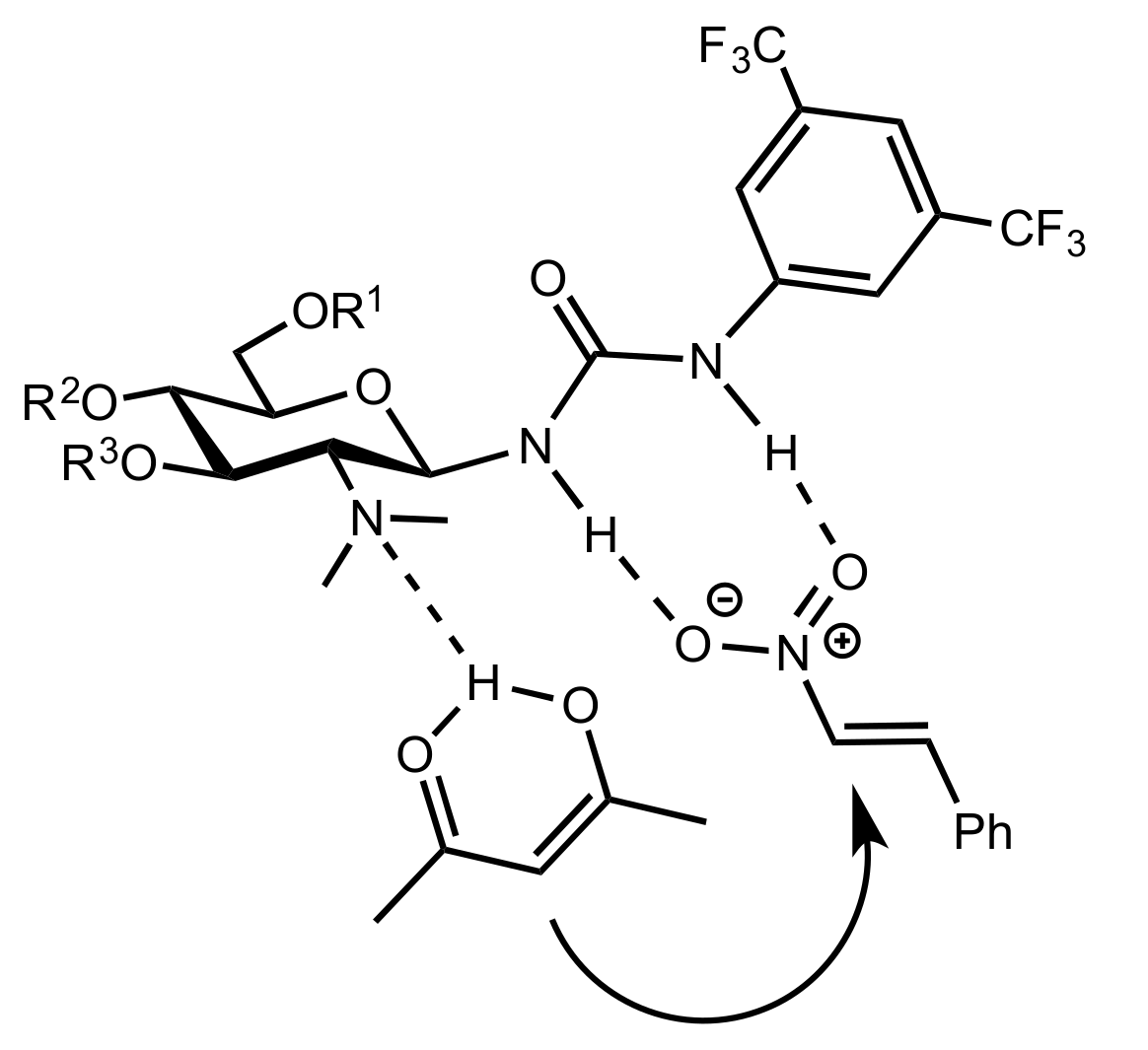
Figure 7.
A proposed transition state explaining the stereoselectivity of the reaction.
Figure 7.
A proposed transition state explaining the stereoselectivity of the reaction.
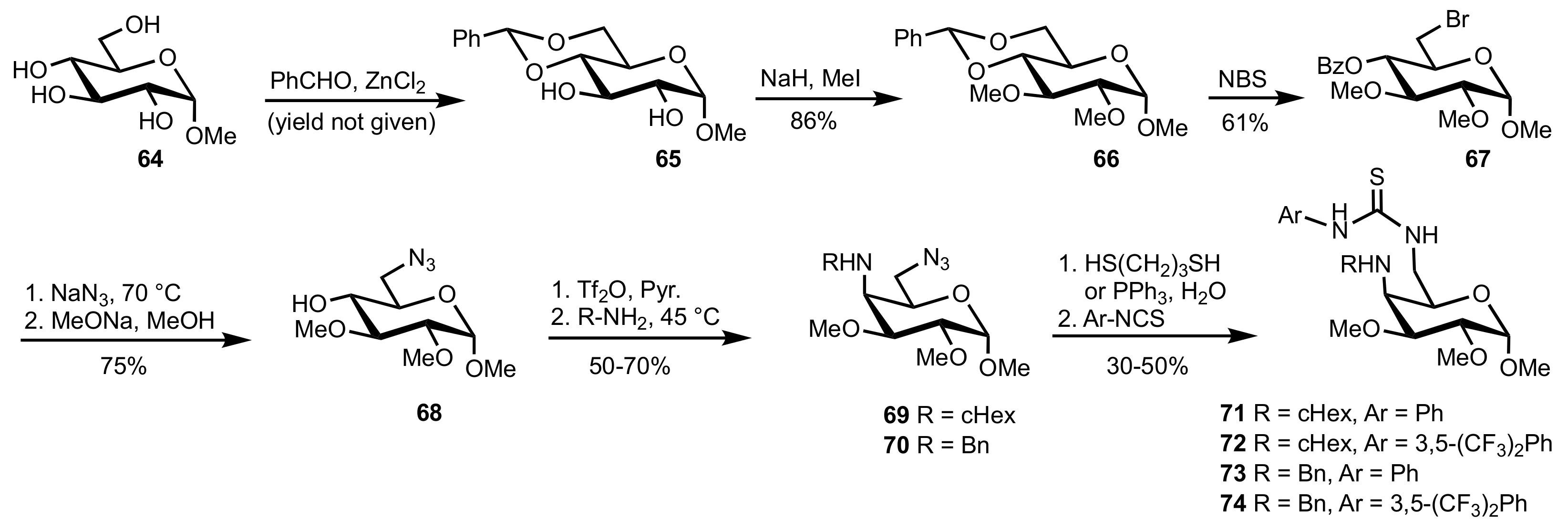
Scheme 16.
The synthesis of organocatalysts 71–74.
Scheme 16.
The synthesis of organocatalysts 71–74.
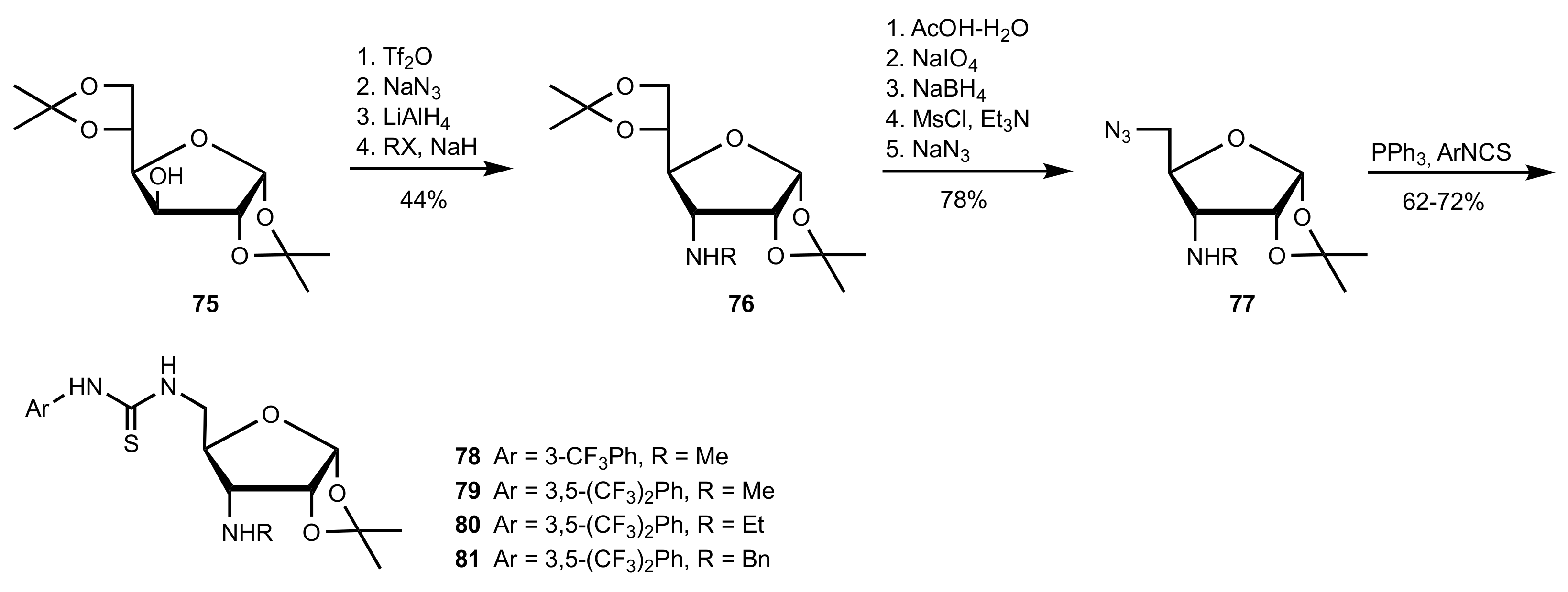
Scheme 17.
The synthesis of the organocatalysts 78–81.
Scheme 17.
The synthesis of the organocatalysts 78–81.
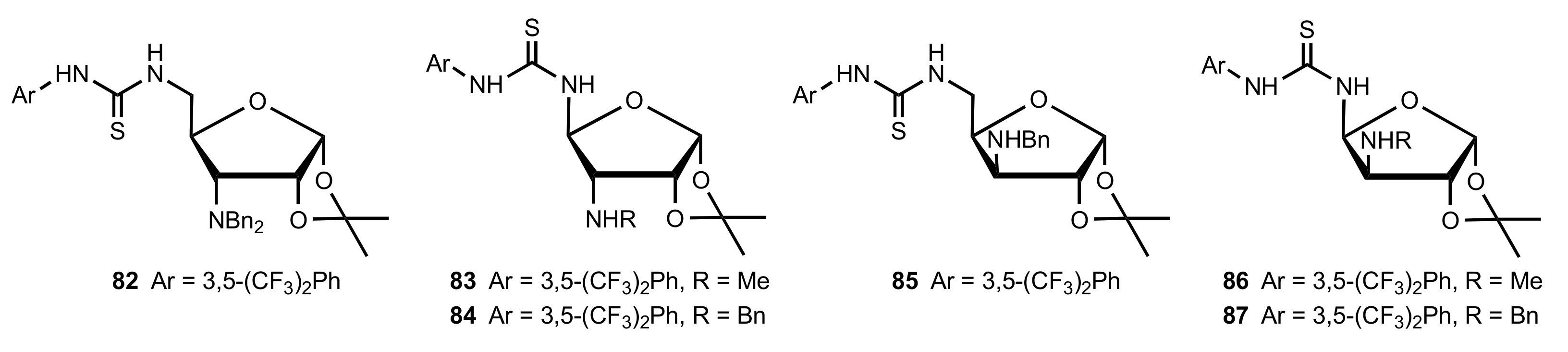
Figure 8.
The structure of organocatalysts 82–87.
Figure 8.
The structure of organocatalysts 82–87.
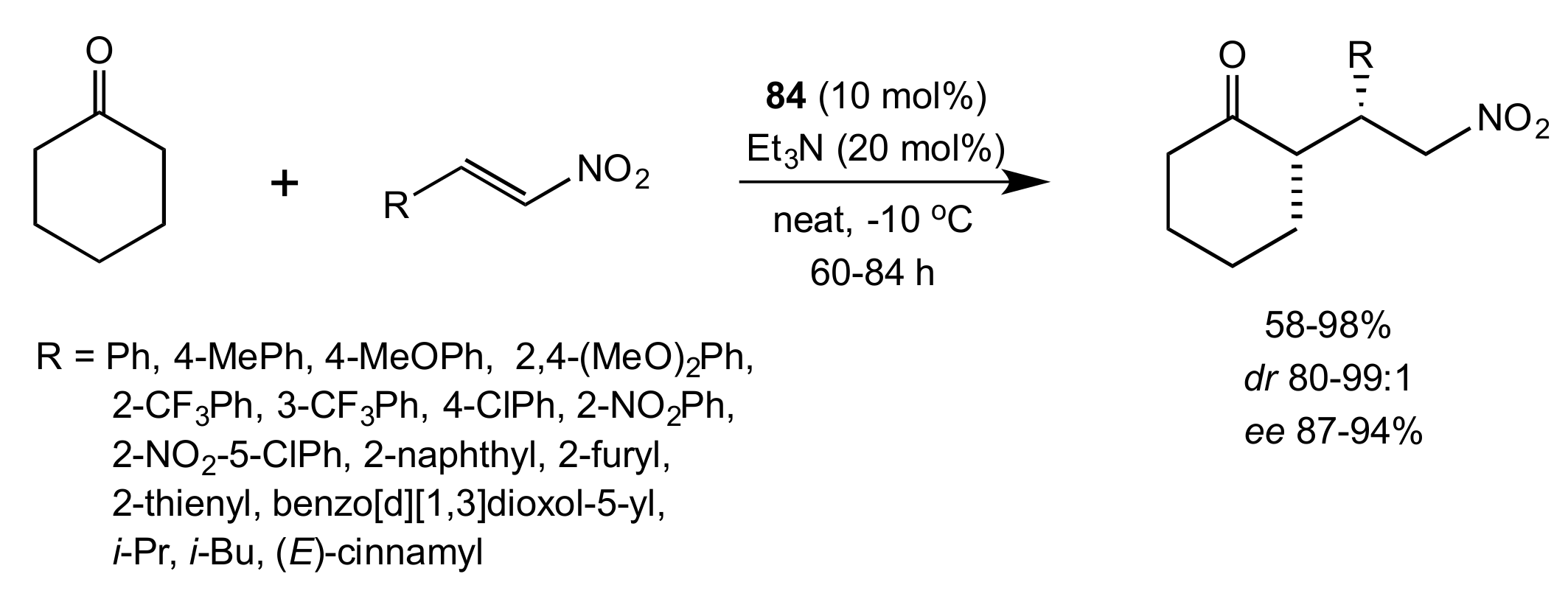
Scheme 18.
The Michael addition catalysed by 84.
Scheme 18.
The Michael addition catalysed by 84.
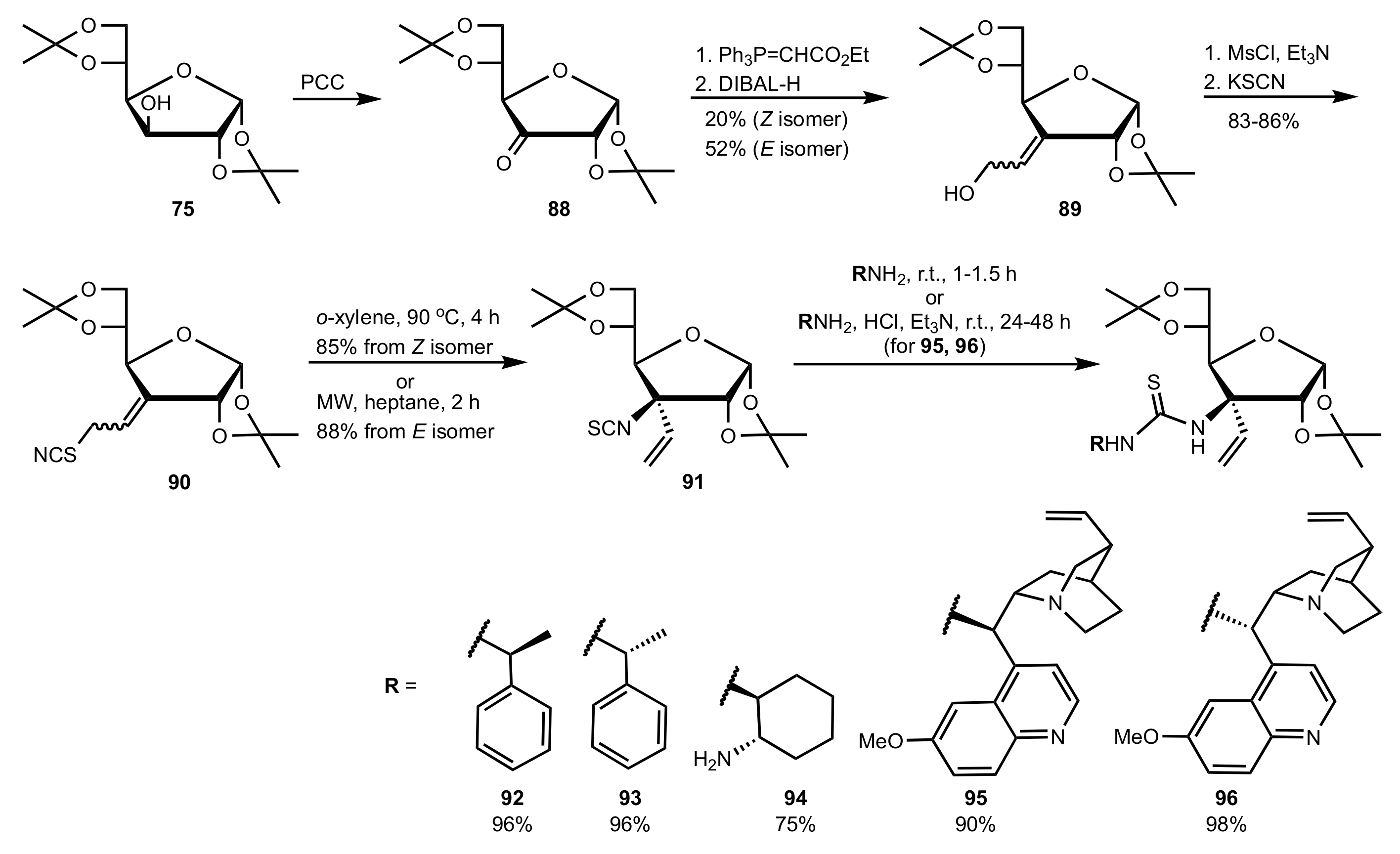
Scheme 19.
Preparation of catalysts 92–96.
Scheme 19.
Preparation of catalysts 92–96.
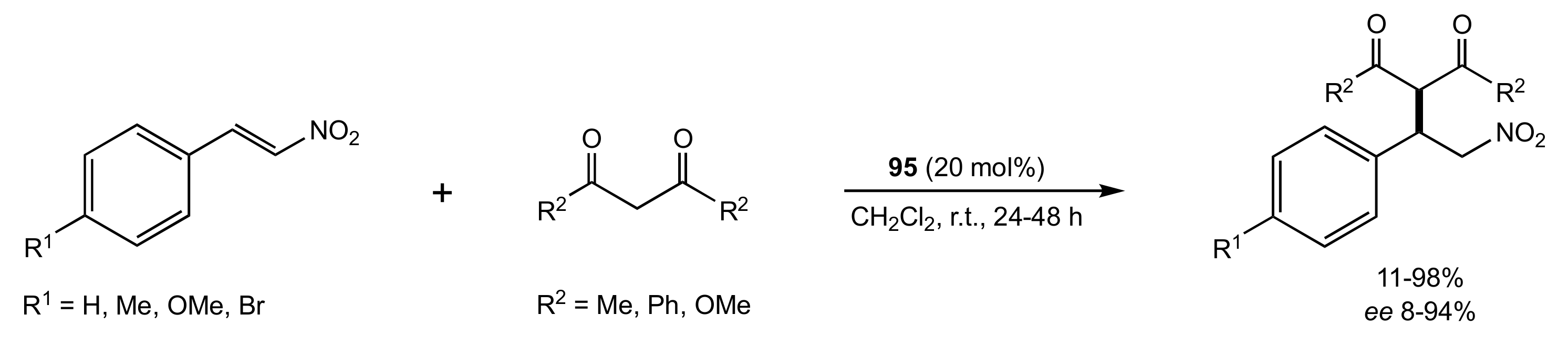
Scheme 20.
The Michael addition catalysed by 95.
Scheme 20.
The Michael addition catalysed by 95.
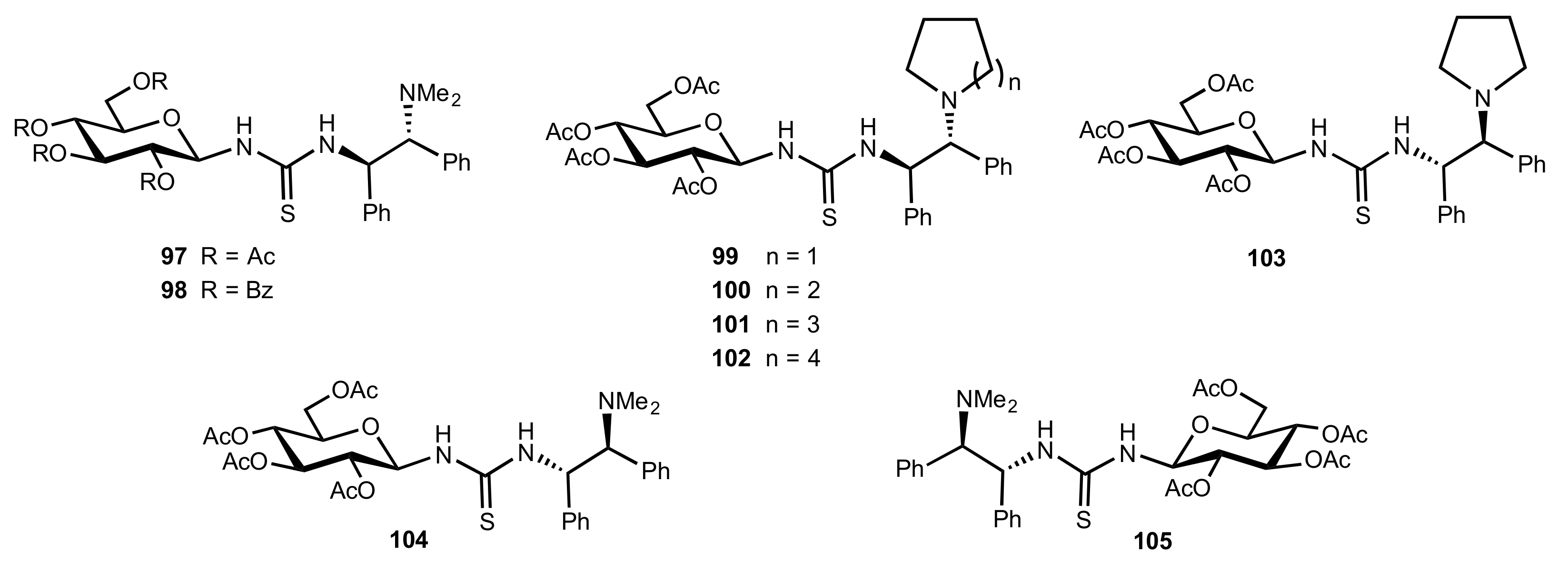
Figure 9.
The structure of organocatalysts 97–105.
Figure 9.
The structure of organocatalysts 97–105.
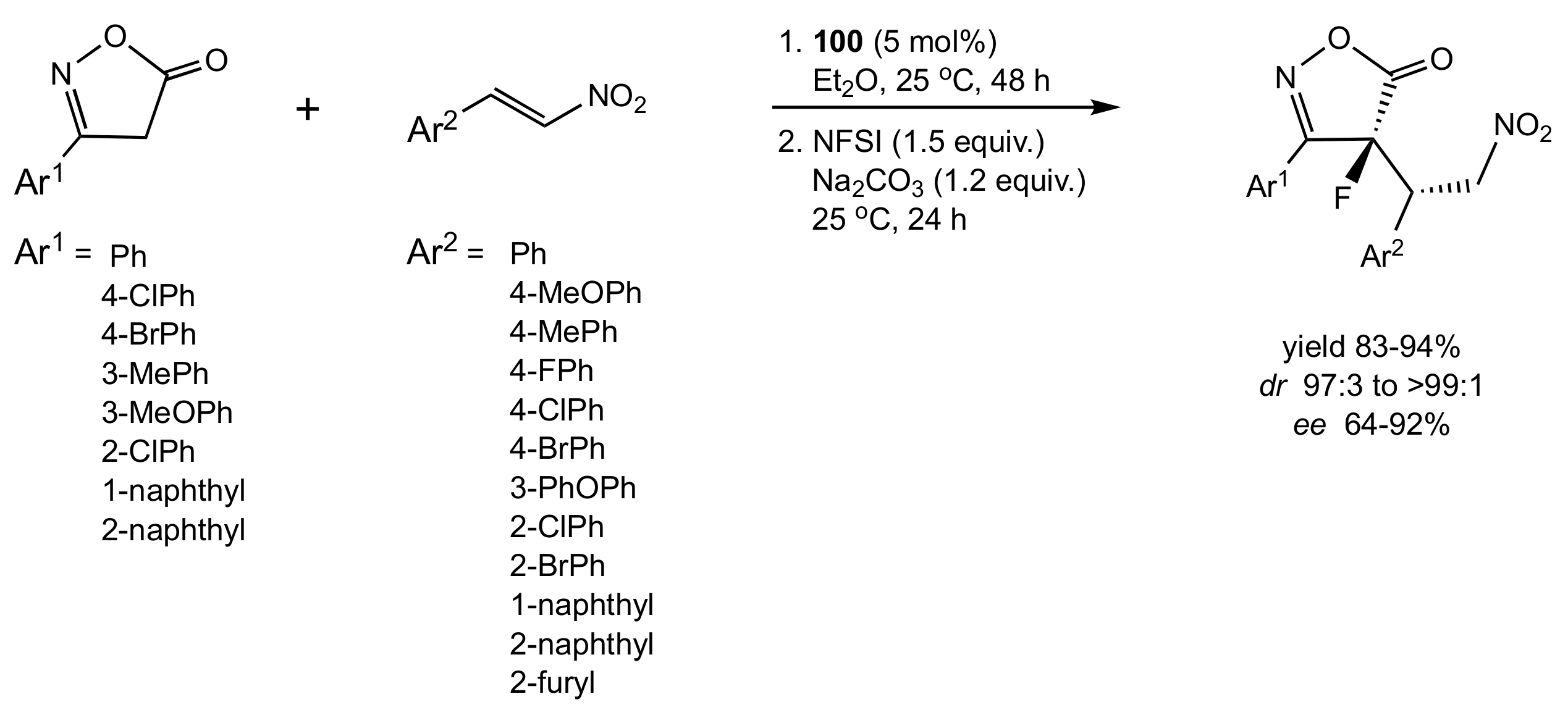
Scheme 21.
The synthesis of chiral fluorinated isoxazol-5(4H)-ones catalysed by 100 (NFSI = N-fluorobenzenesulfonimide).
Scheme 21.
The synthesis of chiral fluorinated isoxazol-5(4H)-ones catalysed by 100 (NFSI = N-fluorobenzenesulfonimide).
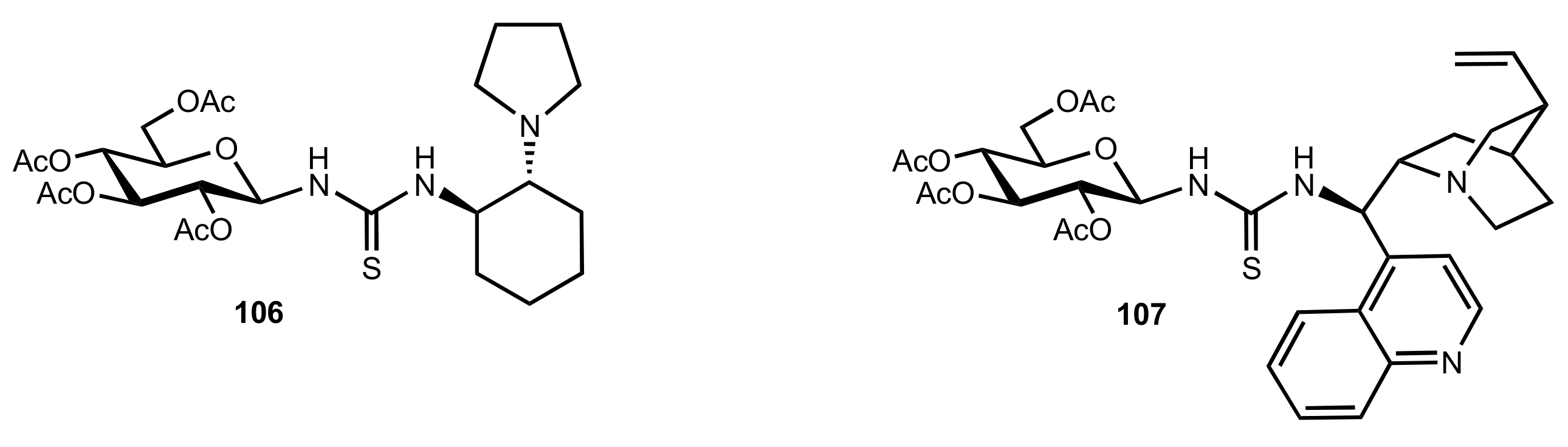
Figure 10.
The structure of organocatalysts 106 and 107.
Figure 10.
The structure of organocatalysts 106 and 107.
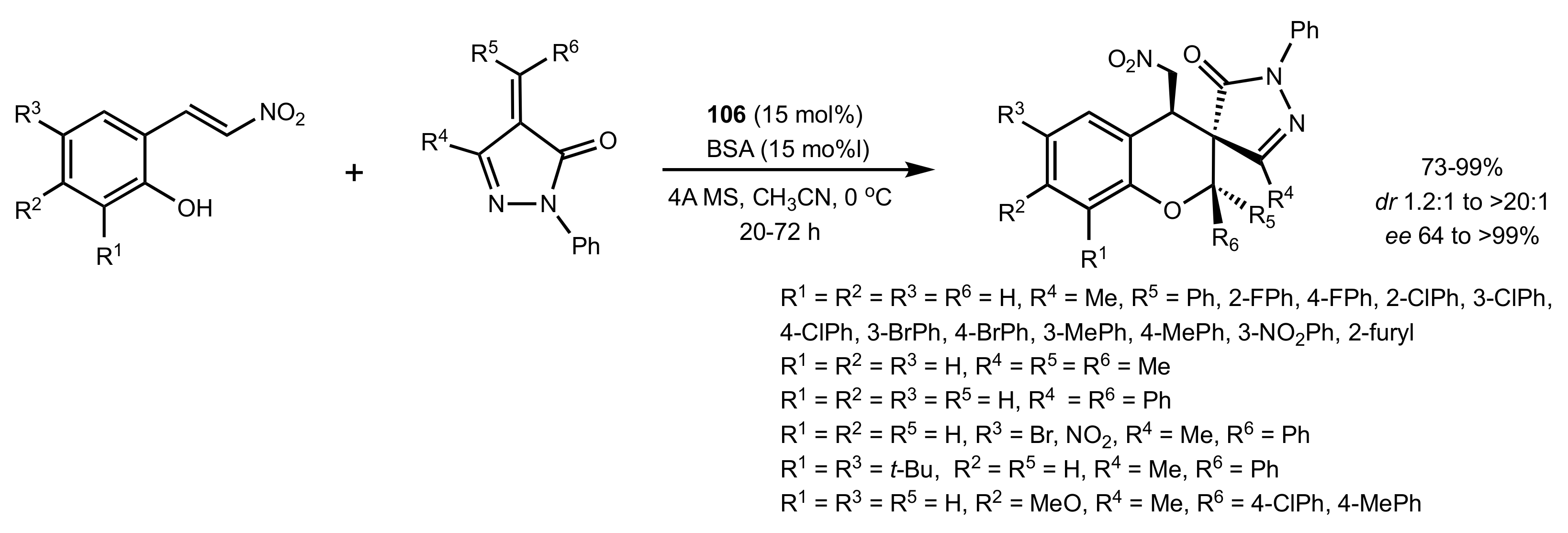
Scheme 22.
The asymmetric synthesis of spiro[chroman-3,3′-pyrazols] catalysed by 106.
Scheme 22.
The asymmetric synthesis of spiro[chroman-3,3′-pyrazols] catalysed by 106.
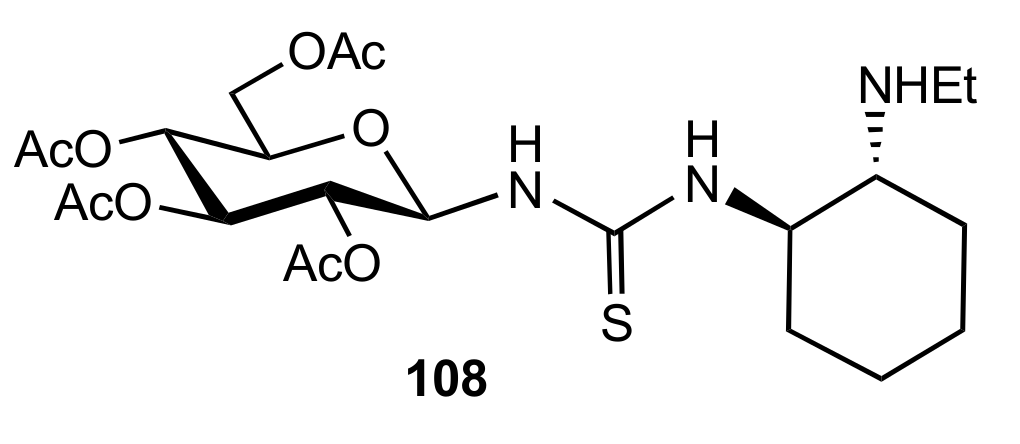
Figure 11.
The structure of organocatalyst 108.
Figure 11.
The structure of organocatalyst 108.
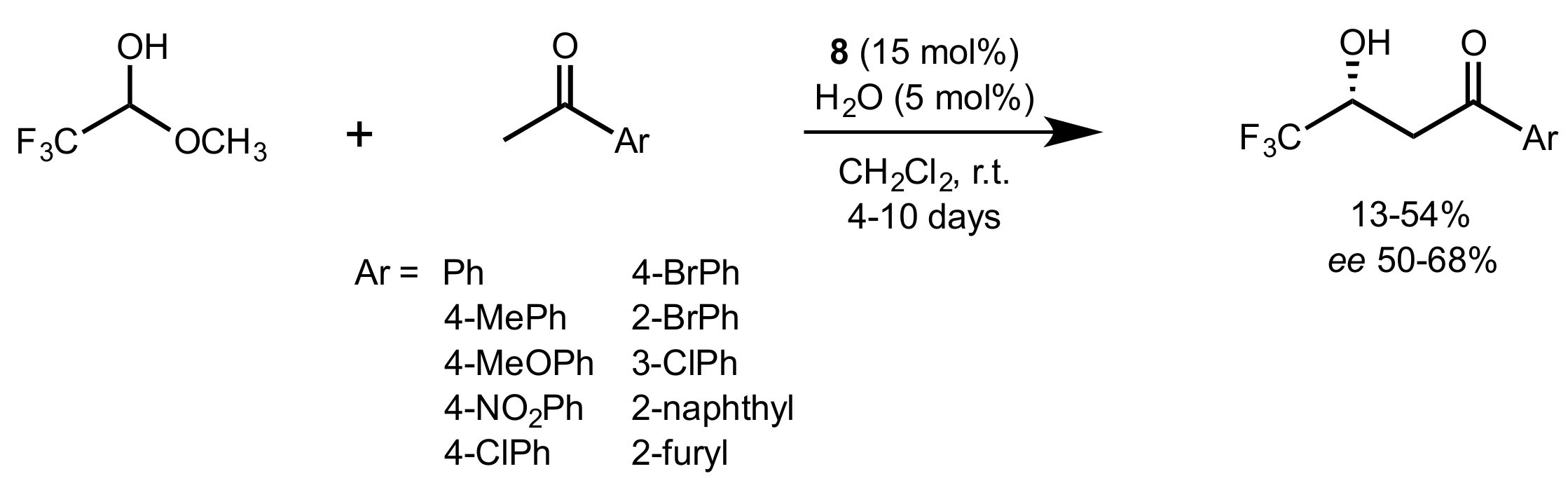
Scheme 23.
The aldol reaction of trifluoroacetaldehyde methyl hemiacetal with aromatic ketones catalysed by 8.
Scheme 23.
The aldol reaction of trifluoroacetaldehyde methyl hemiacetal with aromatic ketones catalysed by 8.
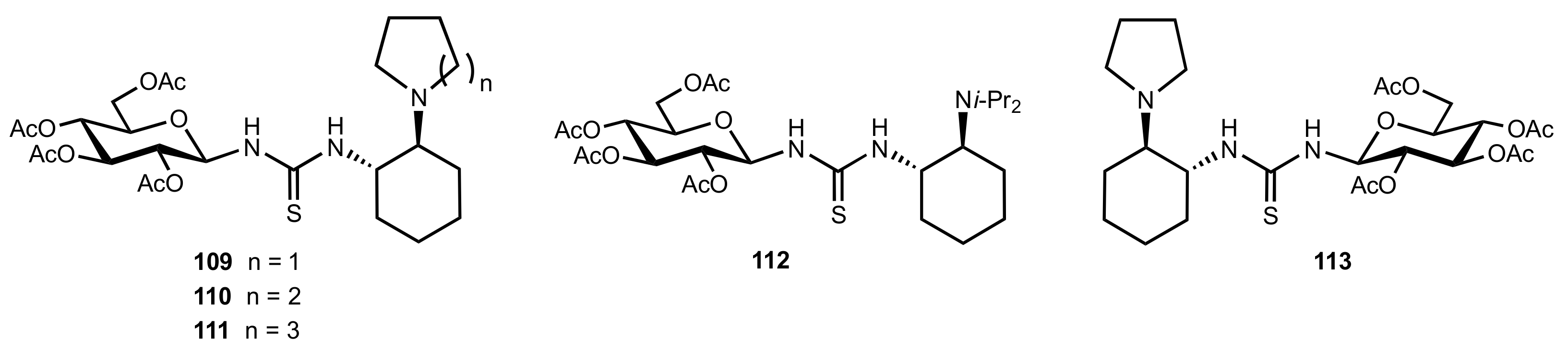
Figure 12.
The structure of catalysts 109–113.
Figure 12.
The structure of catalysts 109–113.
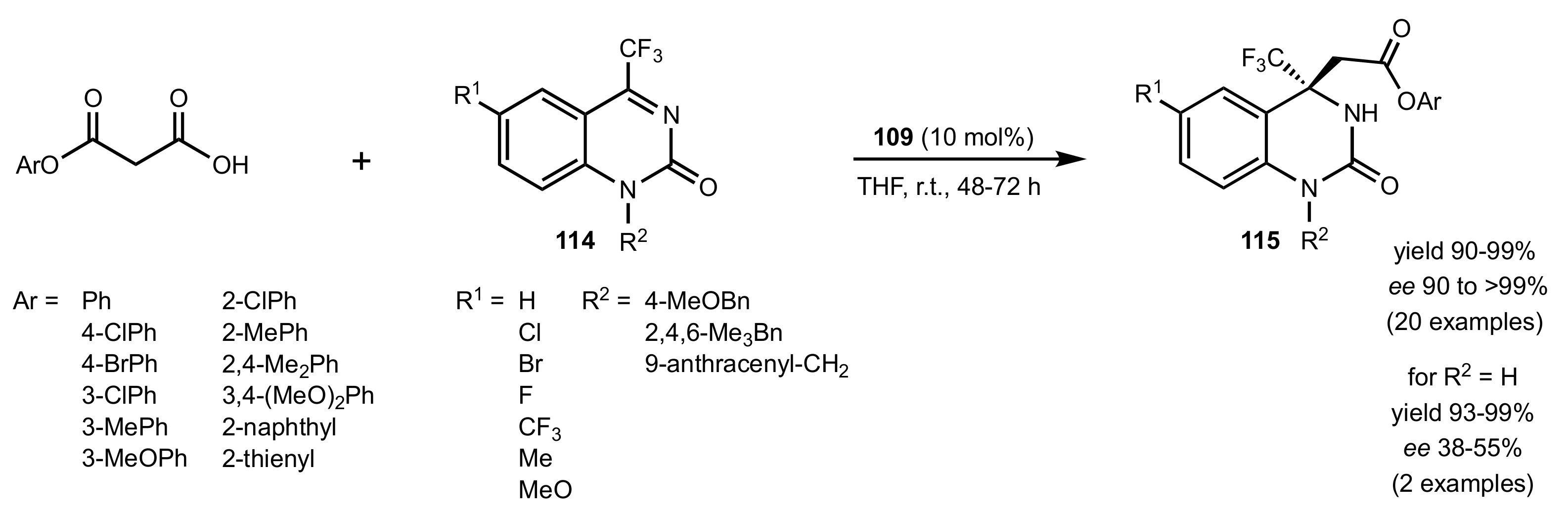
Scheme 24.
The decarboxylative Mannich reaction of malonic acid monoesters with trifluoromethyl cyclic ketimines.
Scheme 24.
The decarboxylative Mannich reaction of malonic acid monoesters with trifluoromethyl cyclic ketimines.
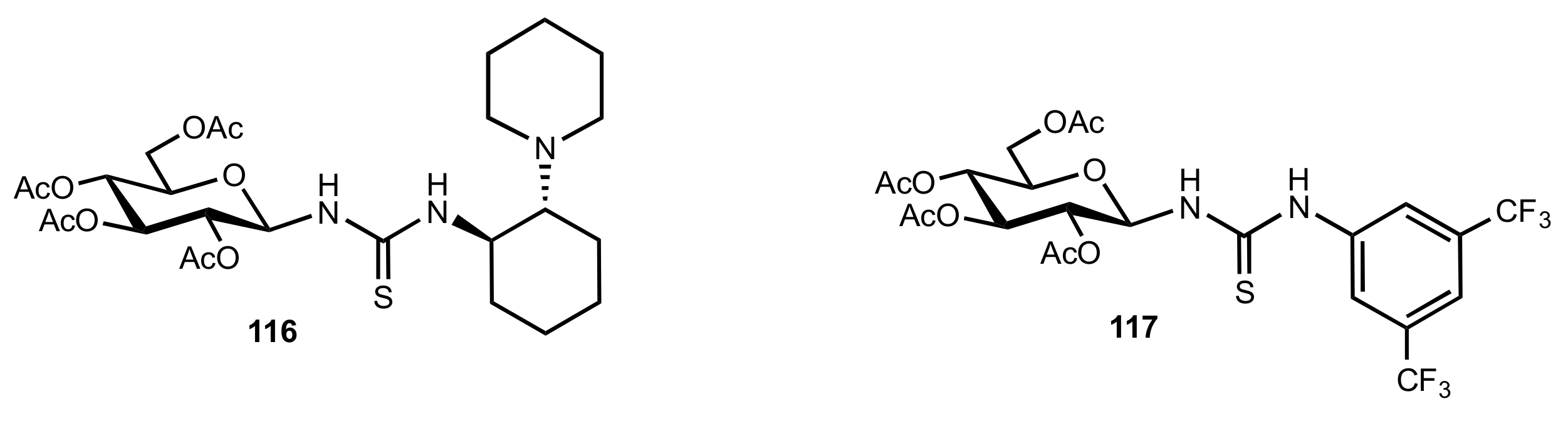
Figure 13.
The structure of organocatalysts 116 and 117.
Figure 13.
The structure of organocatalysts 116 and 117.
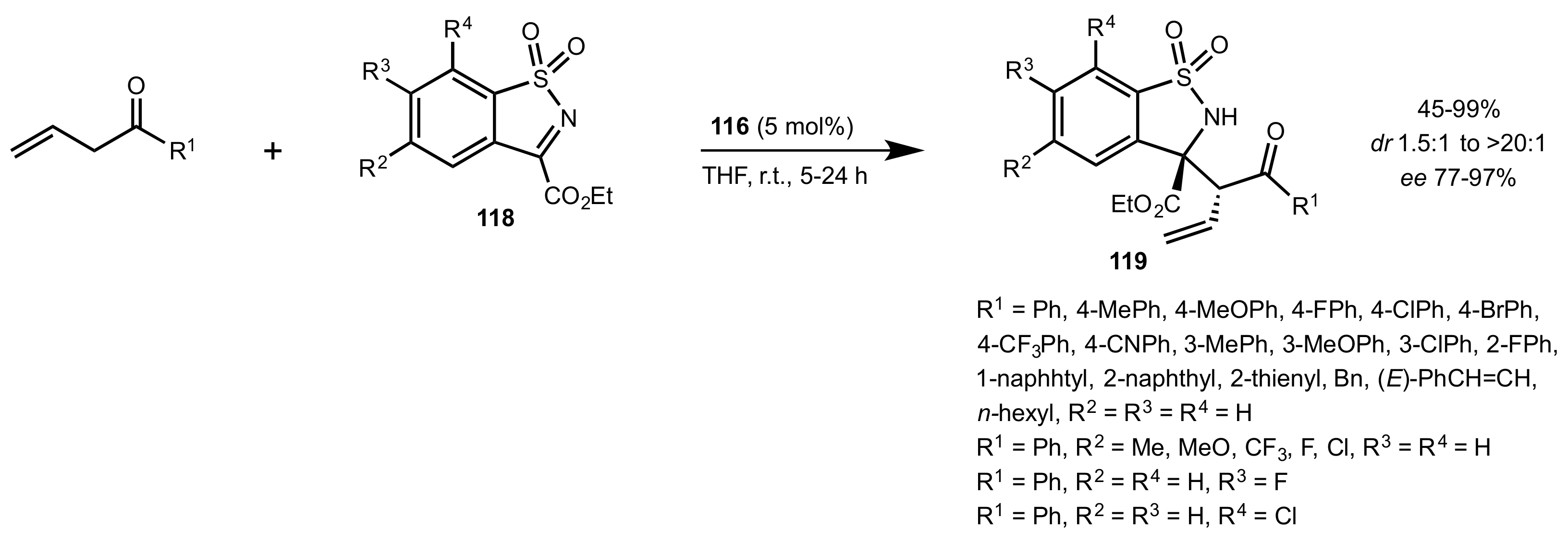
Scheme 25.
Mannich reaction of allylic ketones and N-sulfonyl ketimines.
Scheme 25.
Mannich reaction of allylic ketones and N-sulfonyl ketimines.
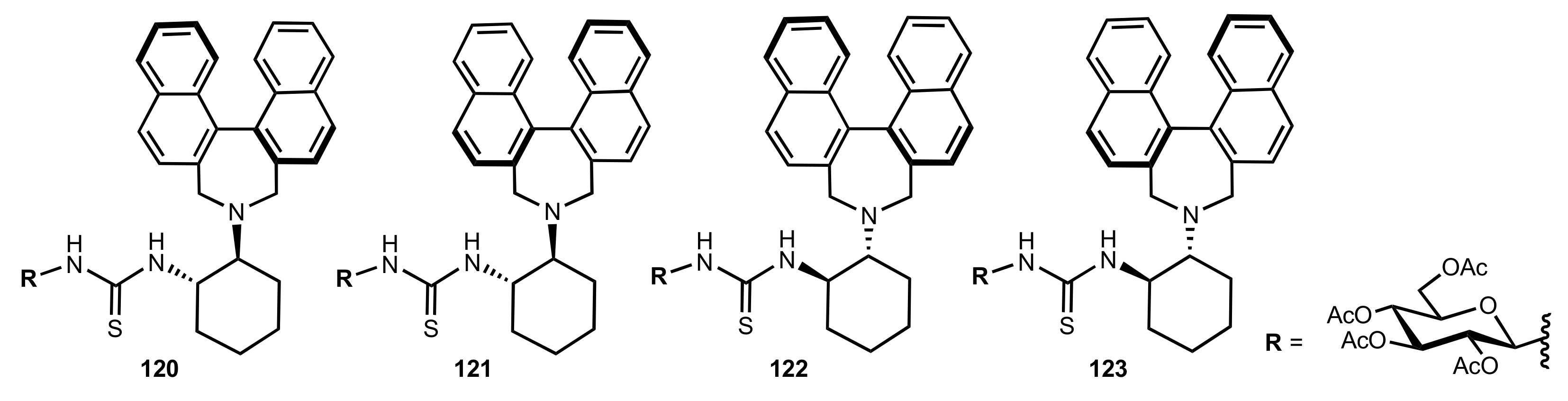
Figure 14.
The structure of organocatalysts 120–123.
Figure 14.
The structure of organocatalysts 120–123.
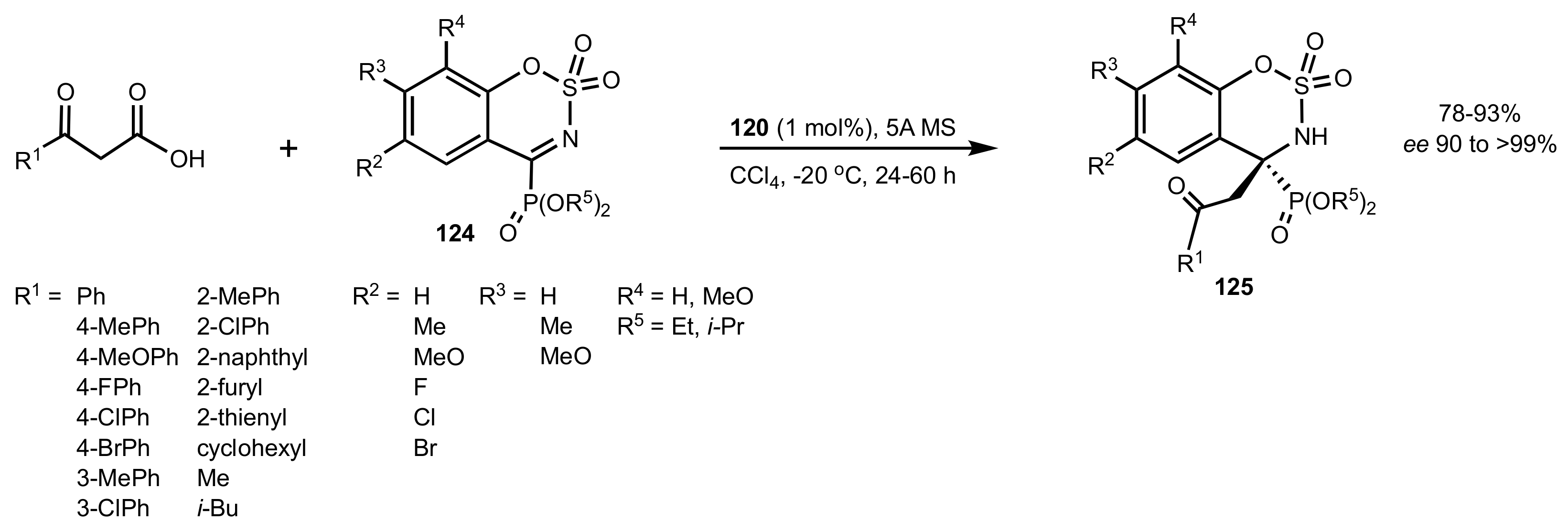
Scheme 26.
The synthesis of chiral phosphonates catalysed by 120.
Scheme 26.
The synthesis of chiral phosphonates catalysed by 120.
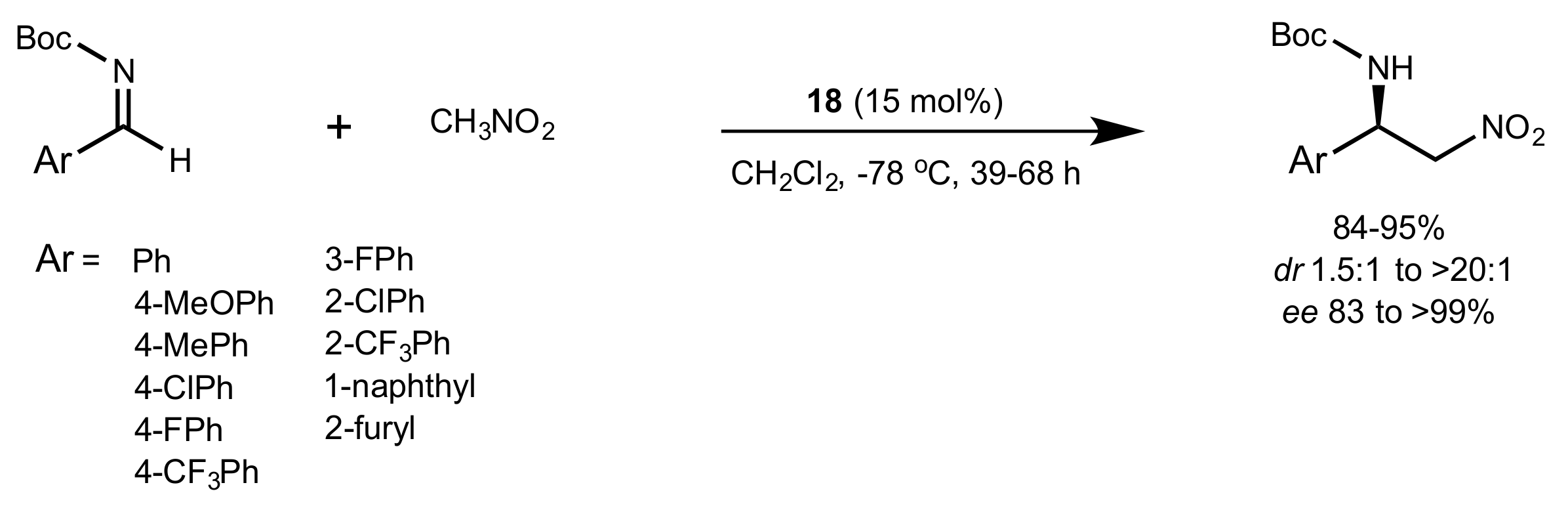
Scheme 27.
The Aza–Henry reaction catalysed by 18.
Scheme 27.
The Aza–Henry reaction catalysed by 18.
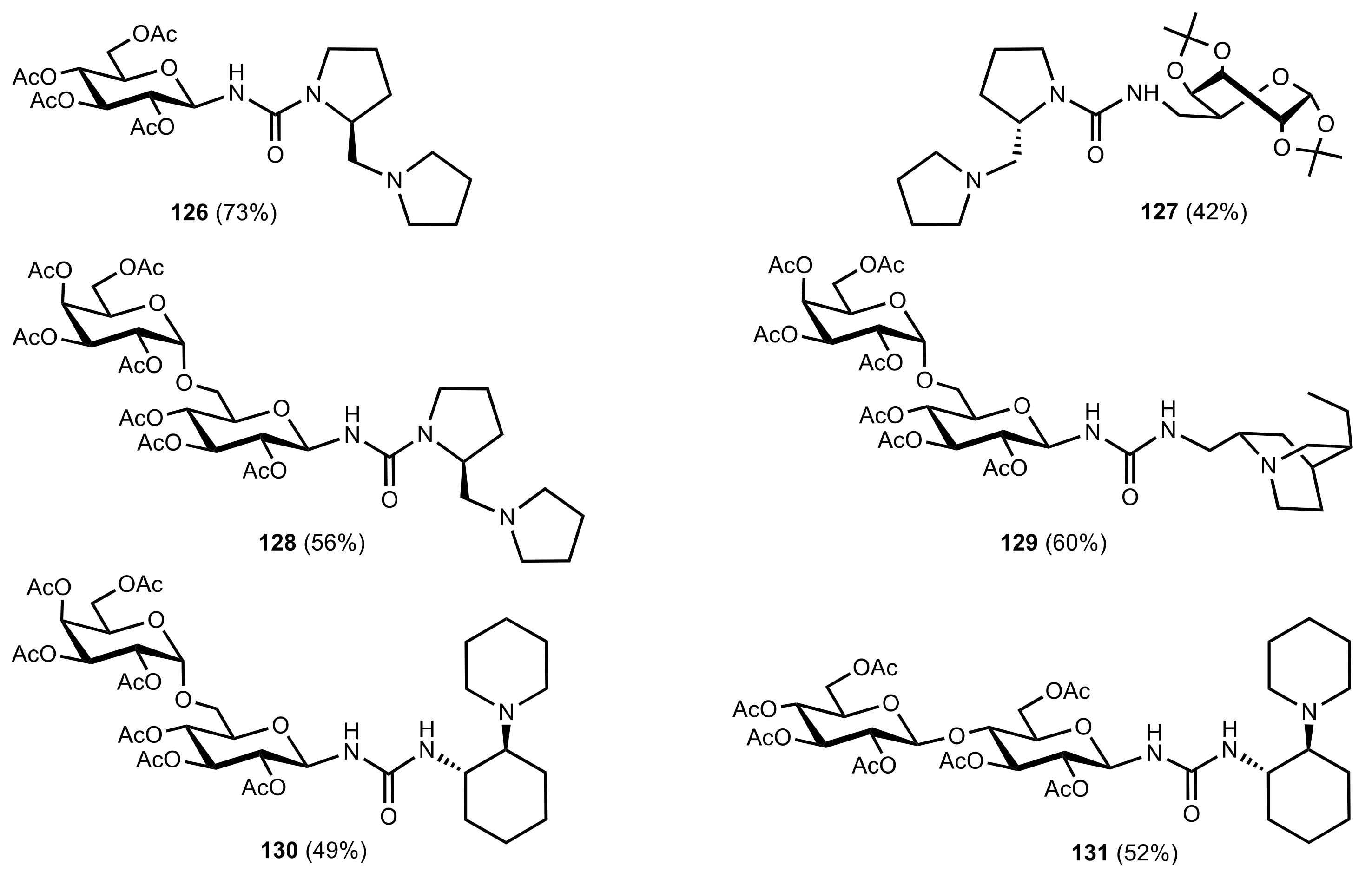
Figure 15.
The structure of organocatalysts 126–131.
Figure 15.
The structure of organocatalysts 126–131.
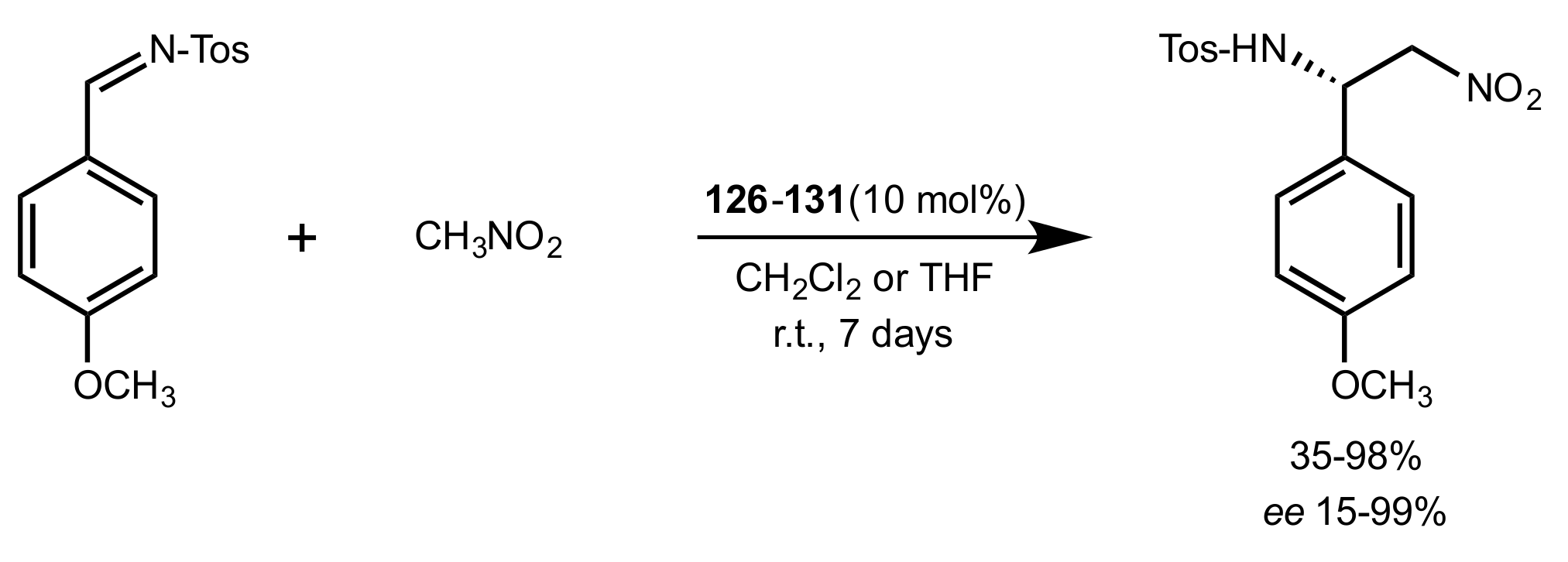
Scheme 28.
The Aza–Henry reaction catalysed by 126–131.
Scheme 28.
The Aza–Henry reaction catalysed by 126–131.
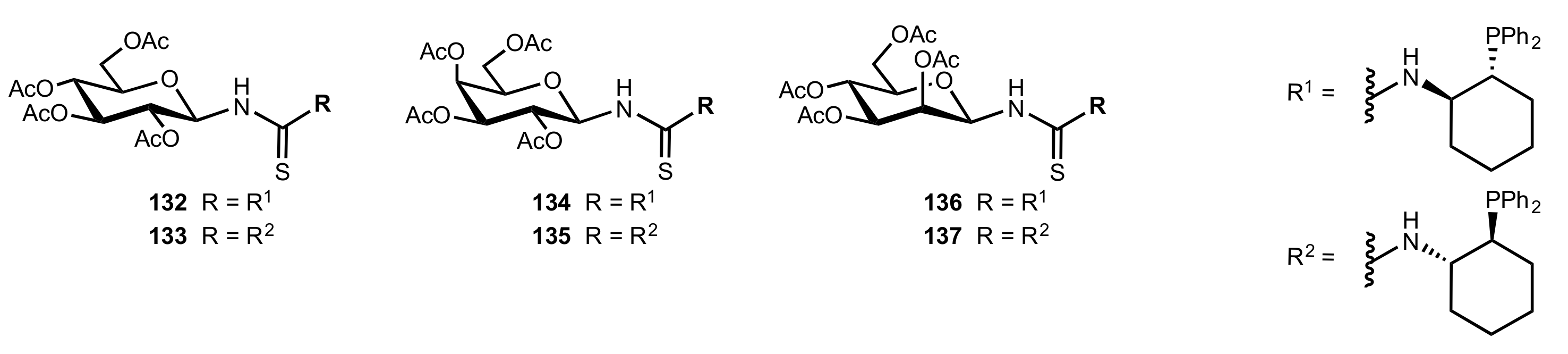
Figure 16.
The structure of organocatalysts 132–137.
Figure 16.
The structure of organocatalysts 132–137.
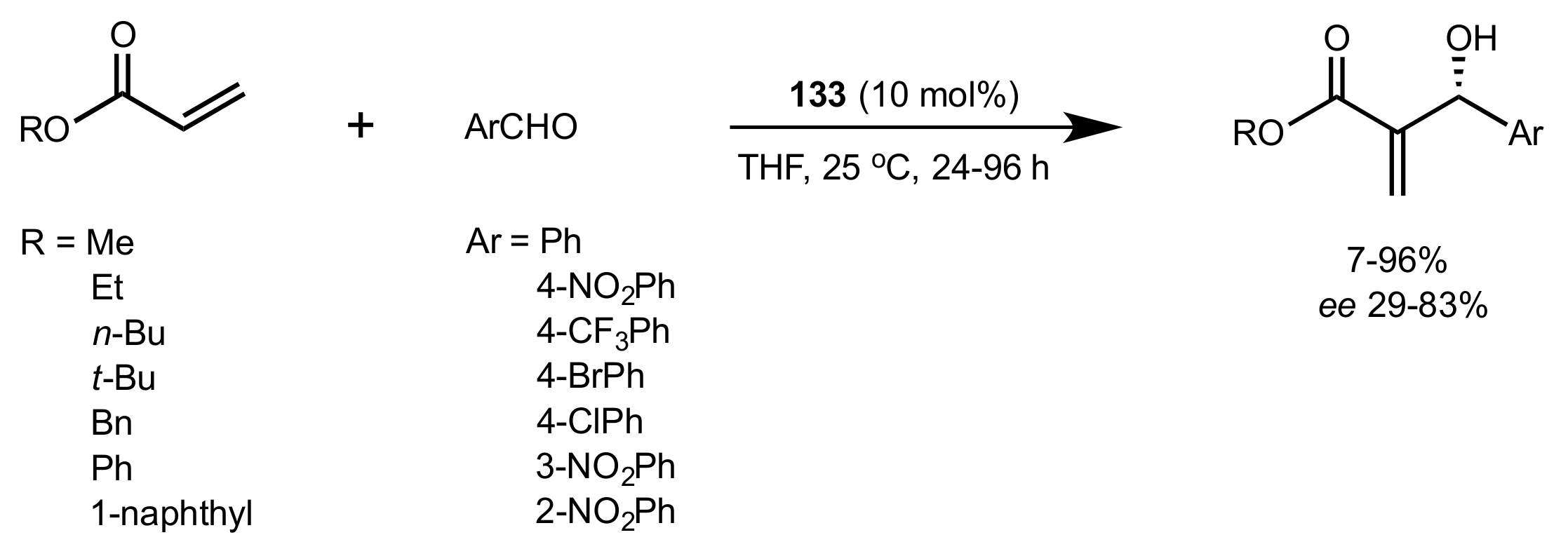
Scheme 29.
Morita–Baylis–Hillman reaction catalysed by 133.
Scheme 29.
Morita–Baylis–Hillman reaction catalysed by 133.
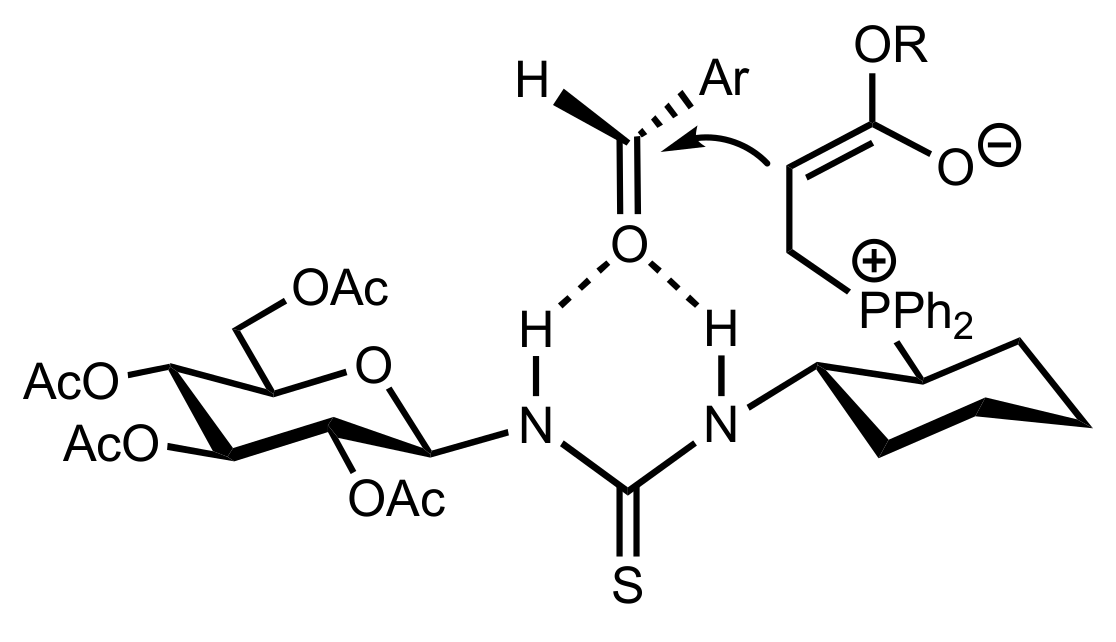
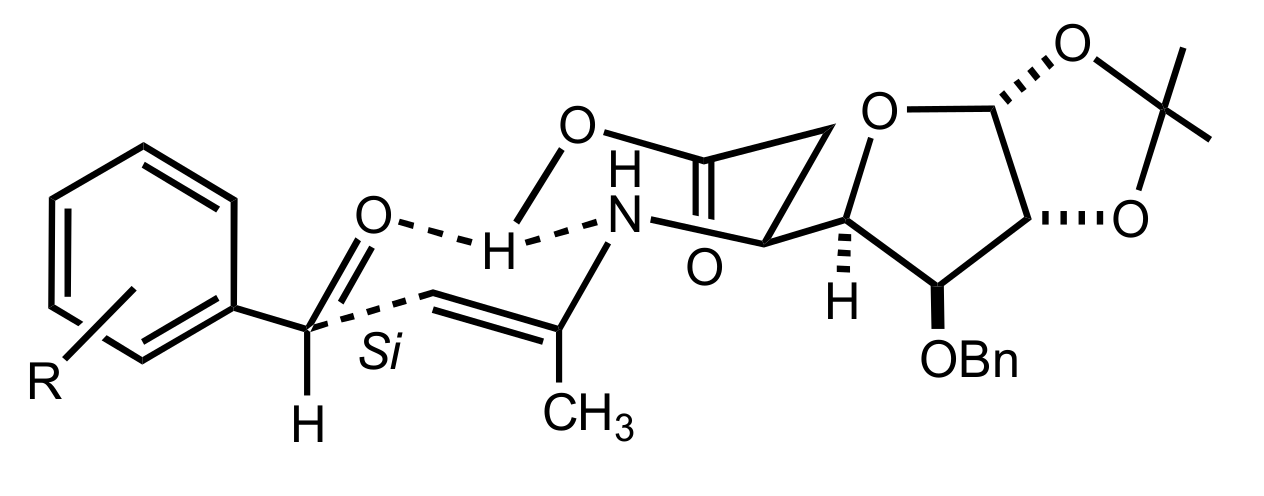
Figure 17.
The proposed transition state for the Morita–Baylis–Hillman reaction catalysed by 133.
Figure 17.
The proposed transition state for the Morita–Baylis–Hillman reaction catalysed by 133.
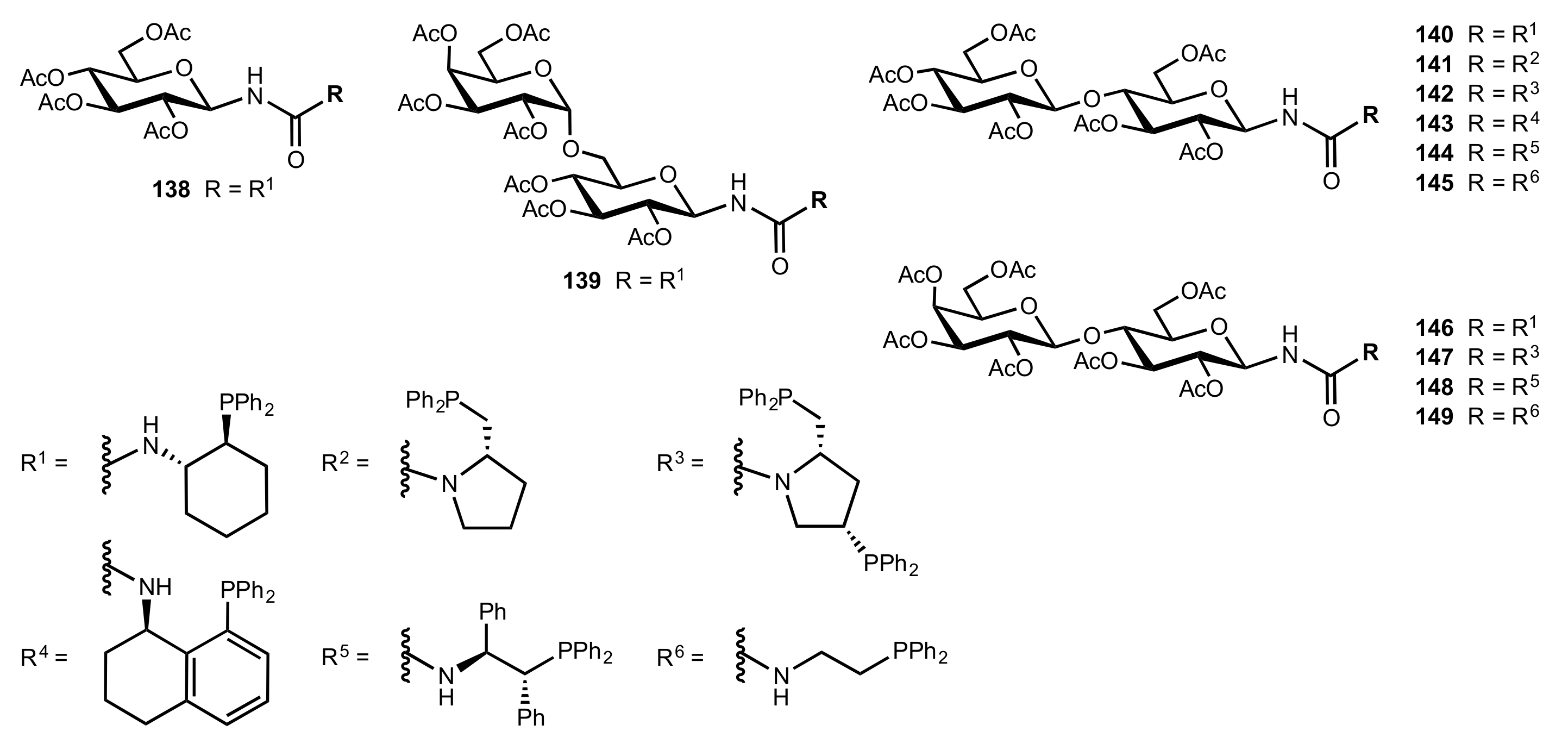
Figure 18.
The structure of organocatalysts 138–149.
Figure 18.
The structure of organocatalysts 138–149.
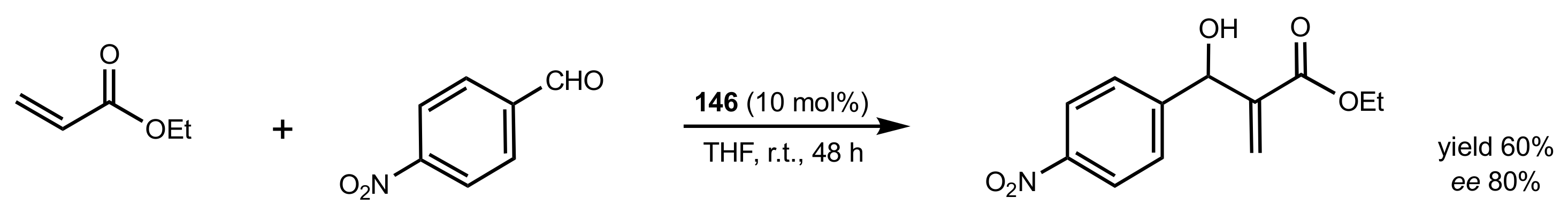
Scheme 30.
Morita–Baylis–Hillman reaction catalysed by 146.
Scheme 30.
Morita–Baylis–Hillman reaction catalysed by 146.
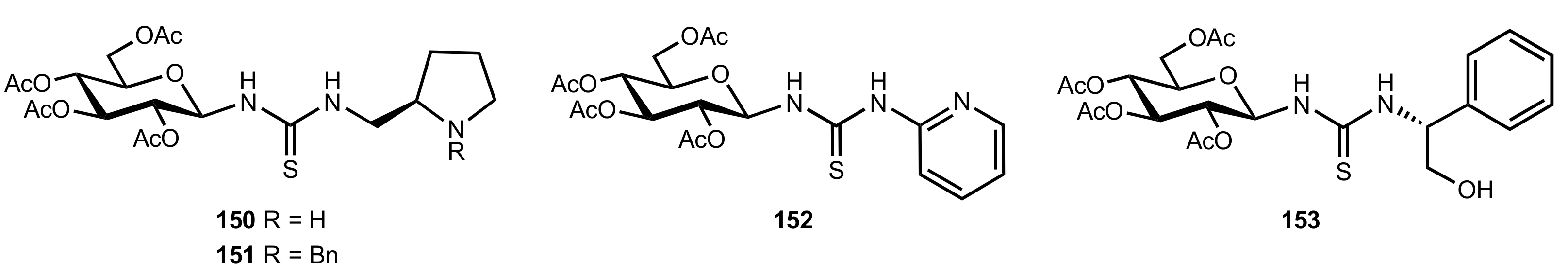
Figure 19.
The structure of organocatalysts 150–153.
Figure 19.
The structure of organocatalysts 150–153.
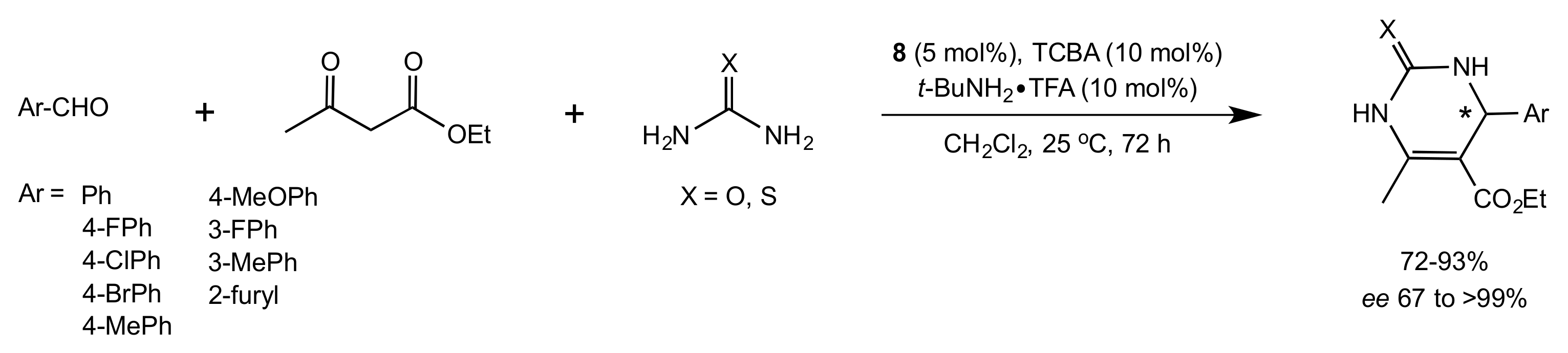
Scheme 31.
The Biginelli reaction catalysed by compound 8.
Scheme 31.
The Biginelli reaction catalysed by compound 8.
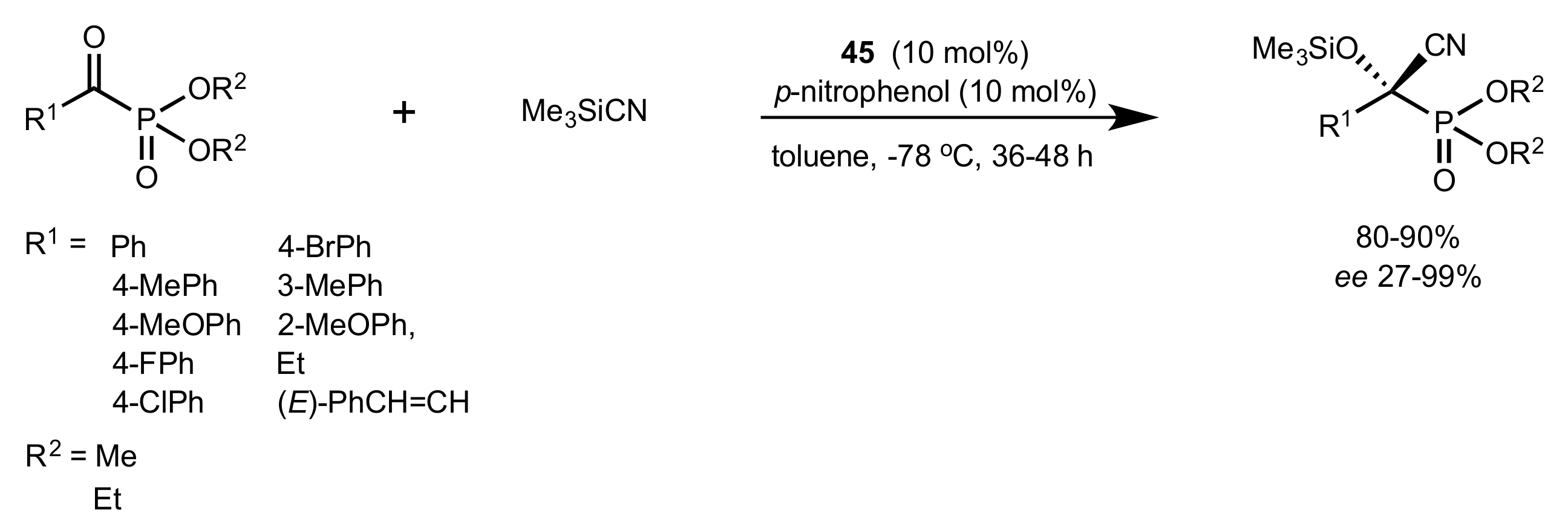
Scheme 32.
The asymmetric cyanation catalysed by 45.
Scheme 32.
The asymmetric cyanation catalysed by 45.
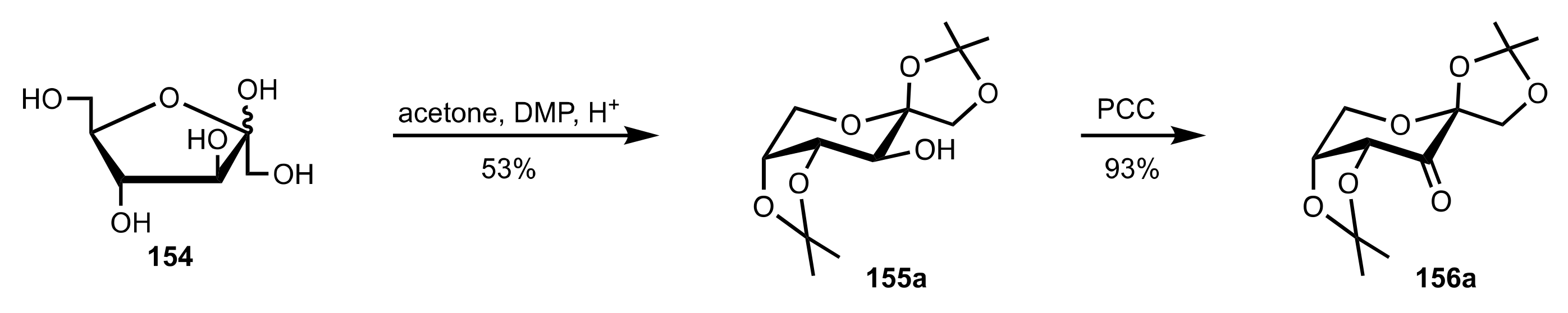
Scheme 33.
The synthesis of sugar ketone organocatalyst 156a from d-fructose.
Scheme 33.
The synthesis of sugar ketone organocatalyst 156a from d-fructose.
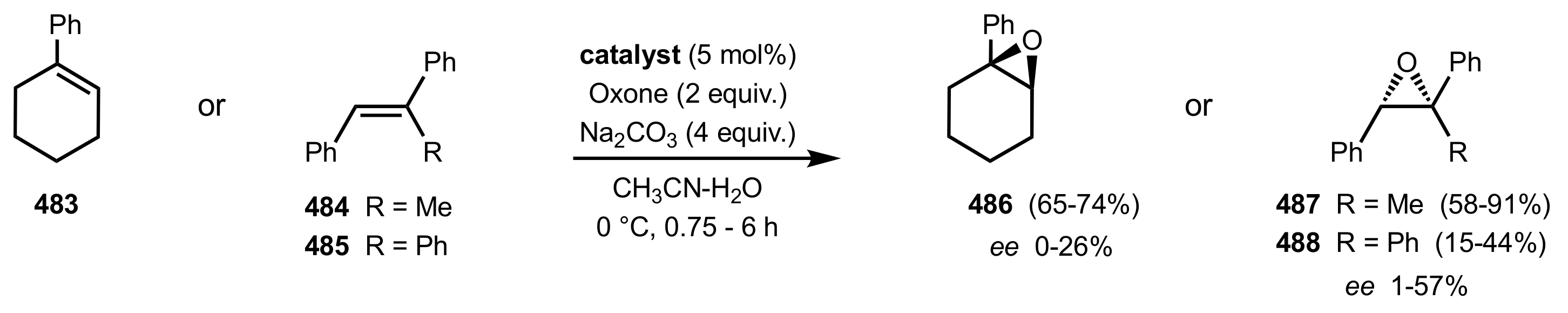
Scheme 34.
The first asymmetric epoxidations using sugar ketone organocatalyst 156a reported by Shi and co-workers.
Scheme 34.
The first asymmetric epoxidations using sugar ketone organocatalyst 156a reported by Shi and co-workers.
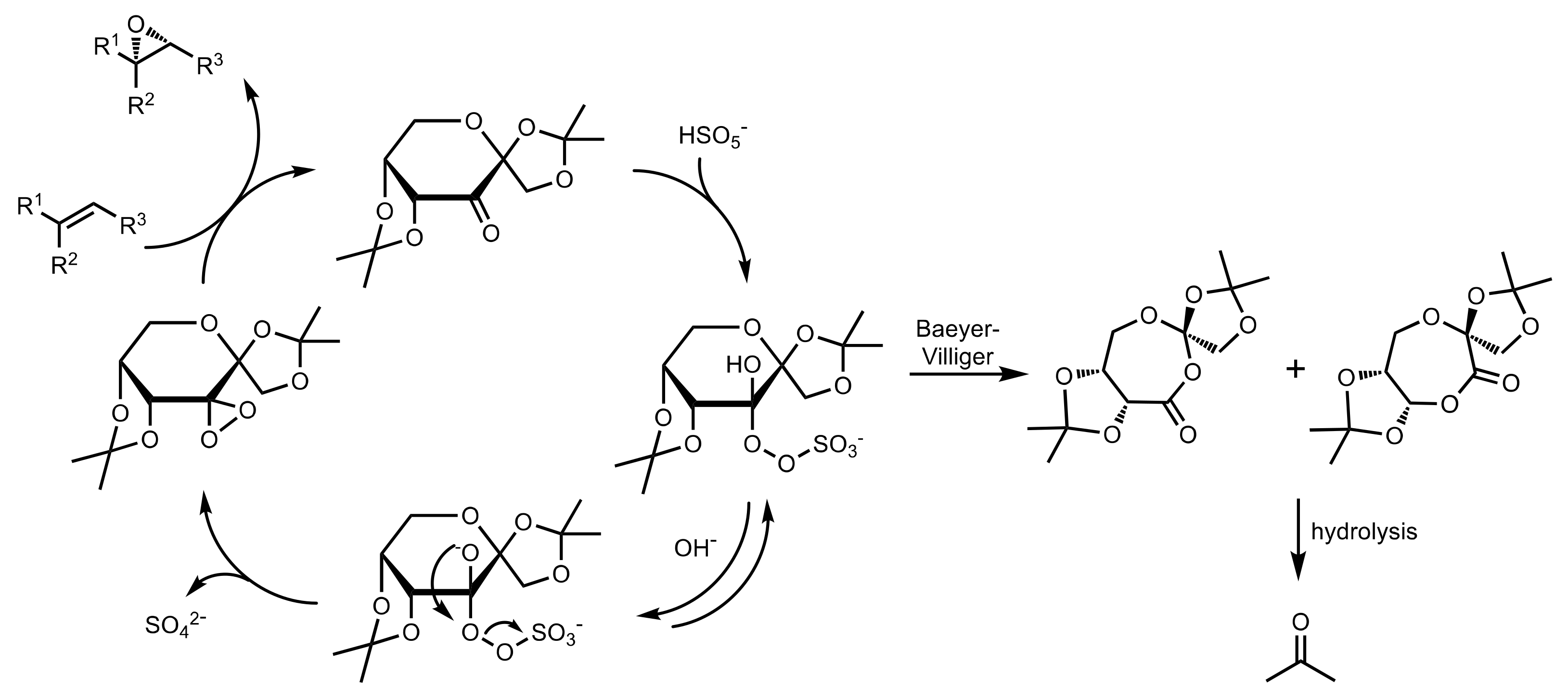
Scheme 35.
The formation of the active dioxirane species from Oxone in the asymmetric epoxidation.
Scheme 35.
The formation of the active dioxirane species from Oxone in the asymmetric epoxidation.
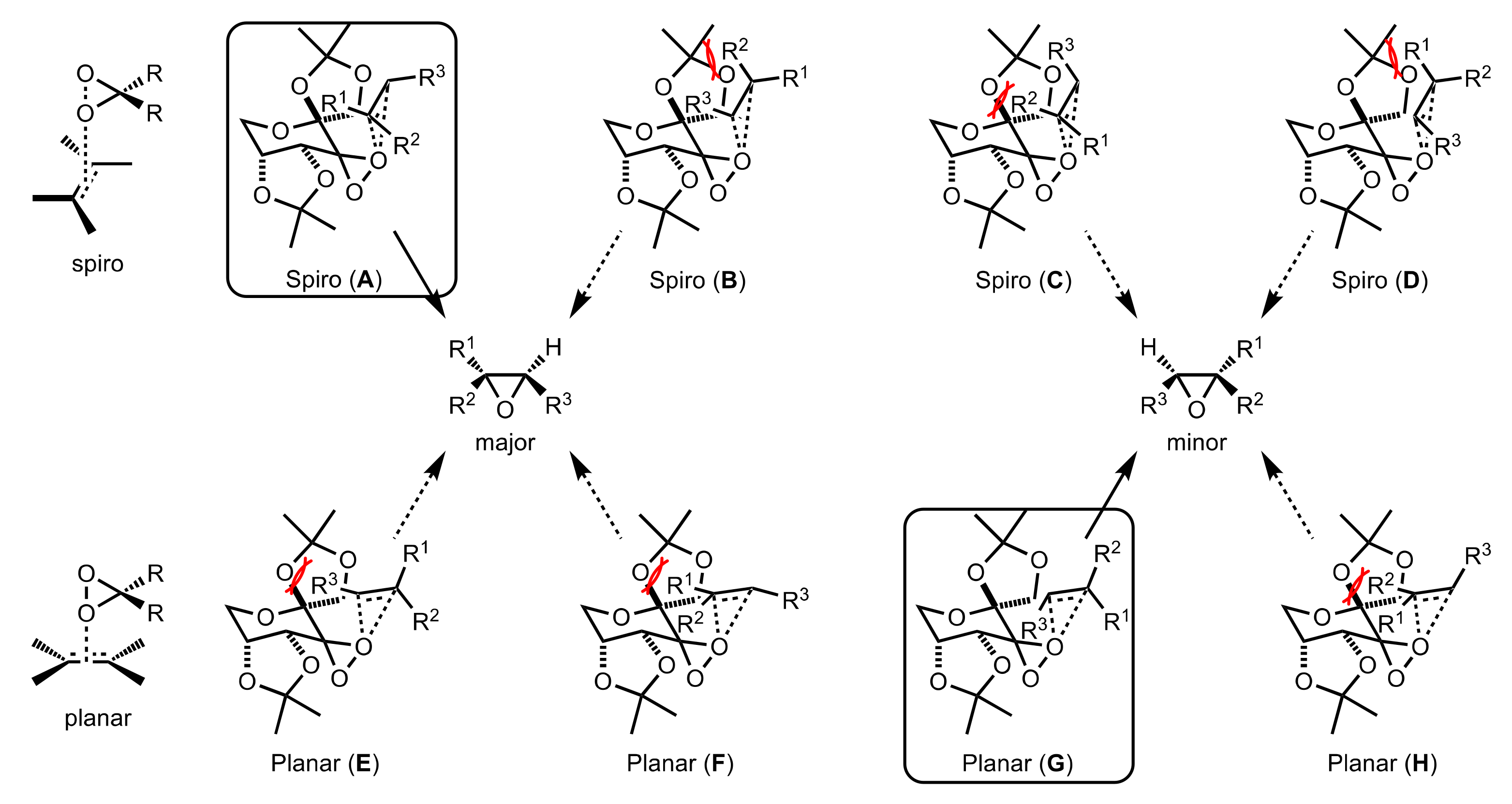
Figure 20.
The proposed transition states A–H for the asymmetric epoxidation catalysed by 156a.
Figure 20.
The proposed transition states A–H for the asymmetric epoxidation catalysed by 156a.
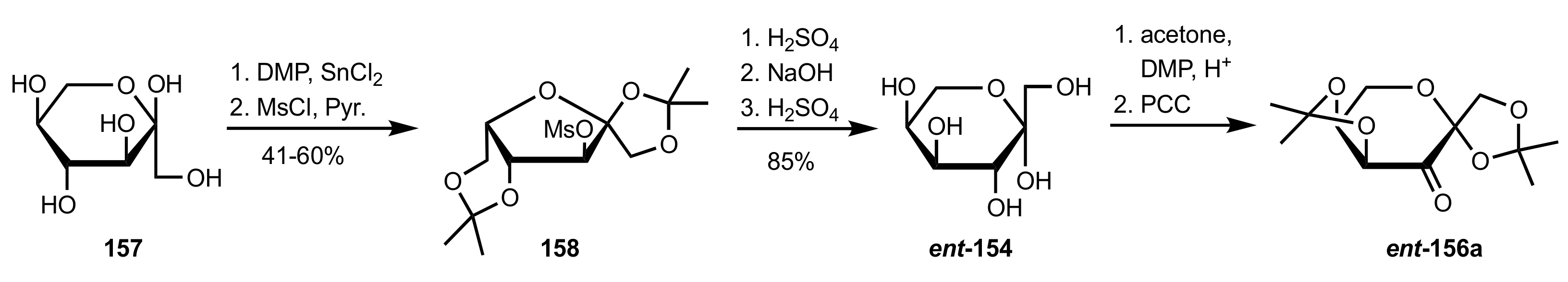
Scheme 36.
The synthesis of sugar ketone organocatalyst ent-156a from l-sorbose.
Scheme 36.
The synthesis of sugar ketone organocatalyst ent-156a from l-sorbose.
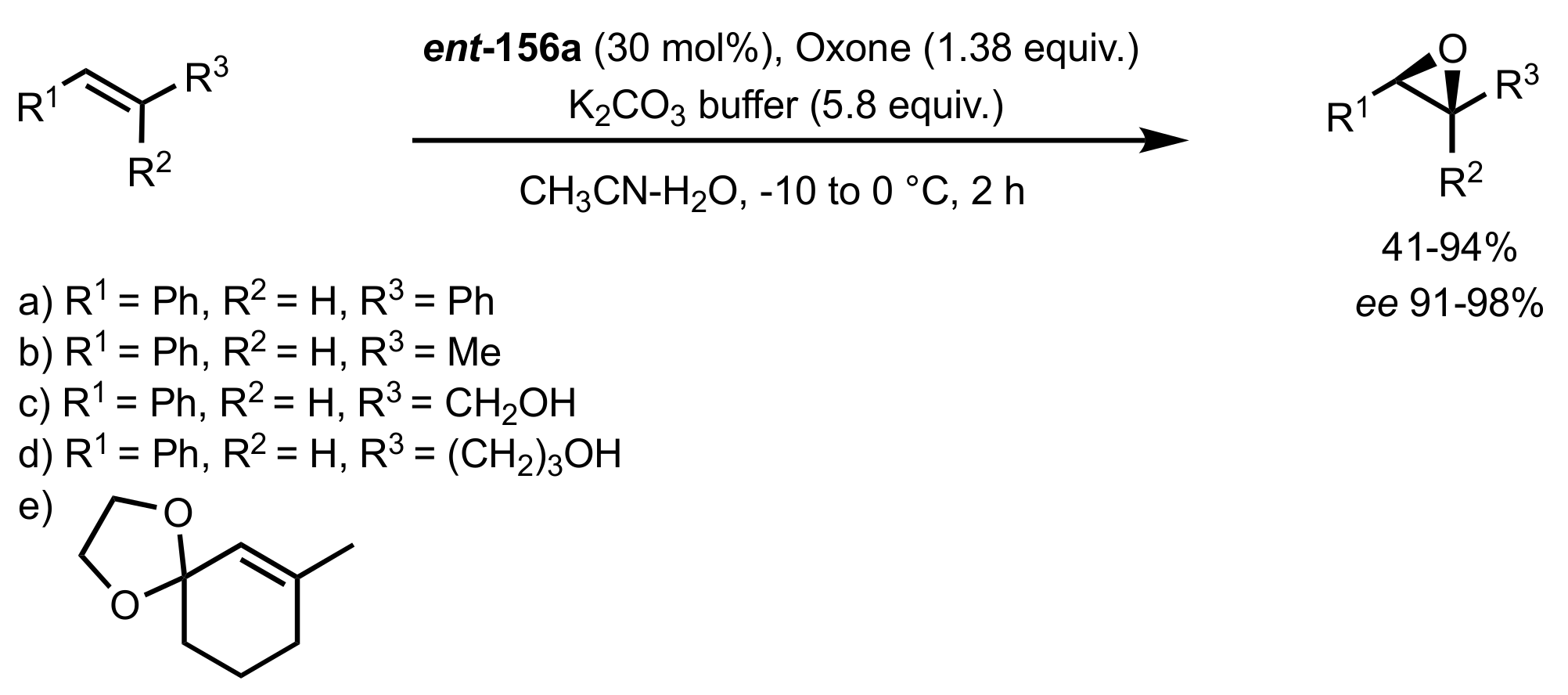
Scheme 37.
The asymmetric epoxidation catalysed by ent-156a.
Scheme 37.
The asymmetric epoxidation catalysed by ent-156a.
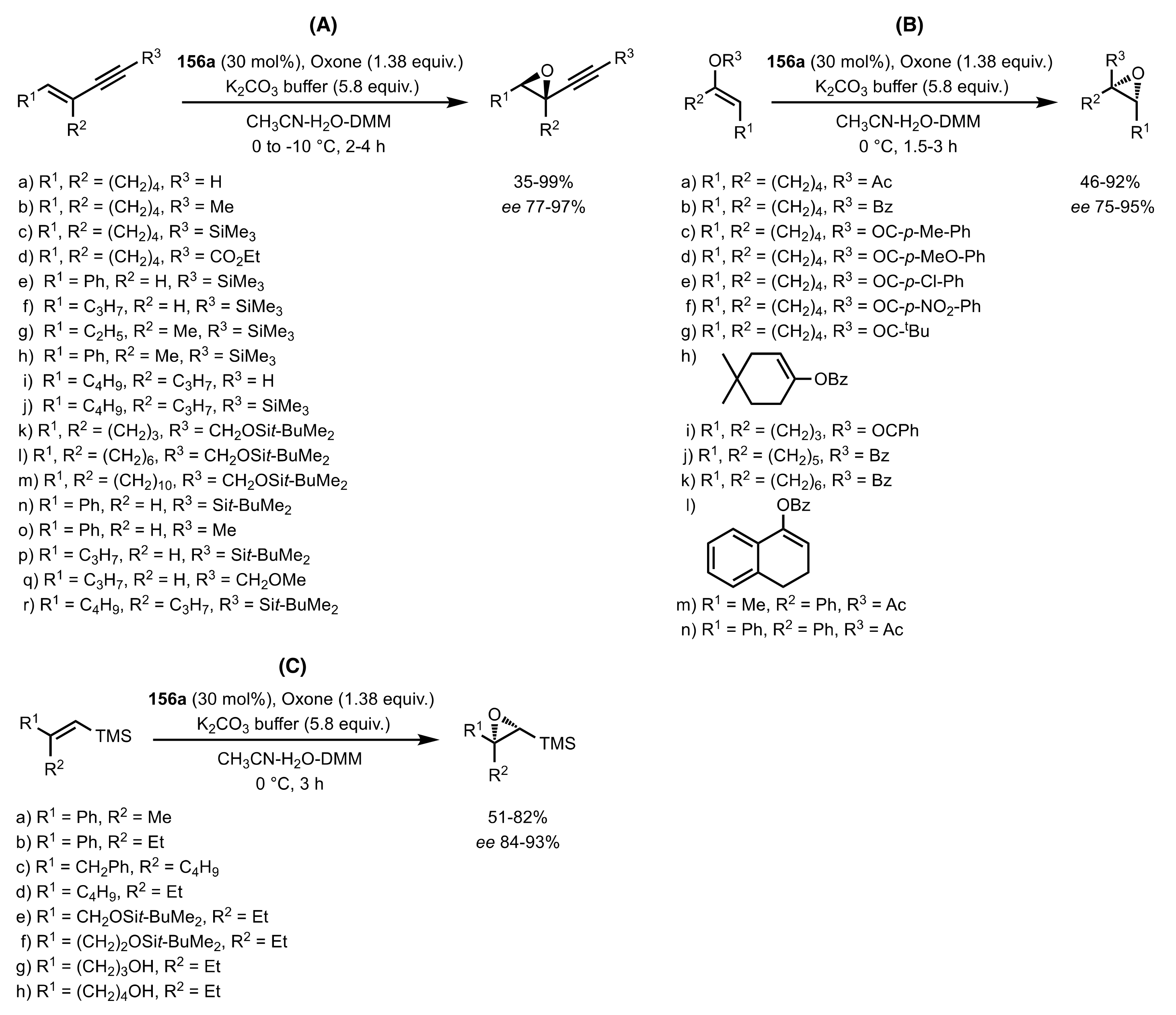
Scheme 38.
The epoxidation of enynes (A), vinylesters (B) and vinylsilanes (C) catalysed by 156a.
Scheme 38.
The epoxidation of enynes (A), vinylesters (B) and vinylsilanes (C) catalysed by 156a.
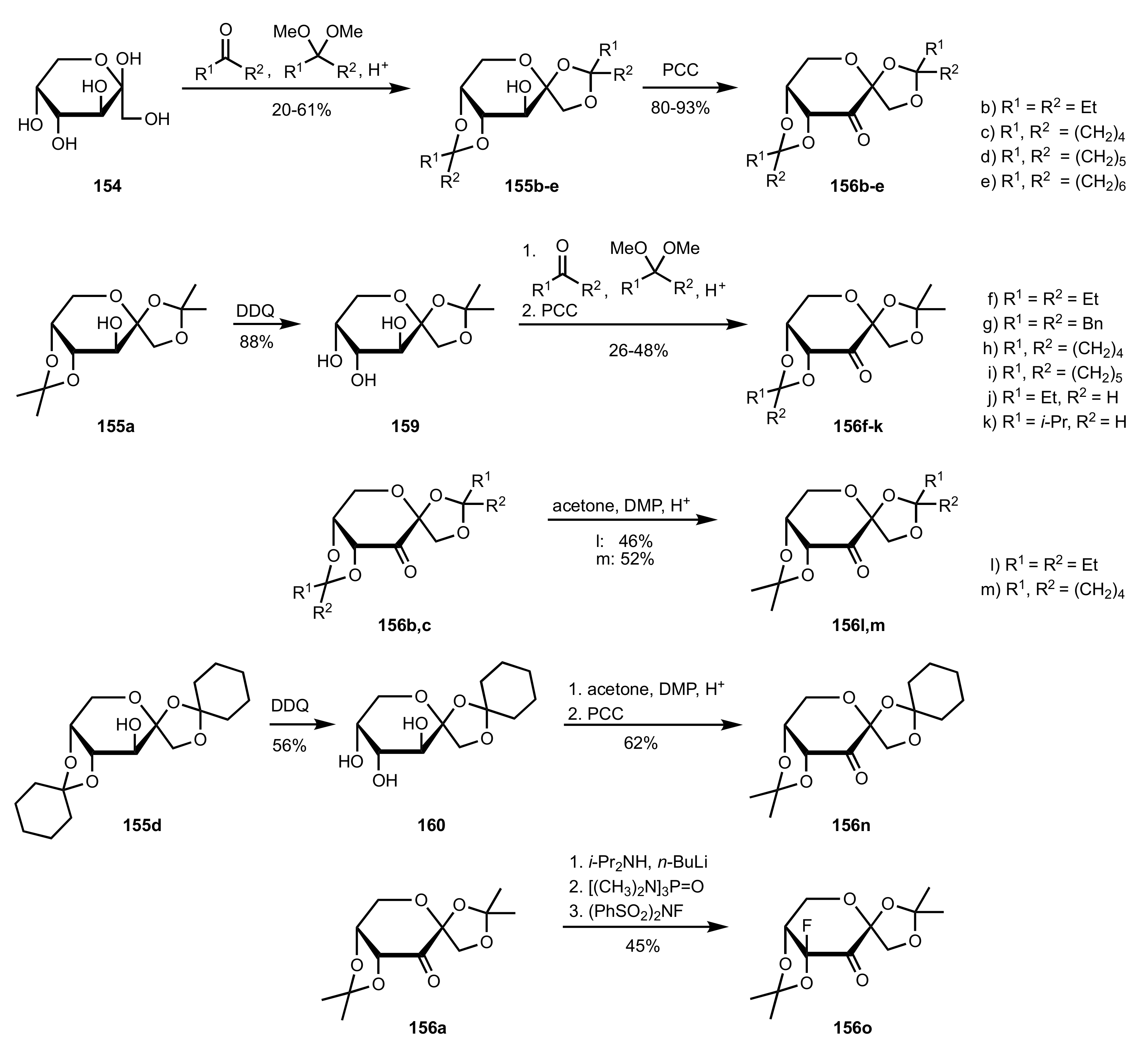
Scheme 39.
The synthesis of sugar ketone organocatalysts 156b-o from d-fructose (154).
Scheme 39.
The synthesis of sugar ketone organocatalysts 156b-o from d-fructose (154).
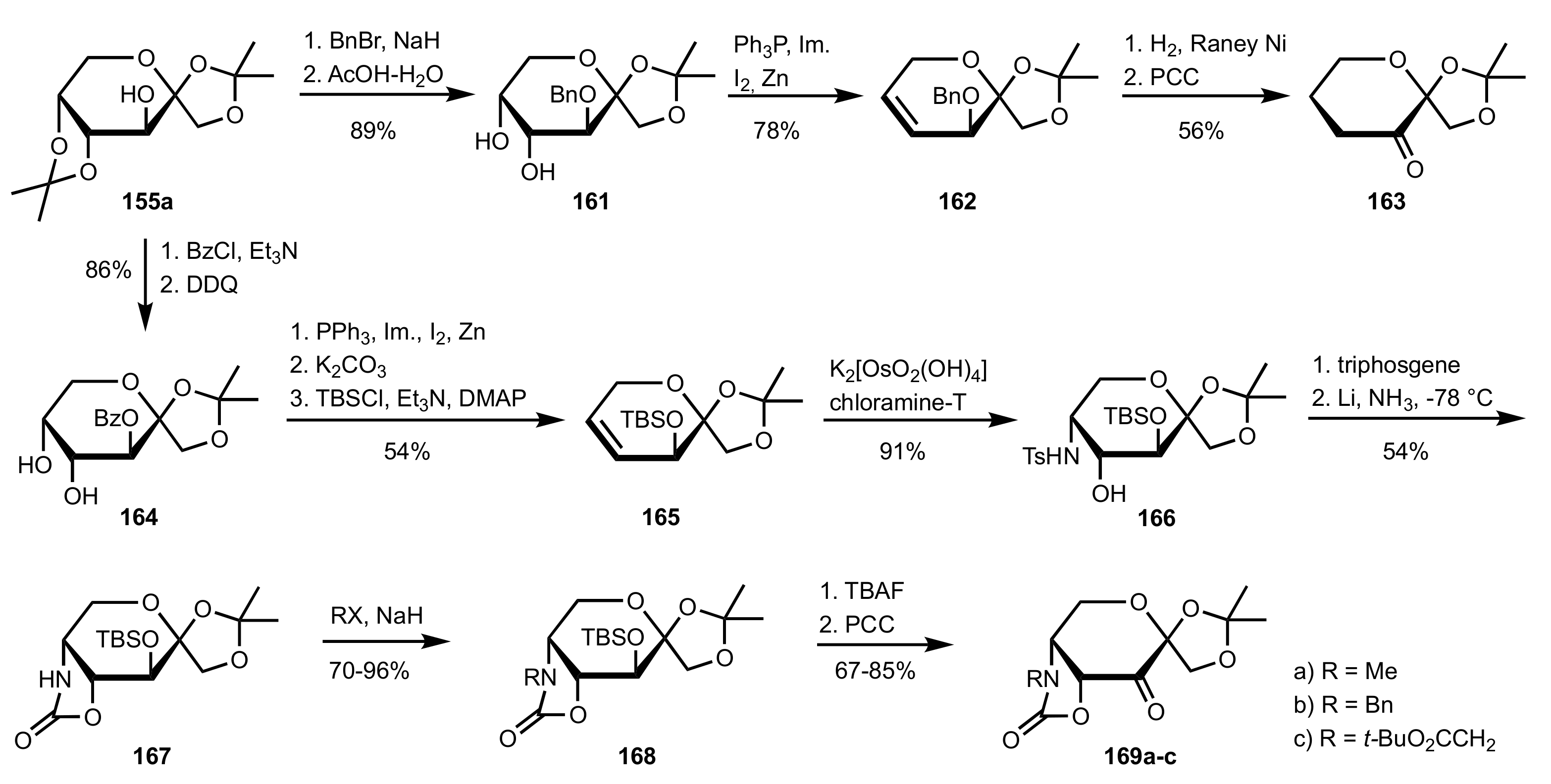
Scheme 40.
The synthesis of sugar ketone organocatalysts 161 and 169a-c from 155a.
Scheme 40.
The synthesis of sugar ketone organocatalysts 161 and 169a-c from 155a.
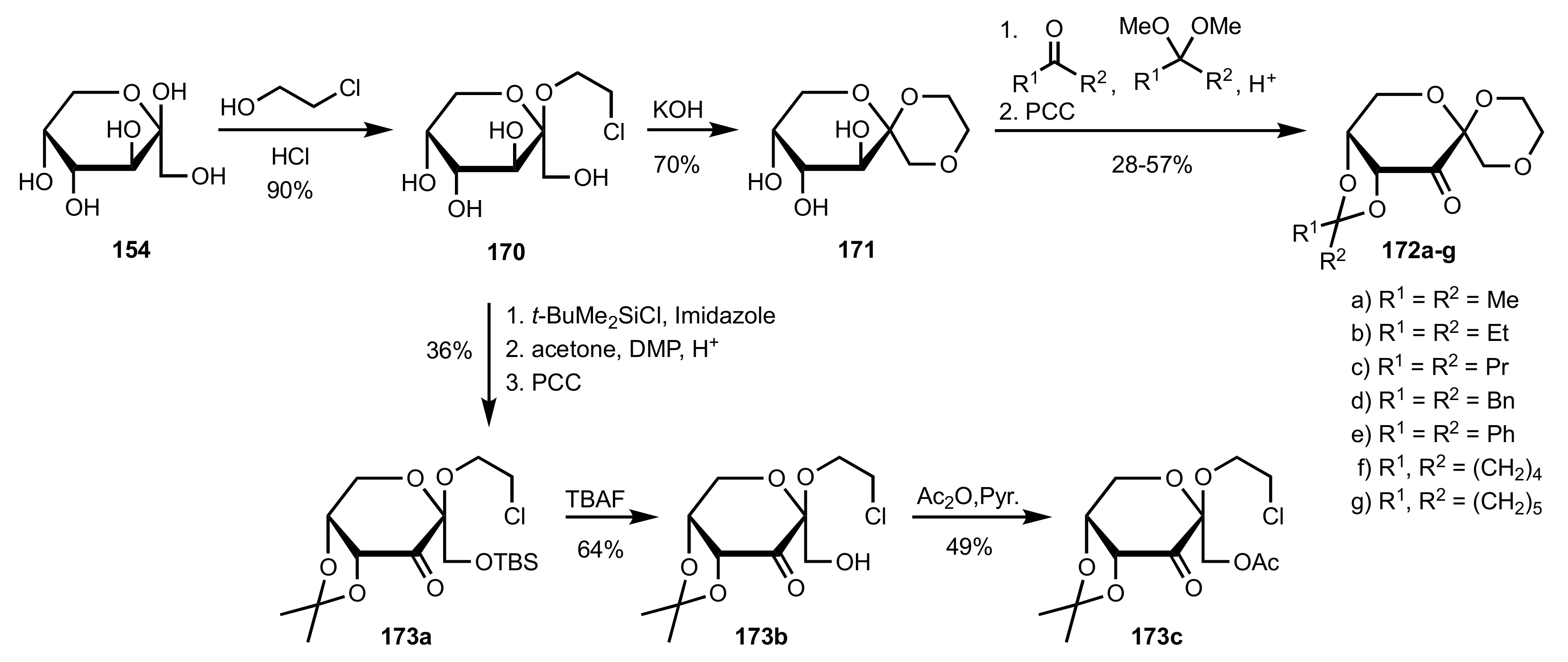
Scheme 41.
The synthesis of sugar ketone organocatalysts 172a-g and 173a-c from d-fructose.
Scheme 41.
The synthesis of sugar ketone organocatalysts 172a-g and 173a-c from d-fructose.
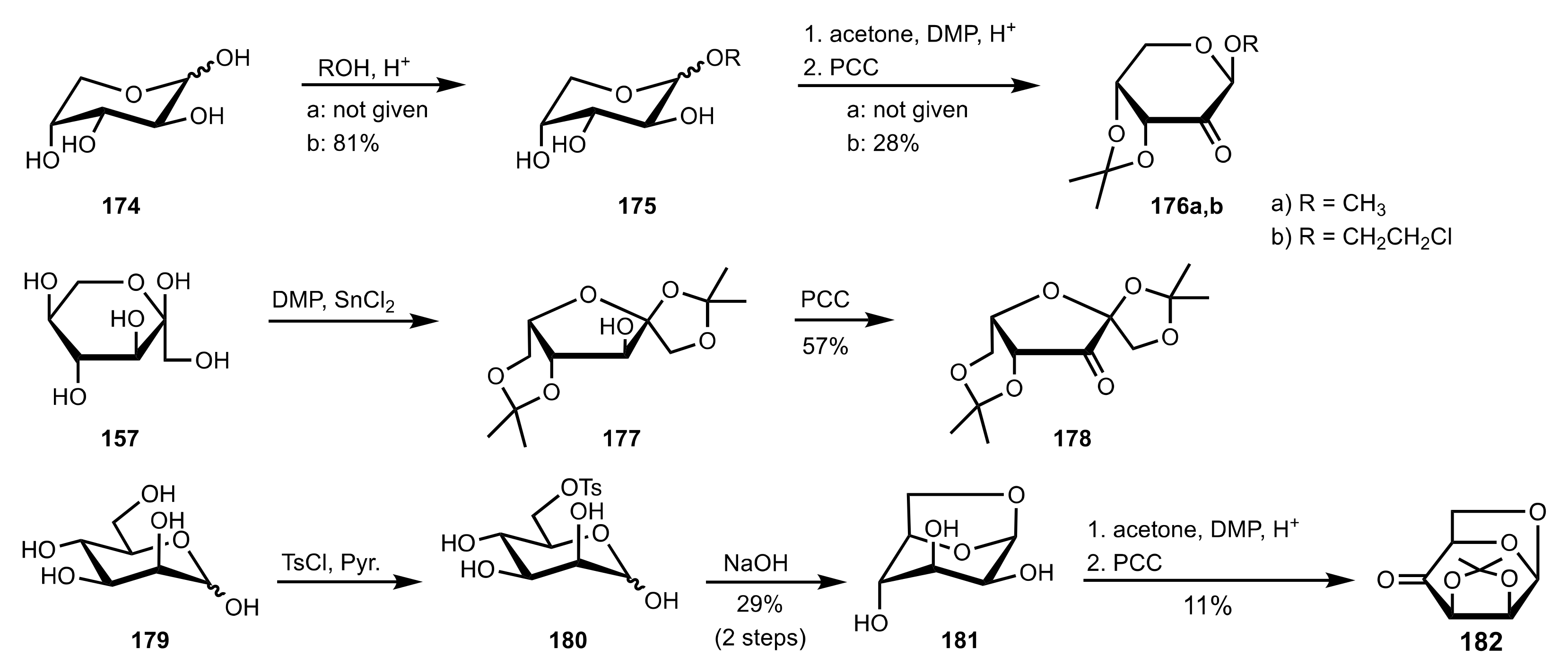
Scheme 42.
The synthesis of sugar ketone organocatalysts 176a-b, 178 and 182.
Scheme 42.
The synthesis of sugar ketone organocatalysts 176a-b, 178 and 182.
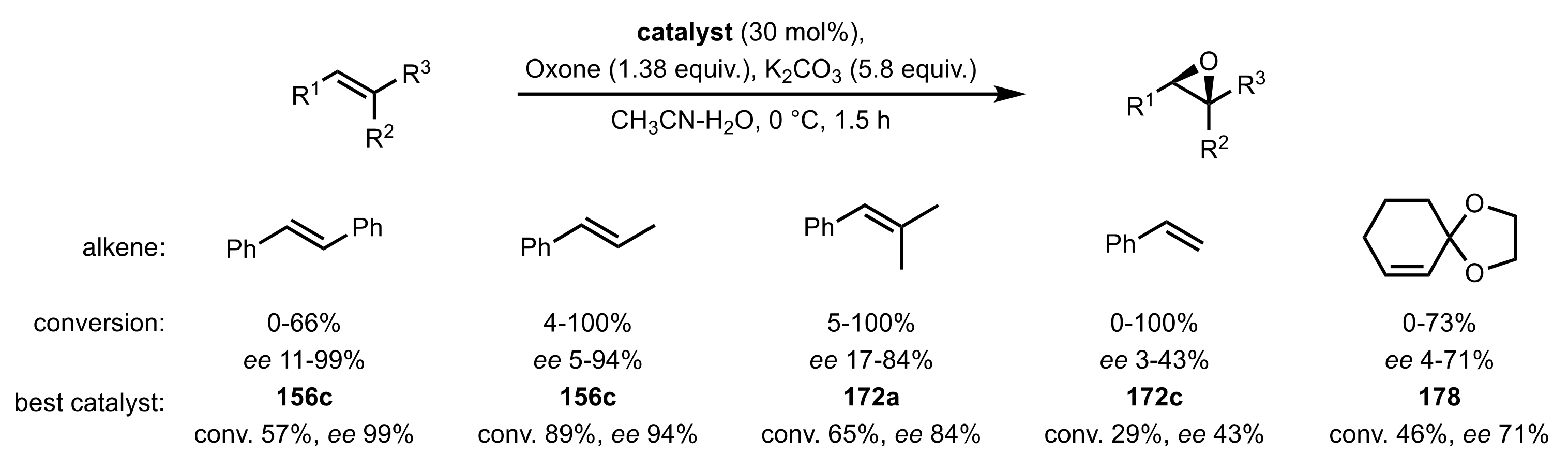
Scheme 43.
Model epoxidation reactions catalysed by the organocatalysts 156a-o, 163, 169a-c, 172a-g, 173a-c, 176a,b, 178, 182 and 88.
Scheme 43.
Model epoxidation reactions catalysed by the organocatalysts 156a-o, 163, 169a-c, 172a-g, 173a-c, 176a,b, 178, 182 and 88.
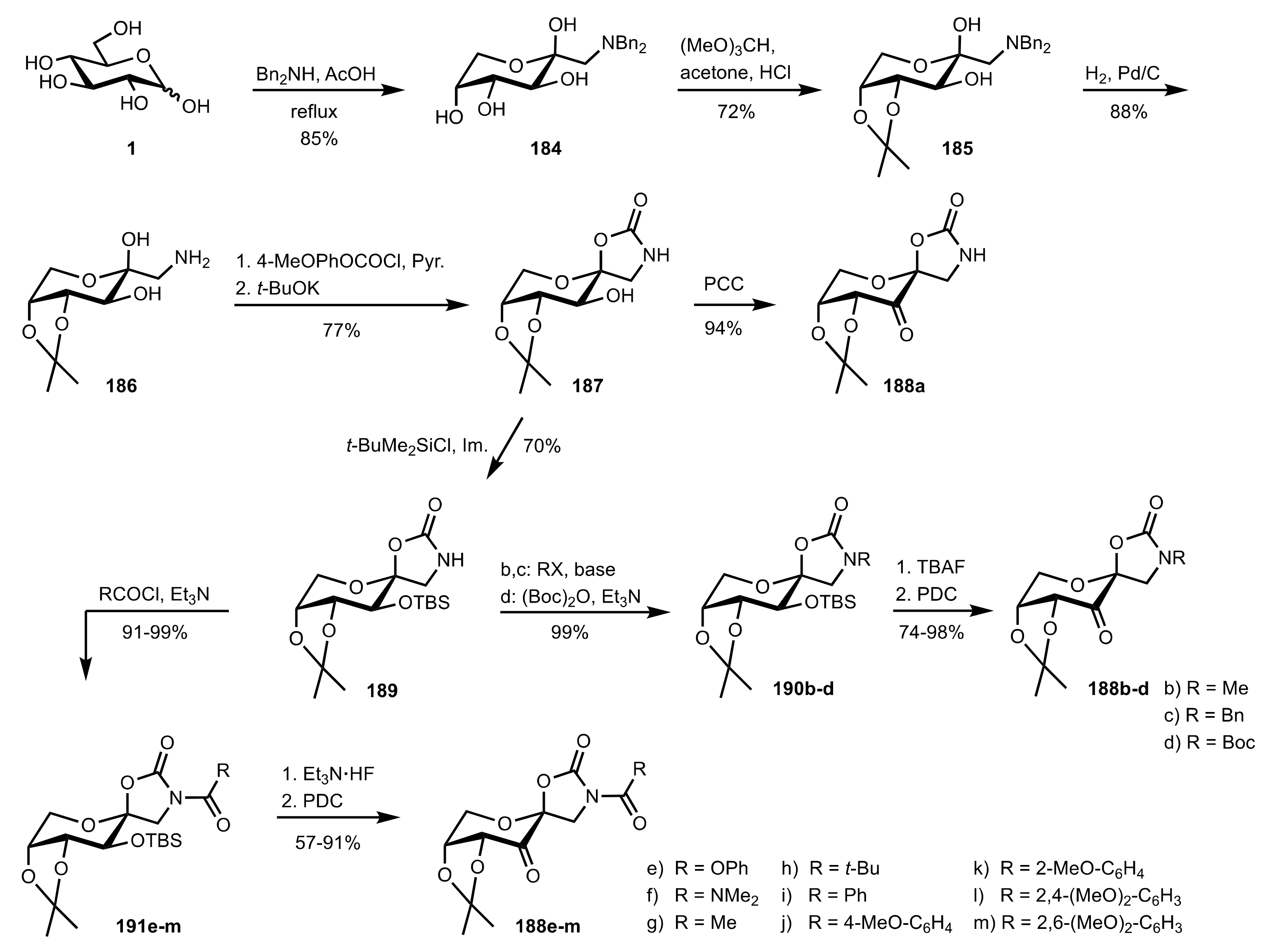
Scheme 44.
The synthesis of sugar ketone organocatalysts 188a-m from d-glucose.
Scheme 44.
The synthesis of sugar ketone organocatalysts 188a-m from d-glucose.
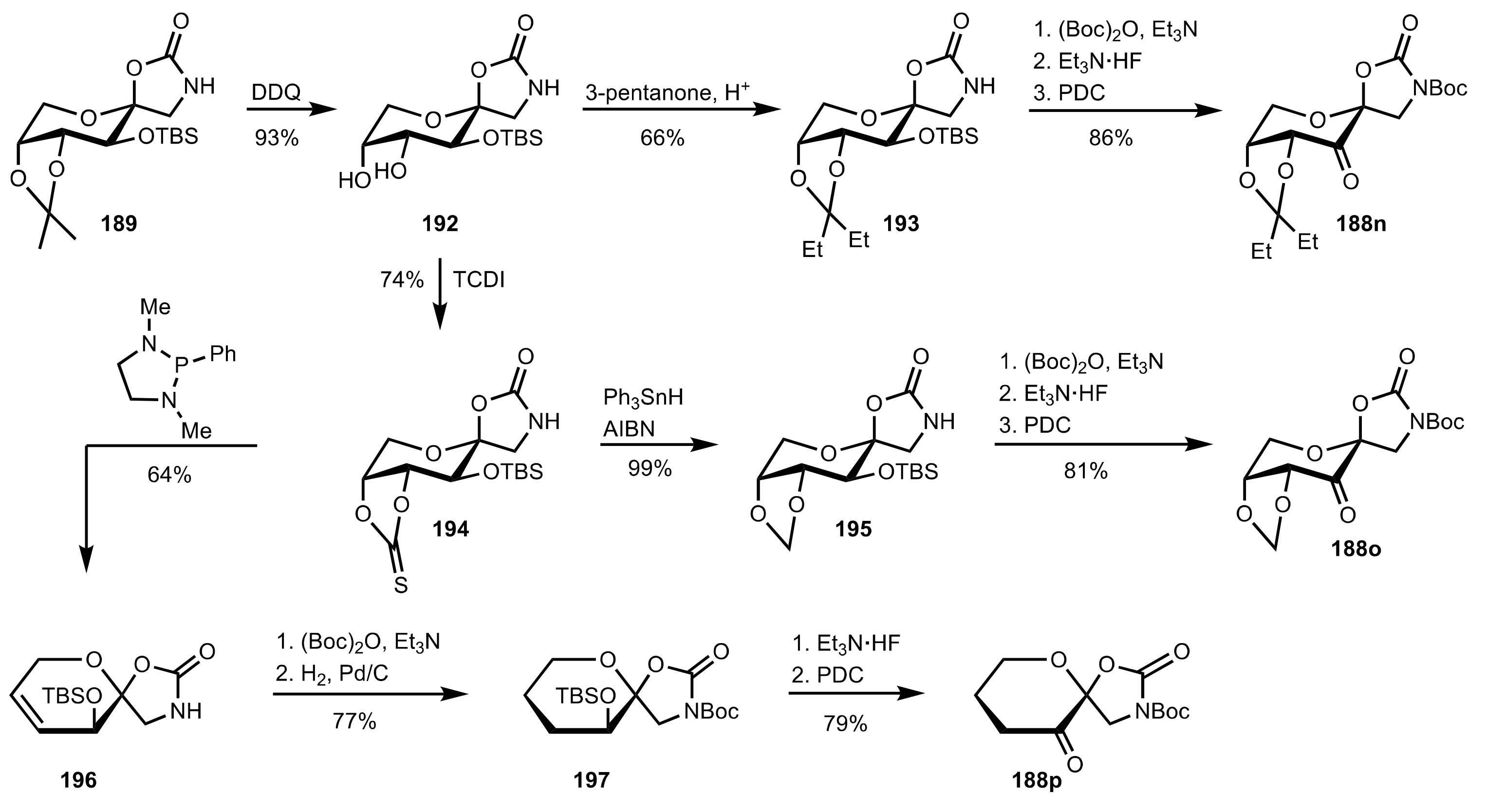
Scheme 45.
The synthesis of sugar ketone organocatalysts 188n-p.
Scheme 45.
The synthesis of sugar ketone organocatalysts 188n-p.
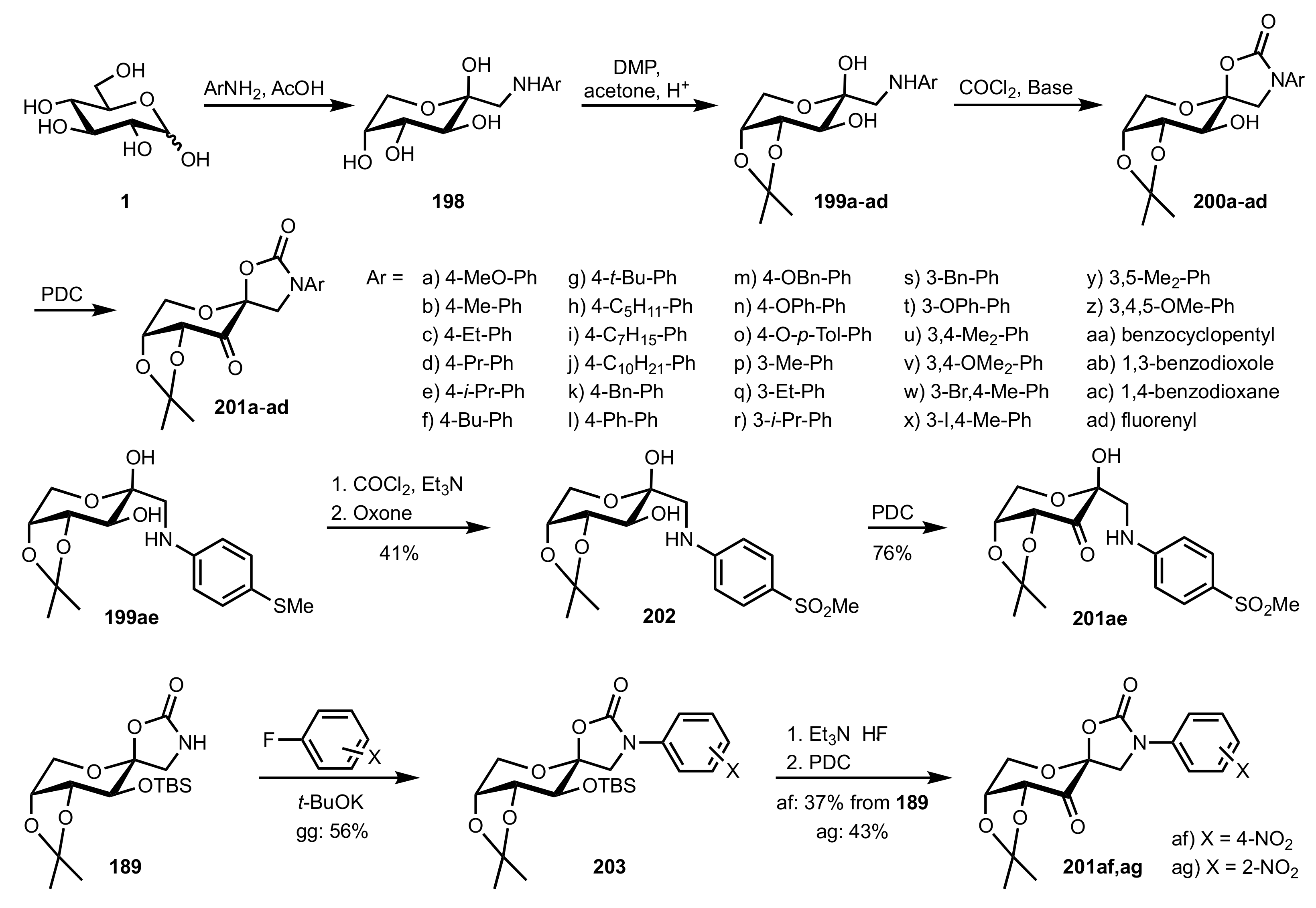
Scheme 46.
The synthesis of sugar ketone organocatalysts 201a-ag from d-glucose.
Scheme 46.
The synthesis of sugar ketone organocatalysts 201a-ag from d-glucose.
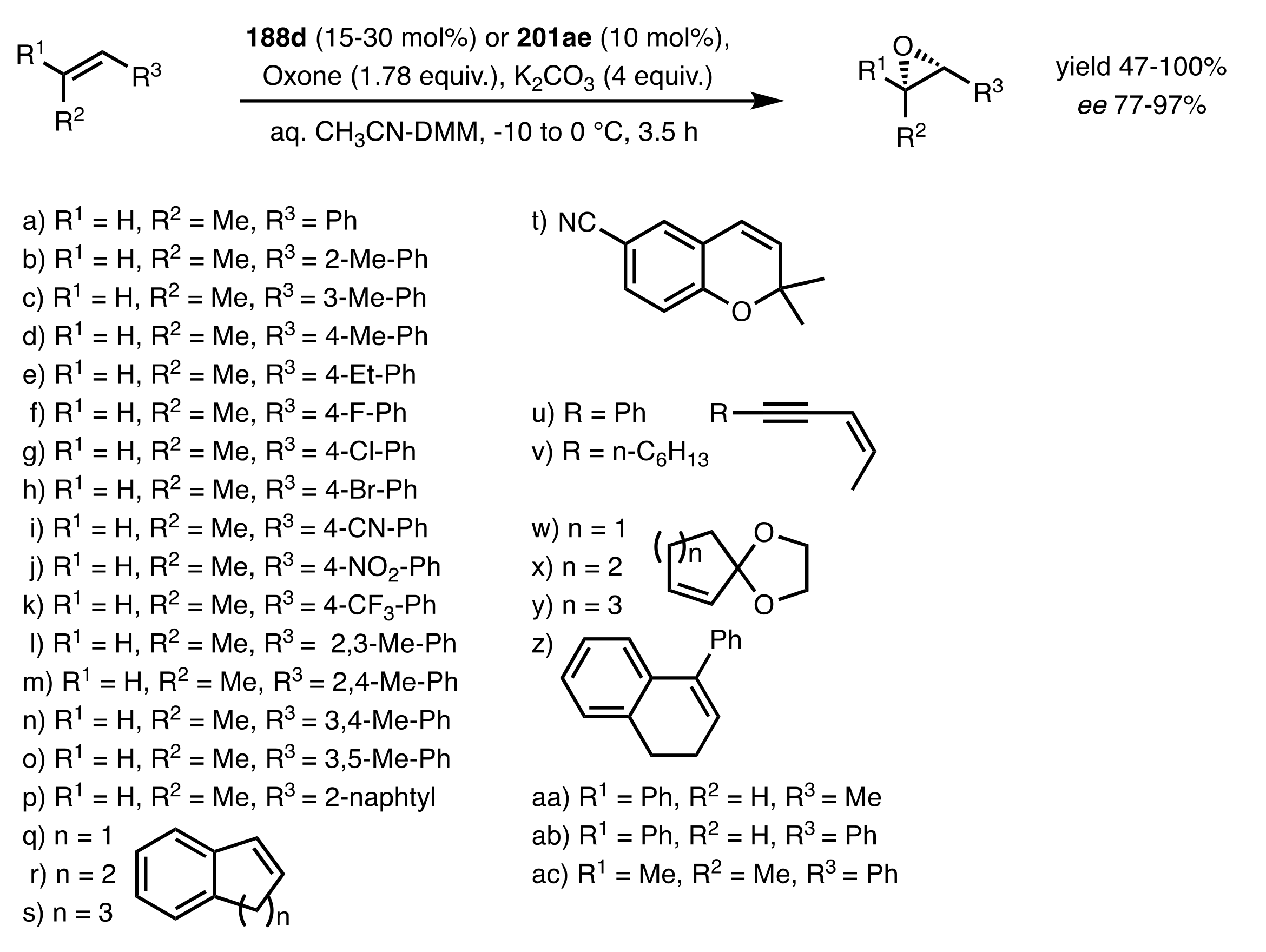
Scheme 47.
The epoxidation of alkenes (mainly cis) catalysed by 188d or 201ae.
Scheme 47.
The epoxidation of alkenes (mainly cis) catalysed by 188d or 201ae.
Scheme 48.
The epoxidation of non-conjugated alkenes catalysed by 188d (A) and 201f (B). DME = dimethoxyethane; DMM = dimethoxymethane.
Scheme 48.
The epoxidation of non-conjugated alkenes catalysed by 188d (A) and 201f (B). DME = dimethoxyethane; DMM = dimethoxymethane.
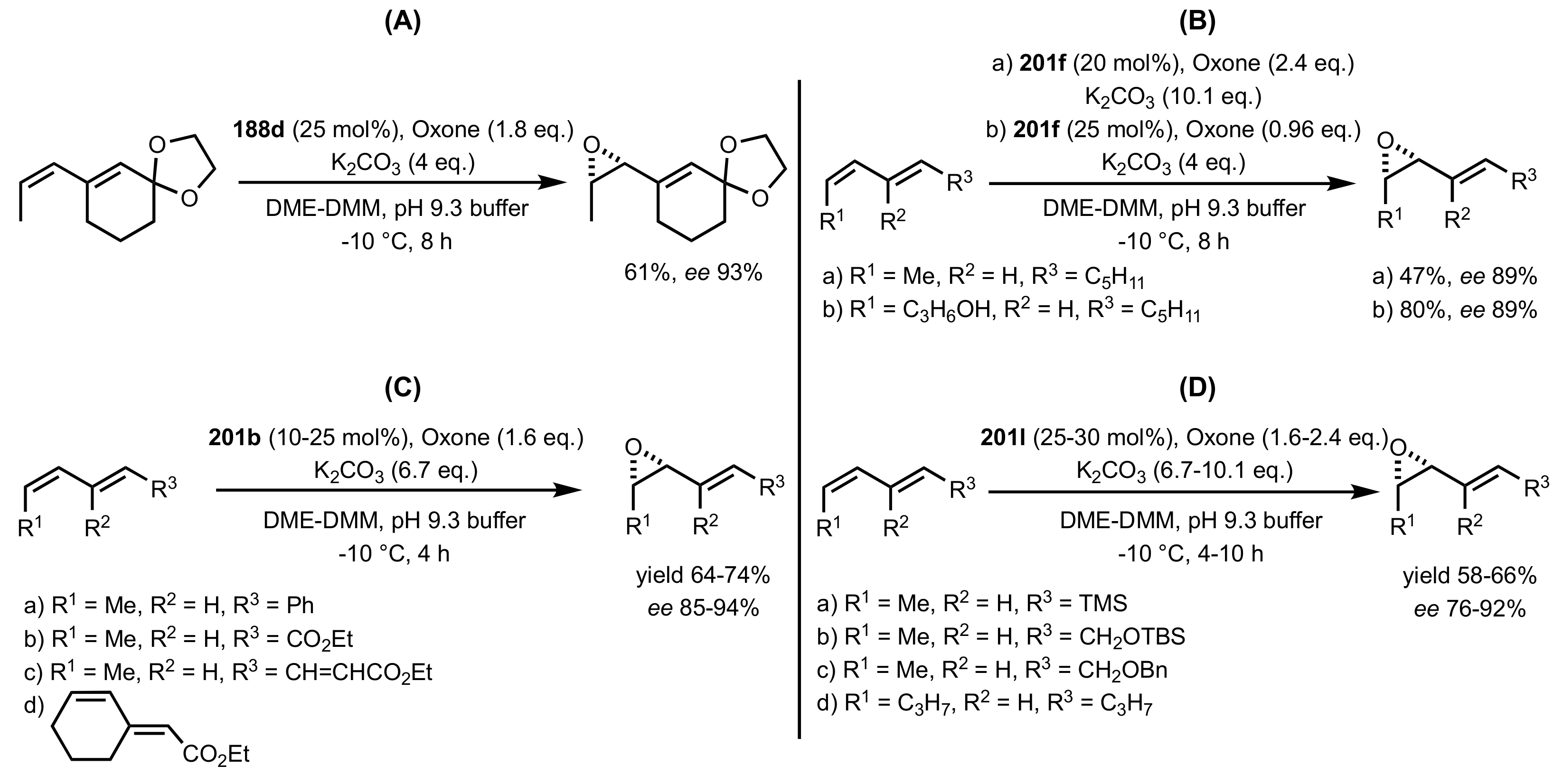
Scheme 49.
The epoxidation of conjugated dienes catalysed by 188d (A), 201f (B), 201b (C) and 201l (D).
Scheme 49.
The epoxidation of conjugated dienes catalysed by 188d (A), 201f (B), 201b (C) and 201l (D).
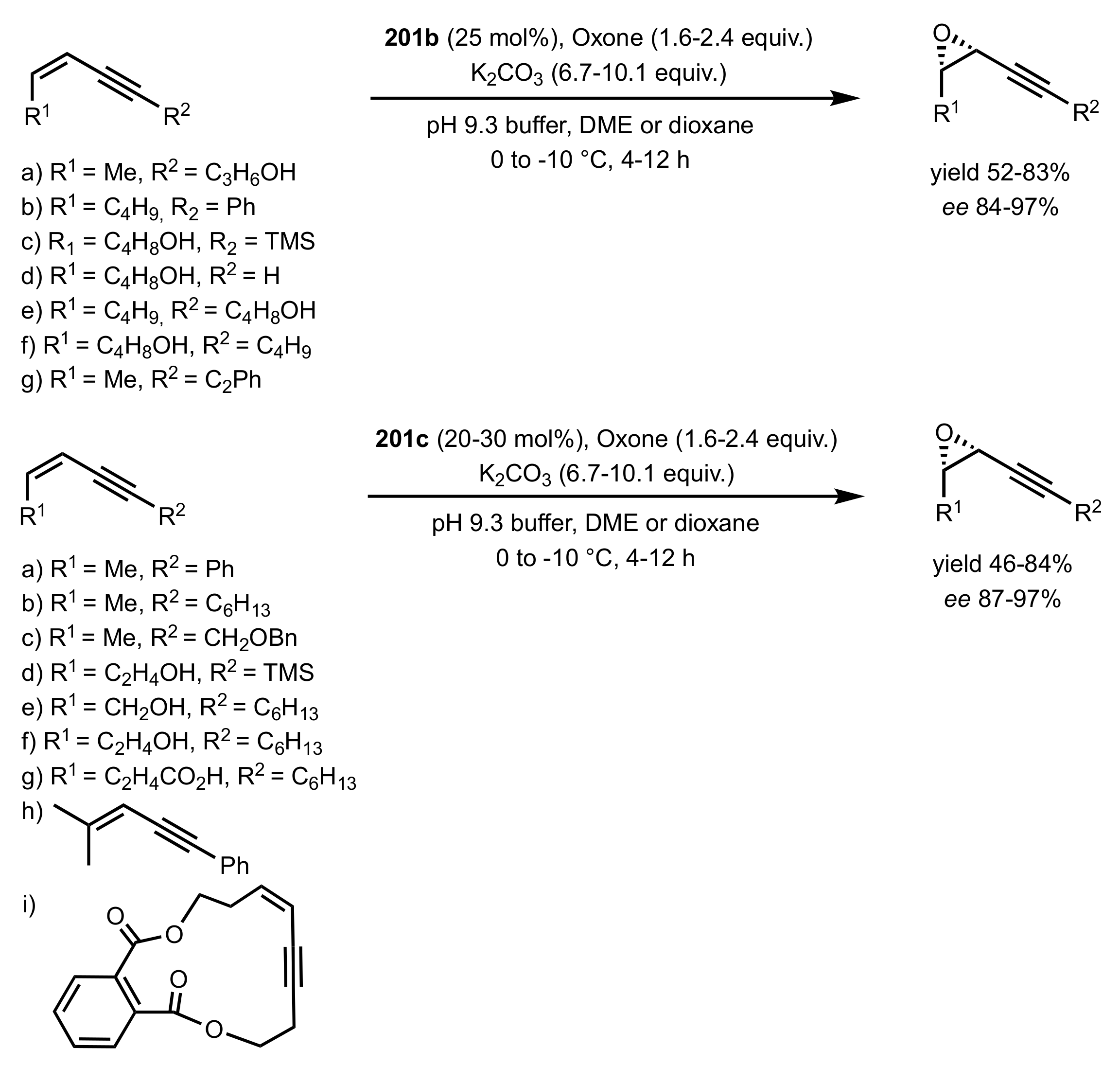
Scheme 50.
The epoxidation of conjugated cis-enynes by sugar ketones 201b,c.
Scheme 50.
The epoxidation of conjugated cis-enynes by sugar ketones 201b,c.
Scheme 51.
The epoxidation of 1-cyclobutylidene-1-phenylethane derivatives catalysed by 201b and epoxide rearrangement.
Scheme 51.
The epoxidation of 1-cyclobutylidene-1-phenylethane derivatives catalysed by 201b and epoxide rearrangement.
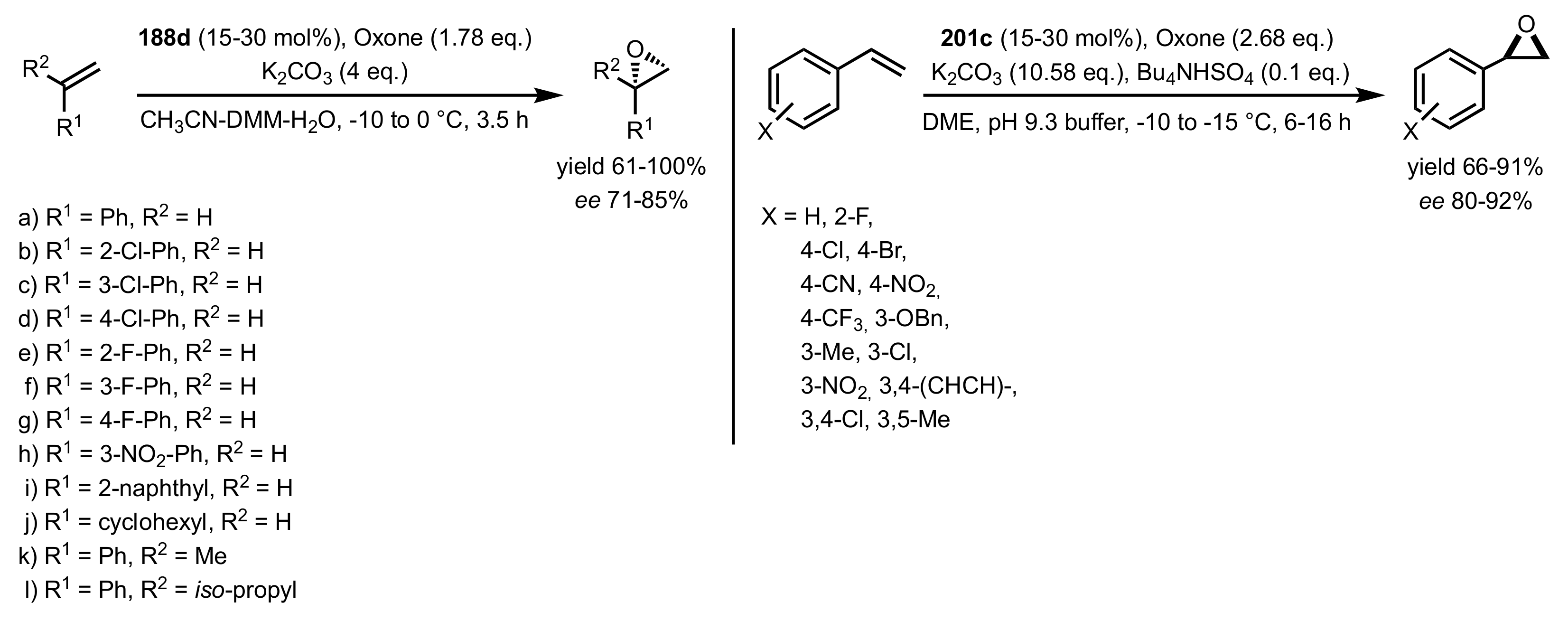
Scheme 52.
The epoxidation of terminal alkenes and styrenes catalysed by 188d and 201c.
Scheme 52.
The epoxidation of terminal alkenes and styrenes catalysed by 188d and 201c.
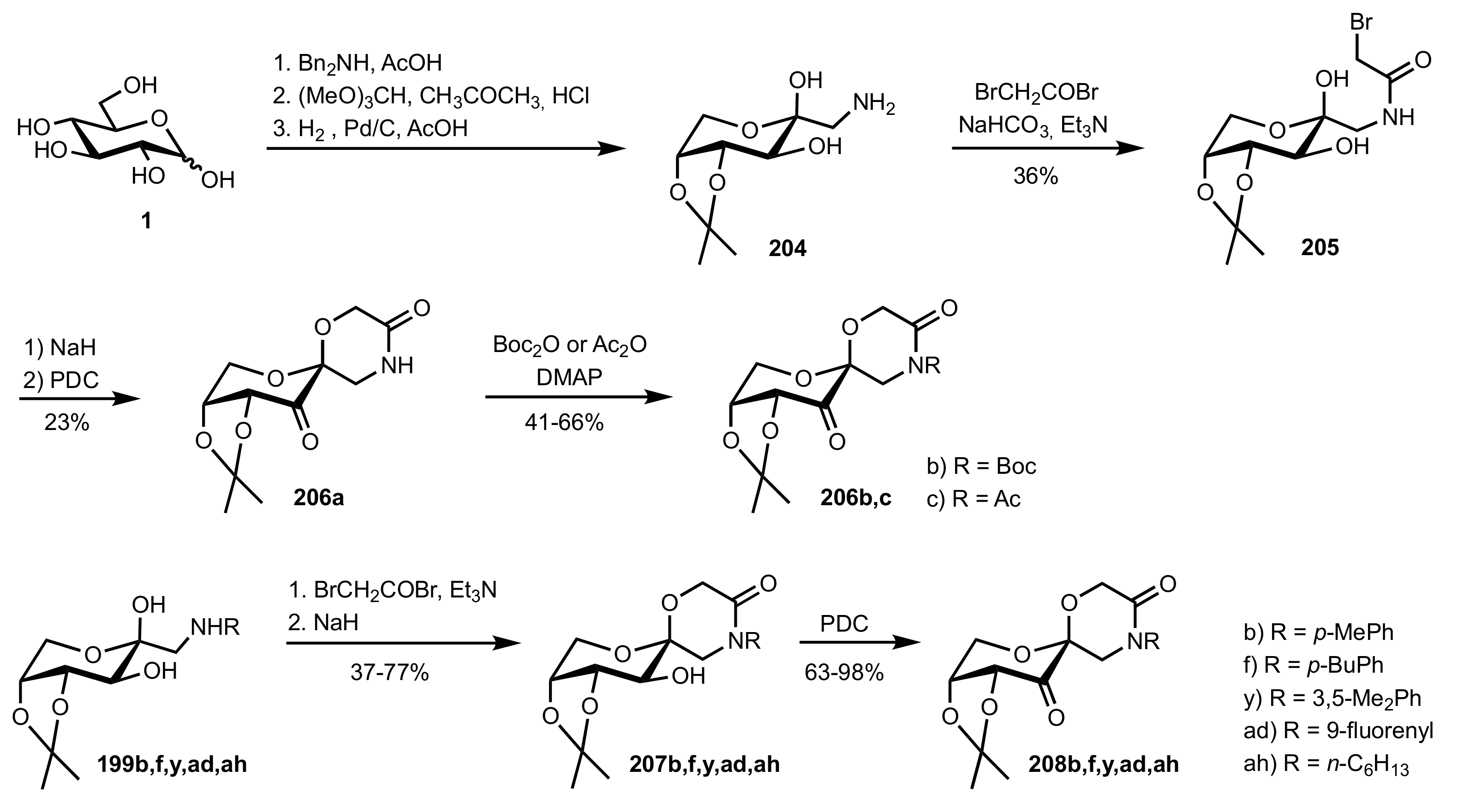
Scheme 53.
The synthesis of sugar ketone organocatalysts 206a-c and 208b,f,y,ad,ah.
Scheme 53.
The synthesis of sugar ketone organocatalysts 206a-c and 208b,f,y,ad,ah.
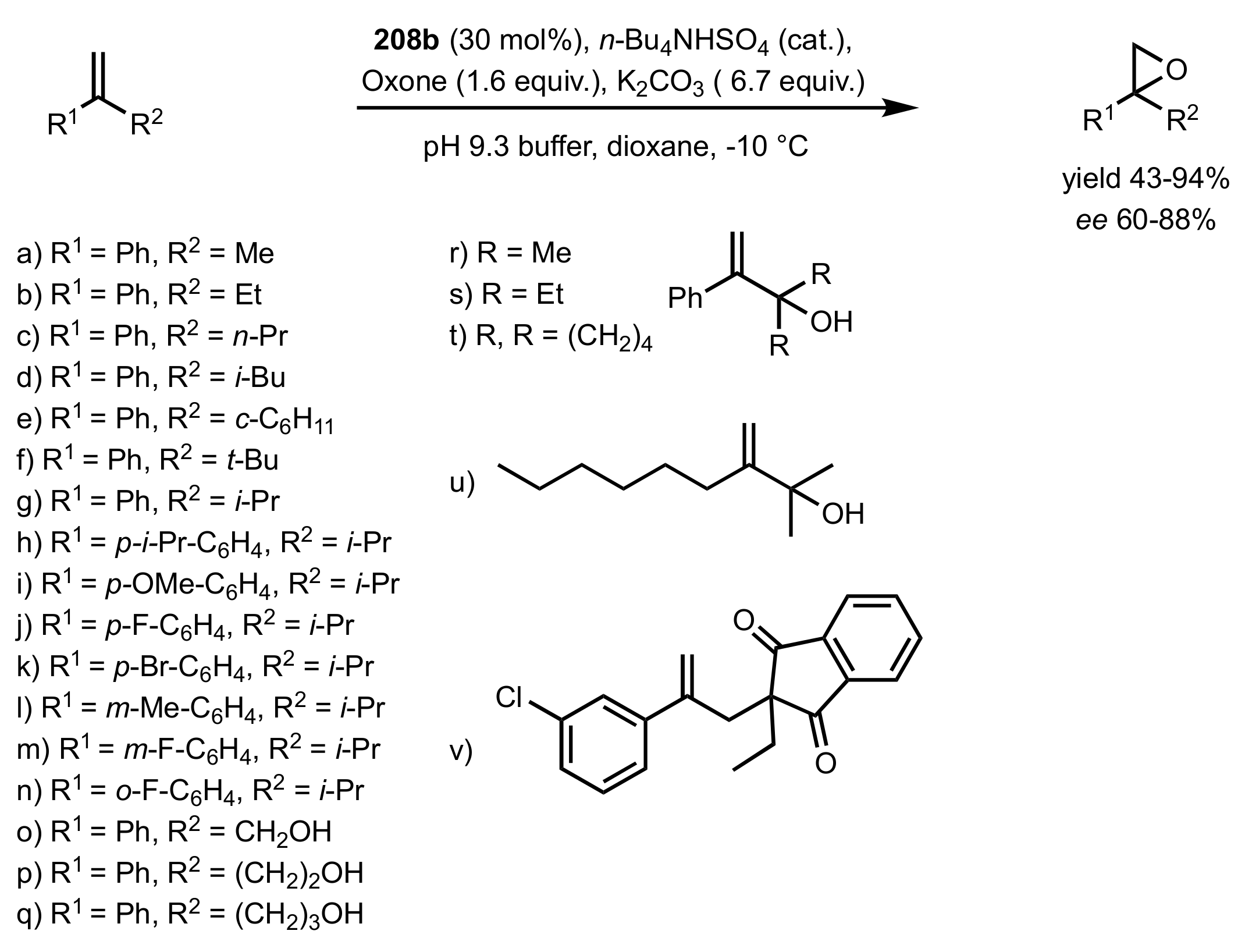
Scheme 54.
The epoxidation of 1,1-disubstituted terminal olefins catalysed by 208b.
Scheme 54.
The epoxidation of 1,1-disubstituted terminal olefins catalysed by 208b.
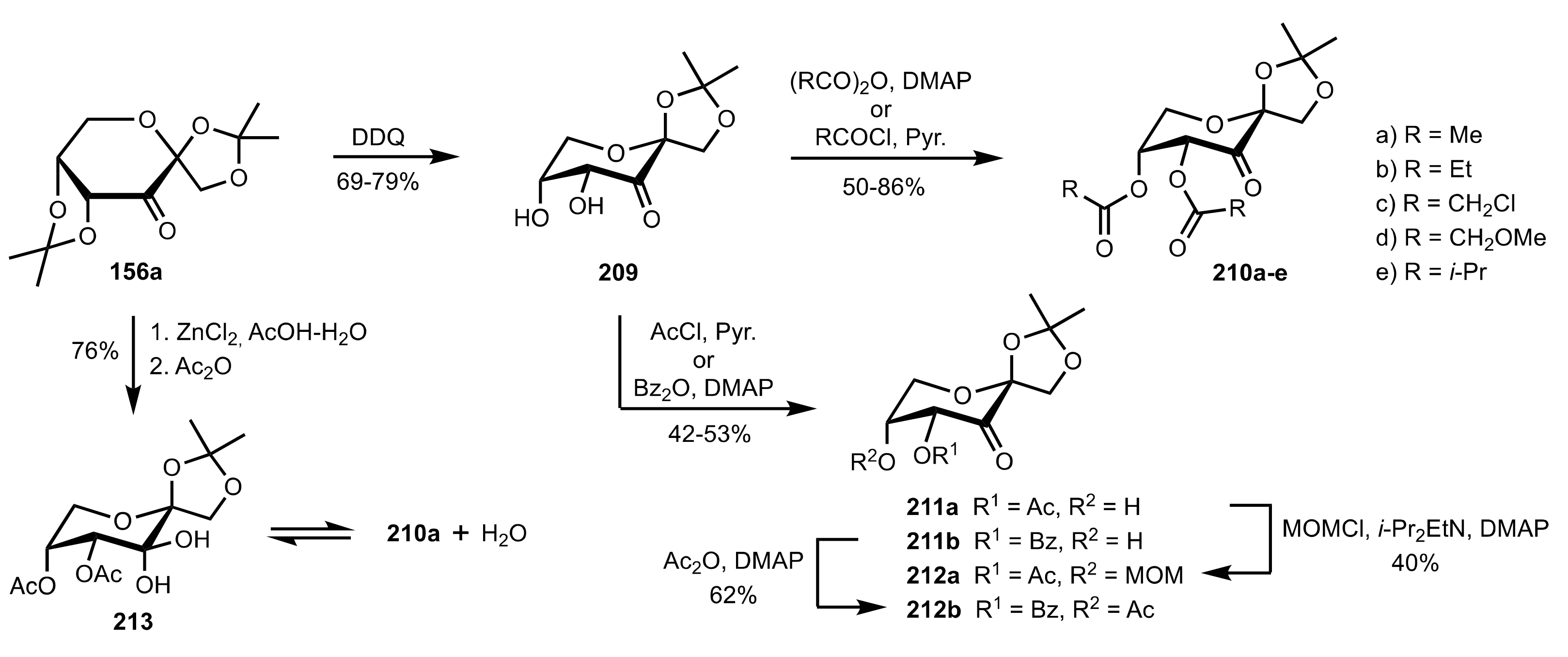
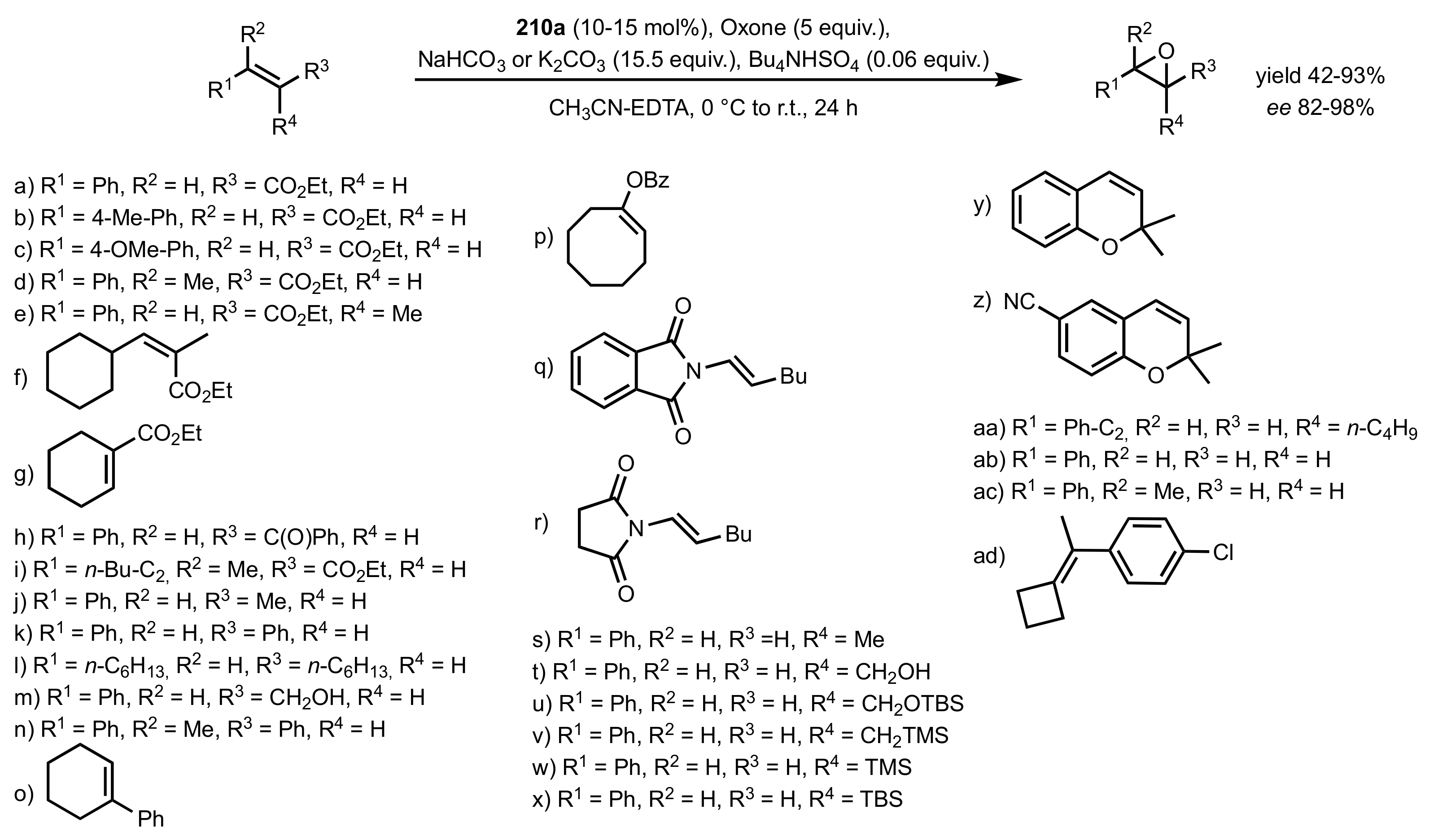
Scheme 55.
The synthesis of d-fructose-derived sugar ketones 210a-e, 211a,b and 212a,b.
Scheme 55.
The synthesis of d-fructose-derived sugar ketones 210a-e, 211a,b and 212a,b.
Scheme 56.
The synthesis of d-fructose-derived sugar ketones 216.
Scheme 56.
The synthesis of d-fructose-derived sugar ketones 216.
Scheme 57.
The epoxidation of alkenes catalysed by sugar ketone 210a.
Scheme 57.
The epoxidation of alkenes catalysed by sugar ketone 210a.
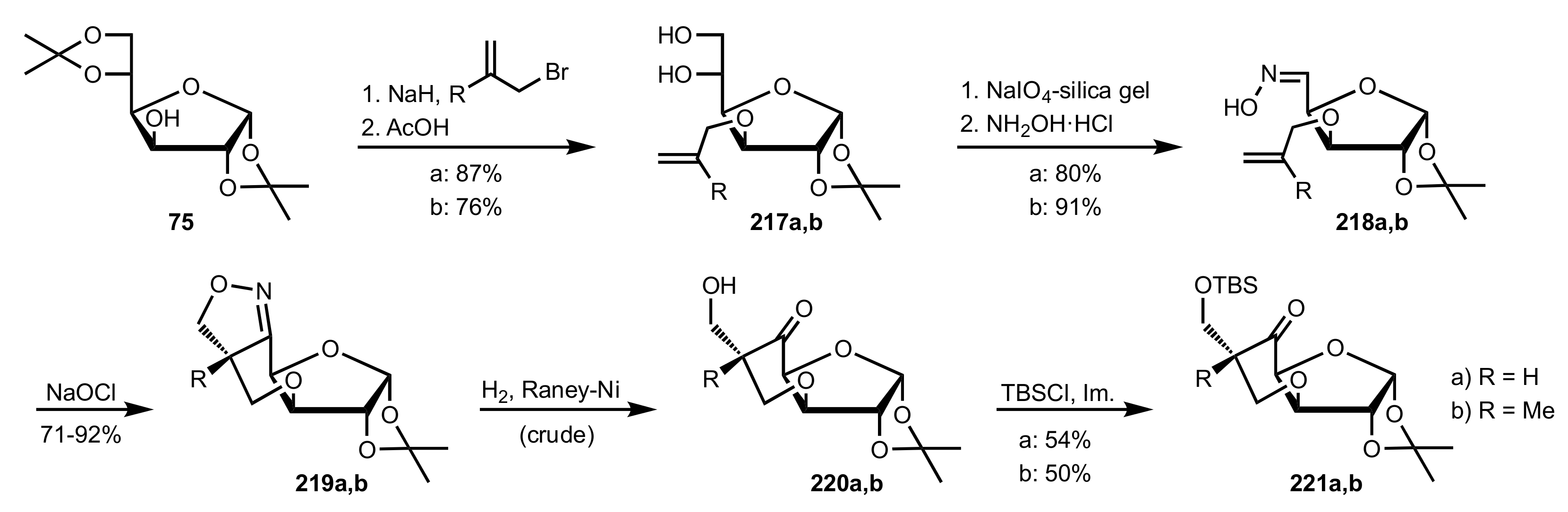
Scheme 58.
The synthesis of sugar organocatalysts 221a,b from d-glucose diacetonide.
Scheme 58.
The synthesis of sugar organocatalysts 221a,b from d-glucose diacetonide.
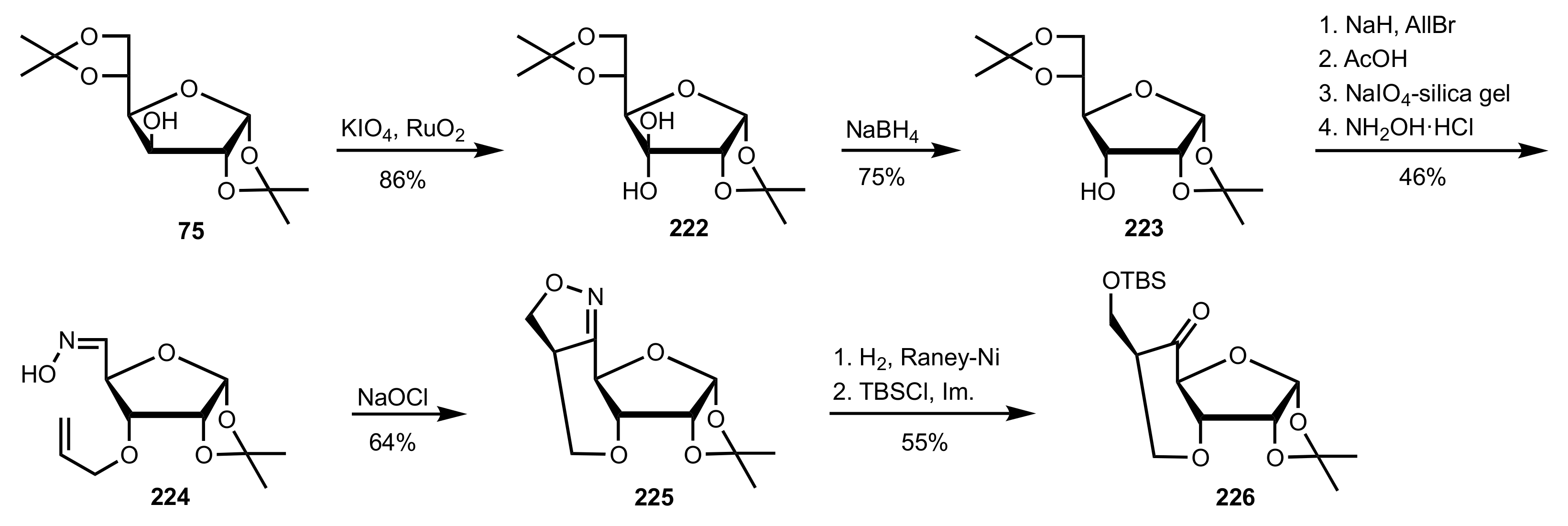
Scheme 59.
The synthesis of organocatalyst 226 from d-glucose diacetonide.
Scheme 59.
The synthesis of organocatalyst 226 from d-glucose diacetonide.
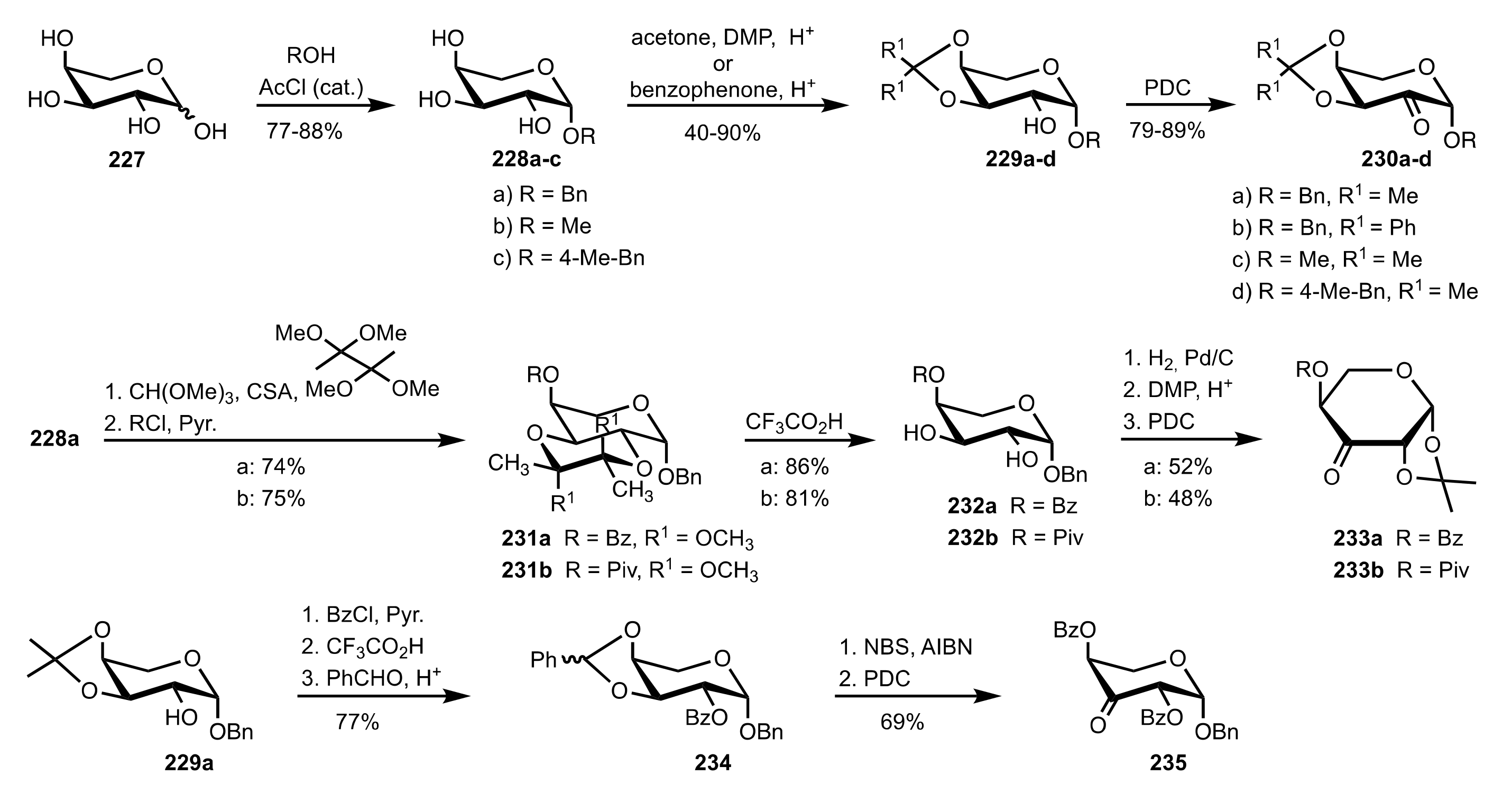
Scheme 60.
The synthesis of sugar ketone organocatalysts 230a-d, 233a,b and 235 from l-arabinose.
Scheme 60.
The synthesis of sugar ketone organocatalysts 230a-d, 233a,b and 235 from l-arabinose.
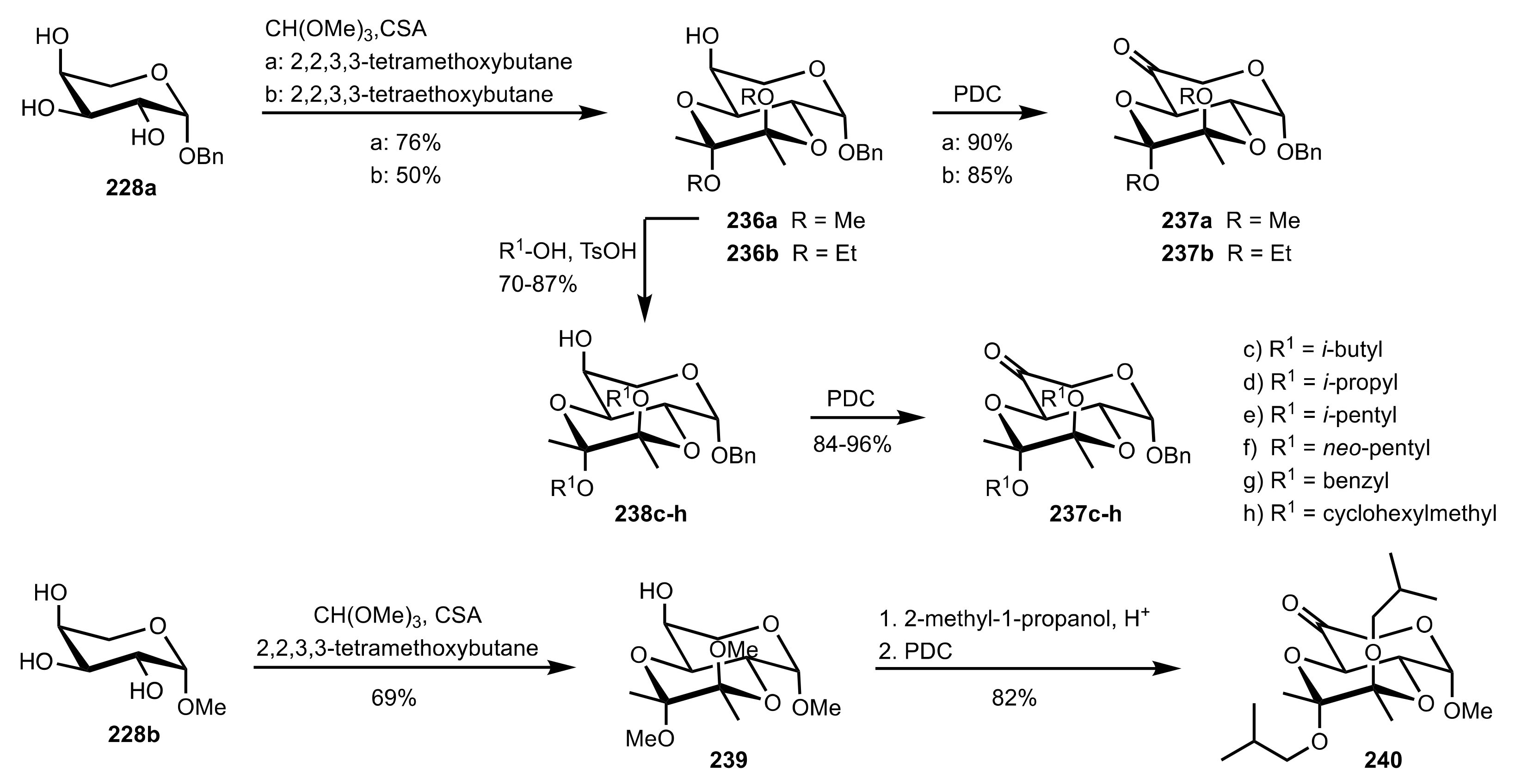
Scheme 61.
The synthesis of sugar ketone organocatalysts 237a-h and 240.
Scheme 61.
The synthesis of sugar ketone organocatalysts 237a-h and 240.
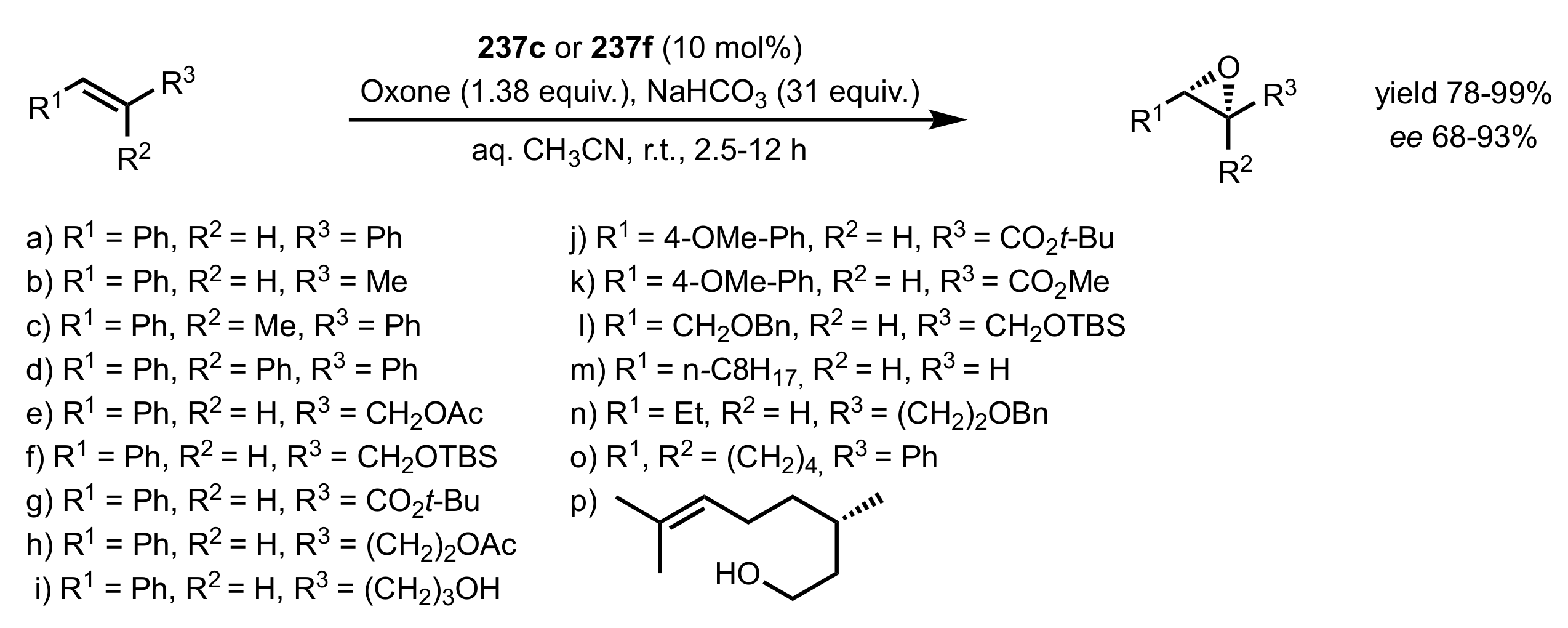
Scheme 62.
The epoxidation of alkenes catalysed by
237c and
237f [
116,
117,
118,
119].
Scheme 62.
The epoxidation of alkenes catalysed by
237c and
237f [
116,
117,
118,
119].
Scheme 63.
The synthesis of the arabinose-derived ketone 242.
Scheme 63.
The synthesis of the arabinose-derived ketone 242.
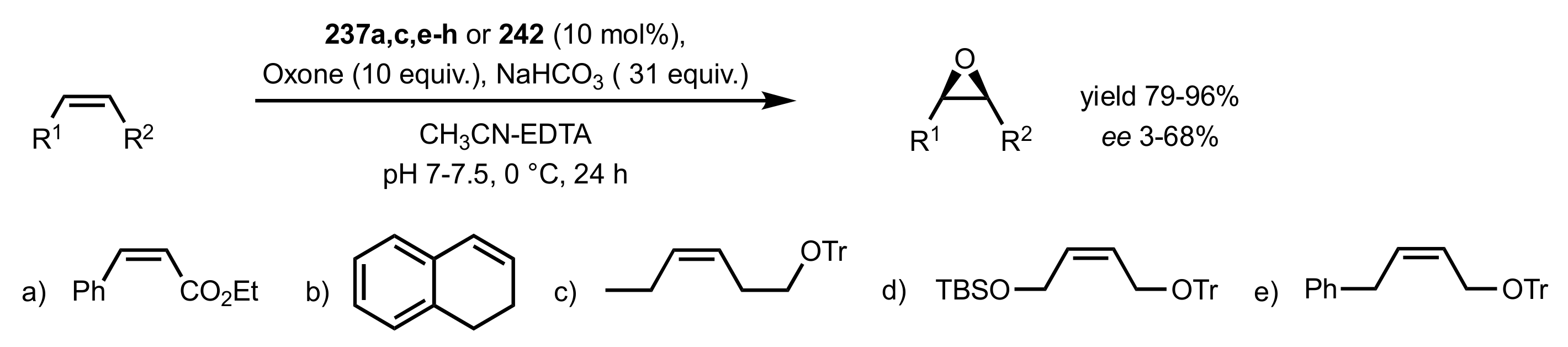
Scheme 64.
The epoxidation of cis-alkenes catalysed by 237a,c,e-h and 242.
Scheme 64.
The epoxidation of cis-alkenes catalysed by 237a,c,e-h and 242.
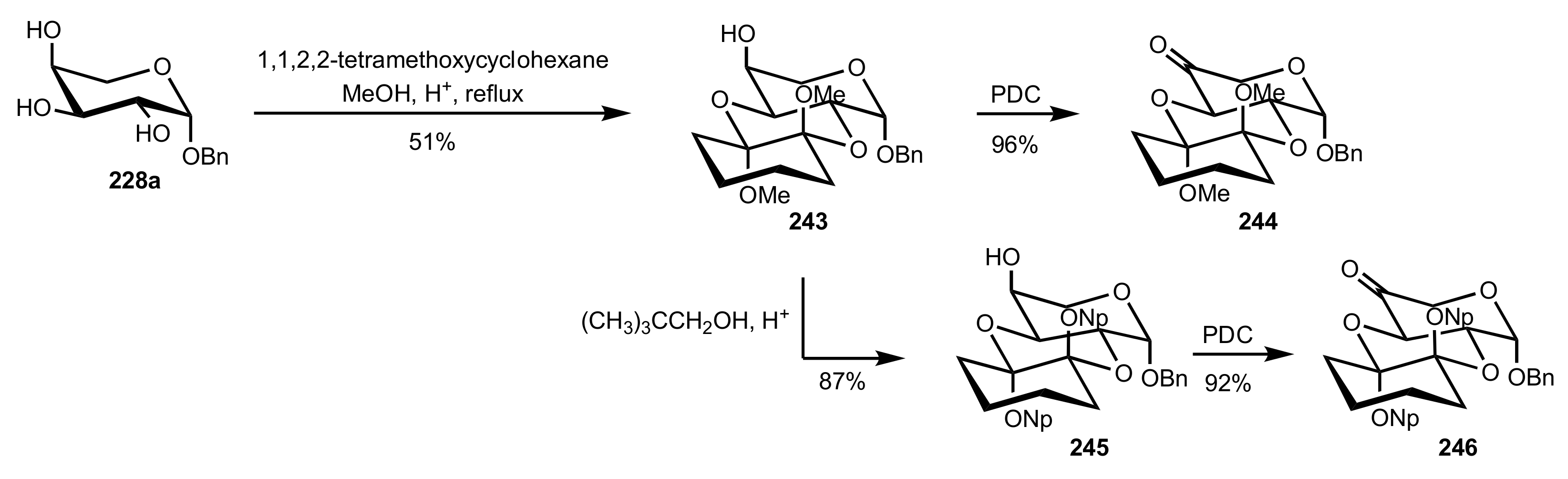
Scheme 65.
The synthesis of the arabinose-based ketones 244 and 246.
Scheme 65.
The synthesis of the arabinose-based ketones 244 and 246.
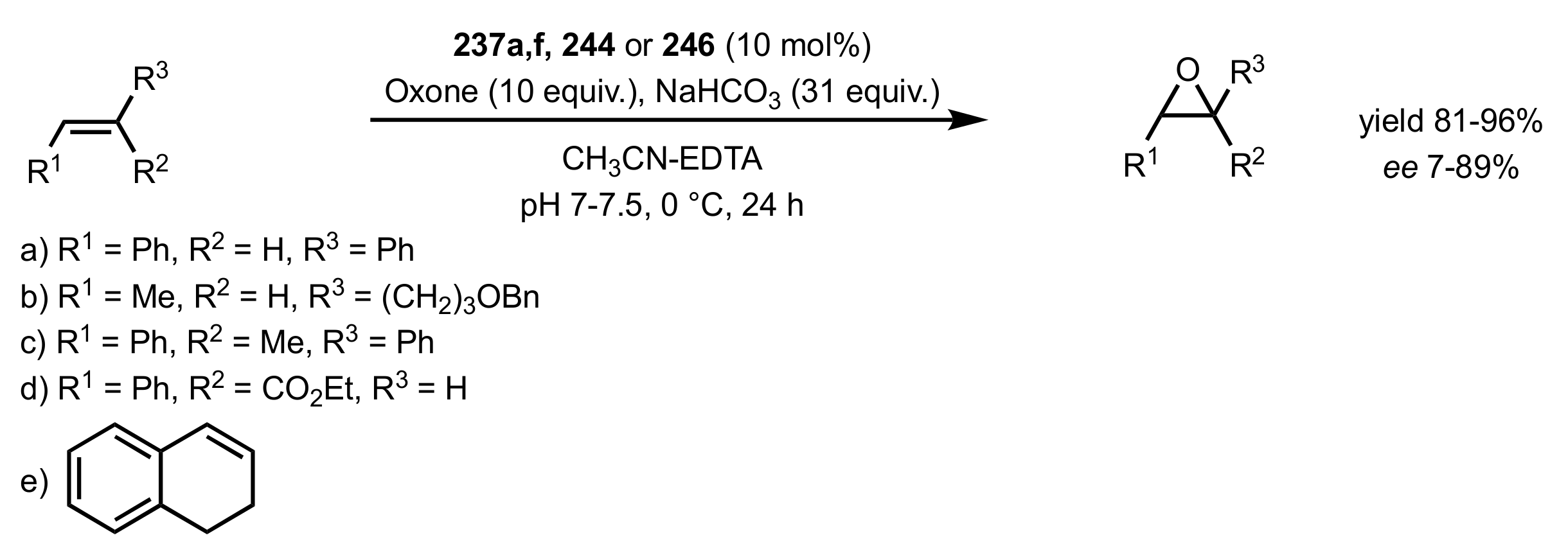
Scheme 66.
The epoxidation of cis- and trans-alkenes catalysed by ketones 237a,f, 244 or 246.
Scheme 66.
The epoxidation of cis- and trans-alkenes catalysed by ketones 237a,f, 244 or 246.
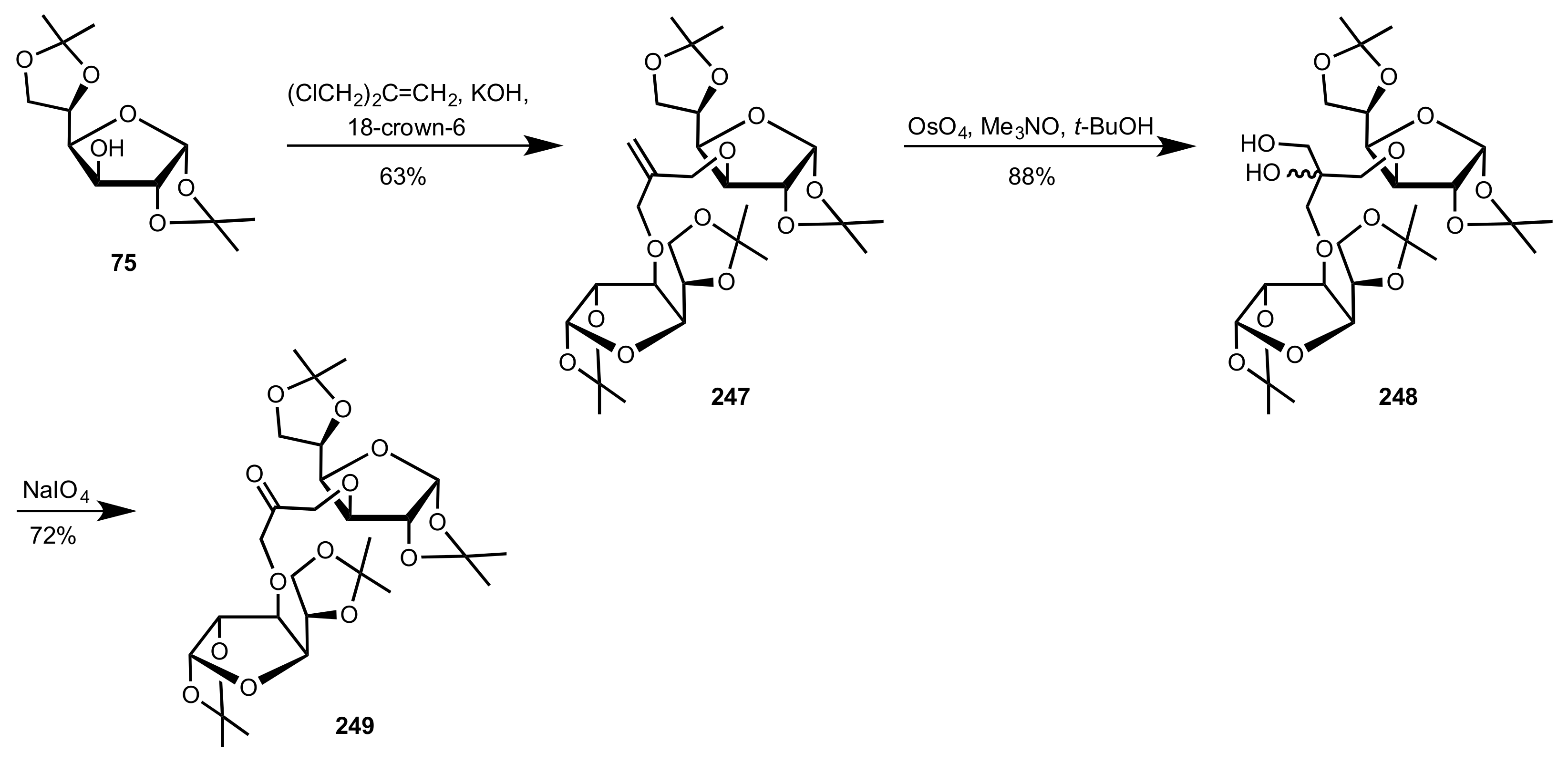
Scheme 67.
The synthesis of the C2-symmetric organocatalyst 249.
Scheme 67.
The synthesis of the C2-symmetric organocatalyst 249.
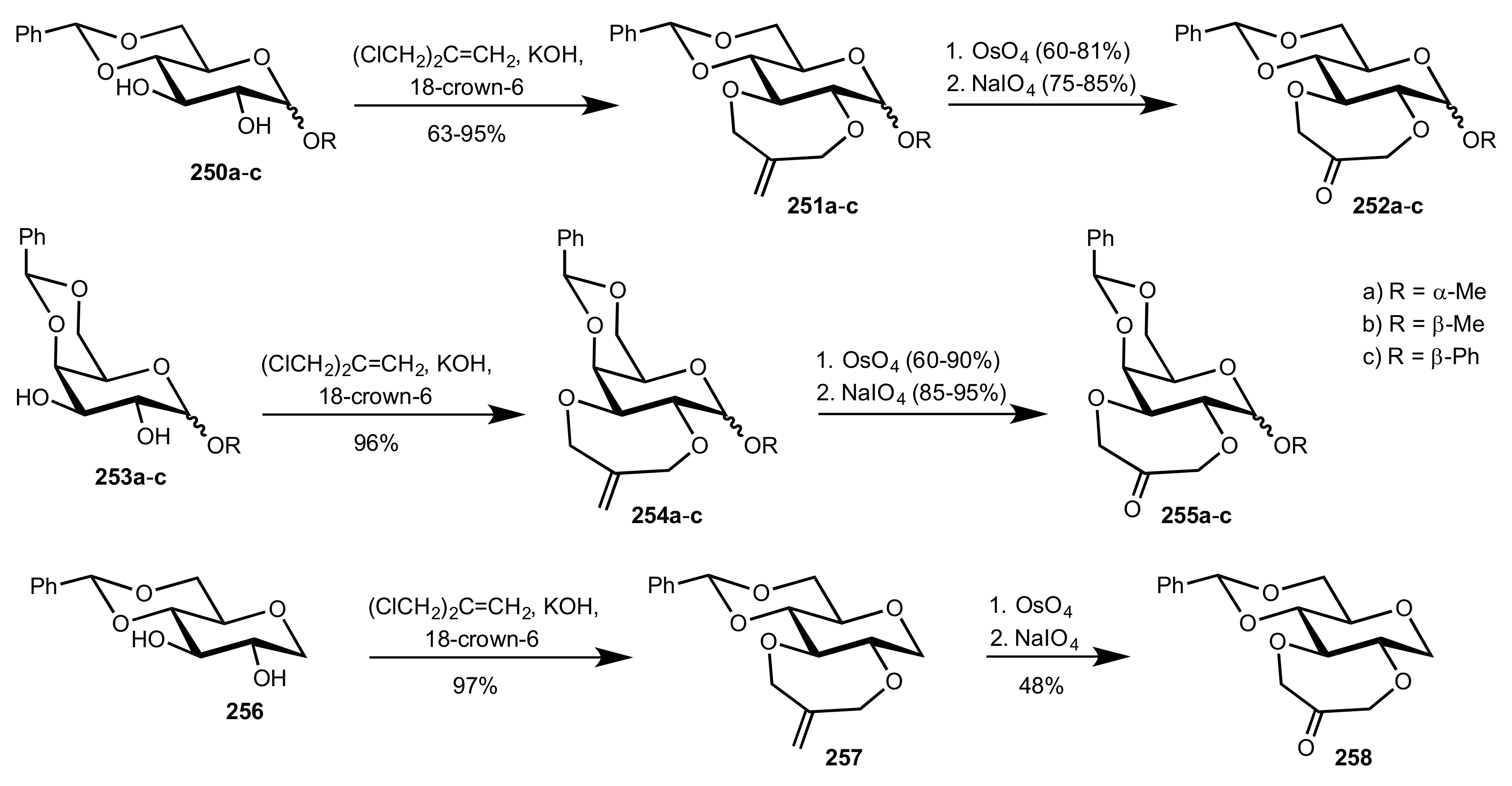
Scheme 68.
The synthesis of the d-gluco (252a-c), d-galacto (255a-c) and 1-deoxy-d-gluco (258) ketone organocatalysts.
Scheme 68.
The synthesis of the d-gluco (252a-c), d-galacto (255a-c) and 1-deoxy-d-gluco (258) ketone organocatalysts.
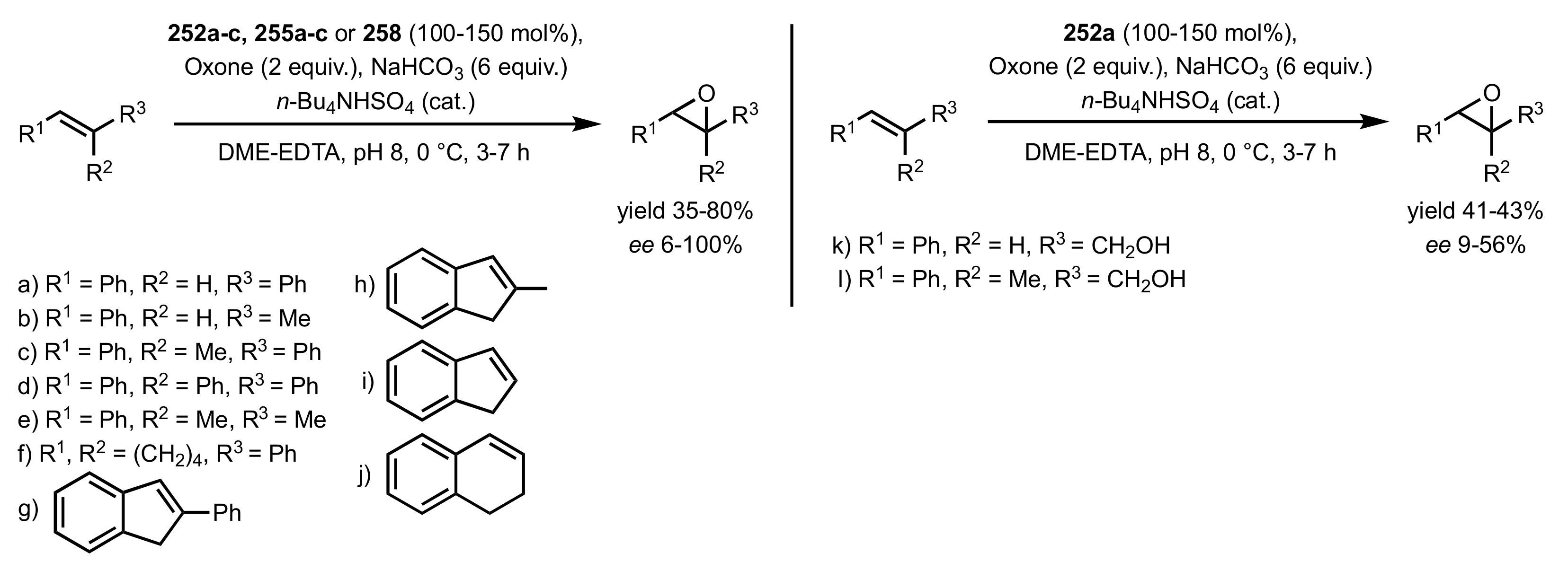
Scheme 69.
The epoxidation of aryl alkenes and allylic alcohols catalysed by 252a-c, 255a-c and 258.
Scheme 69.
The epoxidation of aryl alkenes and allylic alcohols catalysed by 252a-c, 255a-c and 258.
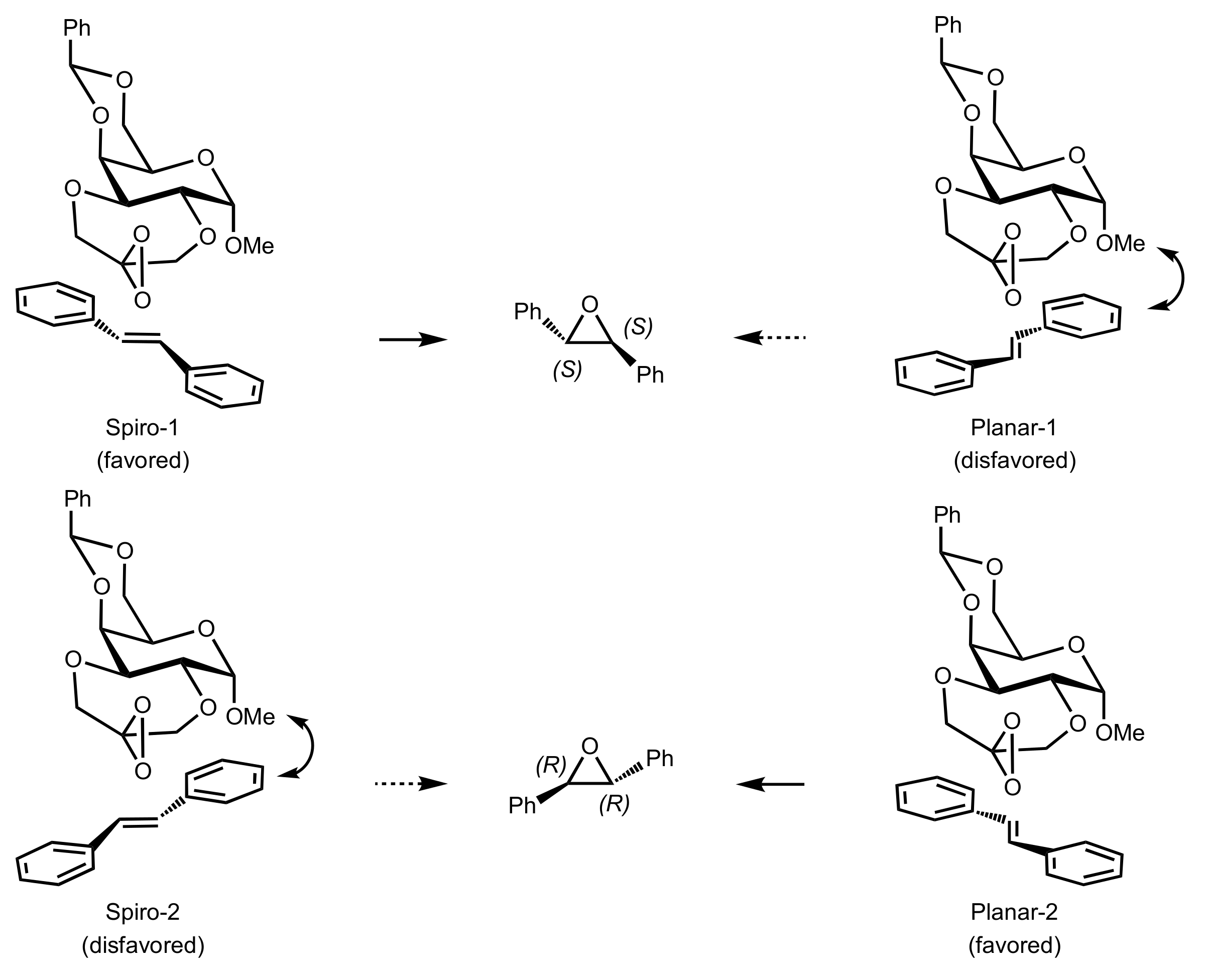
Figure 21.
Competitive transition states.
Figure 21.
Competitive transition states.
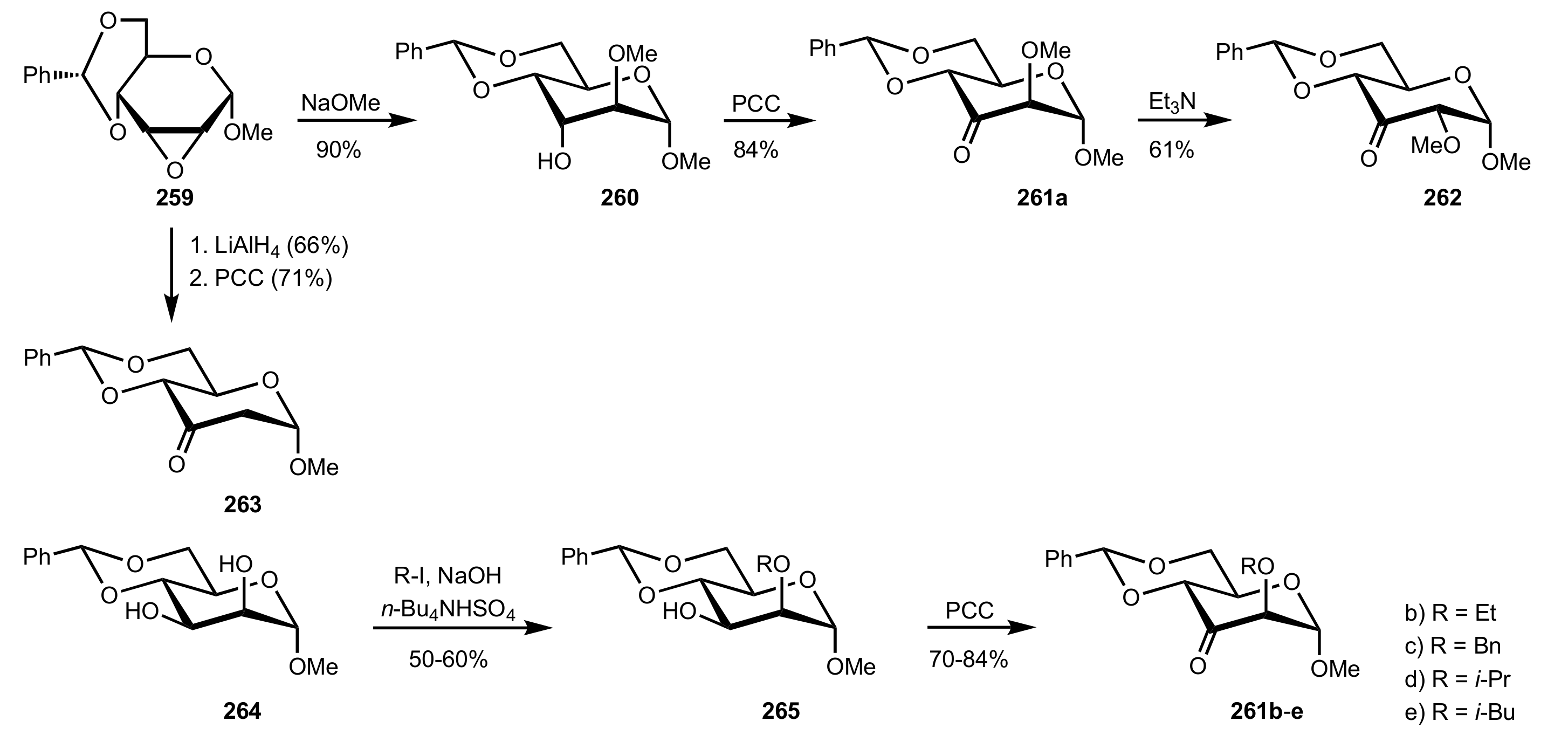
Scheme 70.
The synthesis of mannose-derived ketones 261a-e, 262 and 263.
Scheme 70.
The synthesis of mannose-derived ketones 261a-e, 262 and 263.
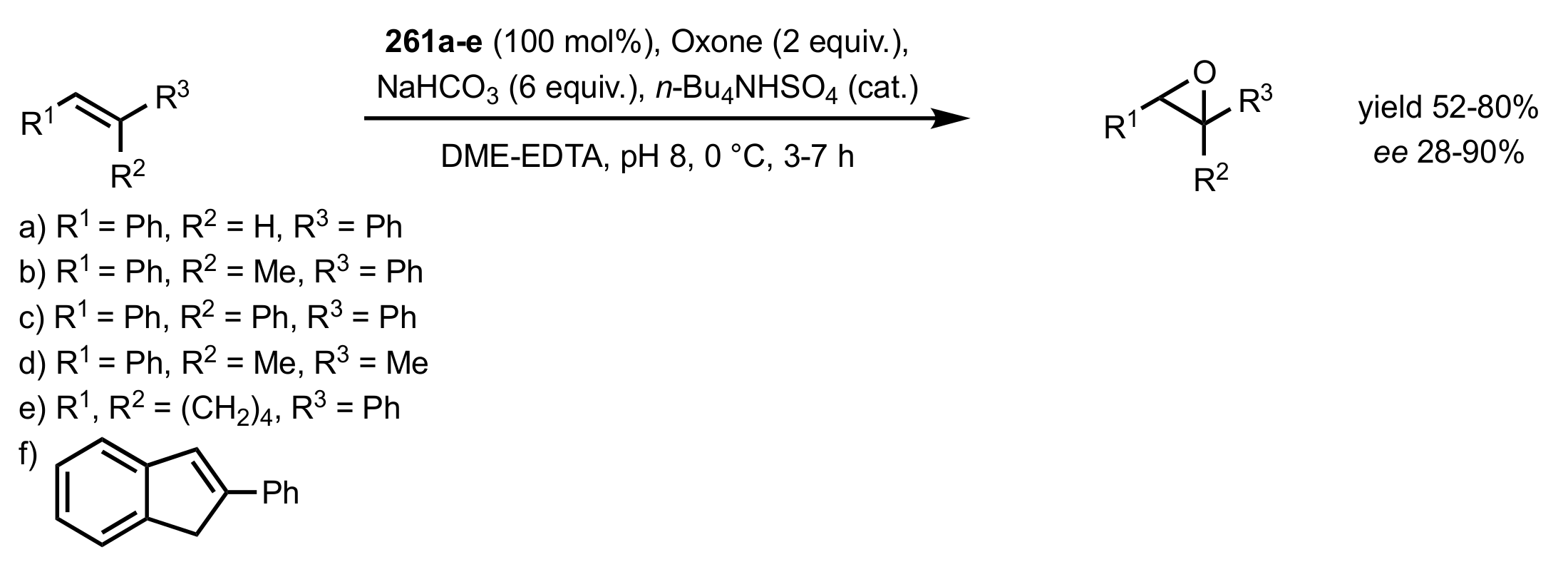
Scheme 71.
The epoxidation of trans- and trisubstituted alkenes catalysed by ketones 261a-e.
Scheme 71.
The epoxidation of trans- and trisubstituted alkenes catalysed by ketones 261a-e.
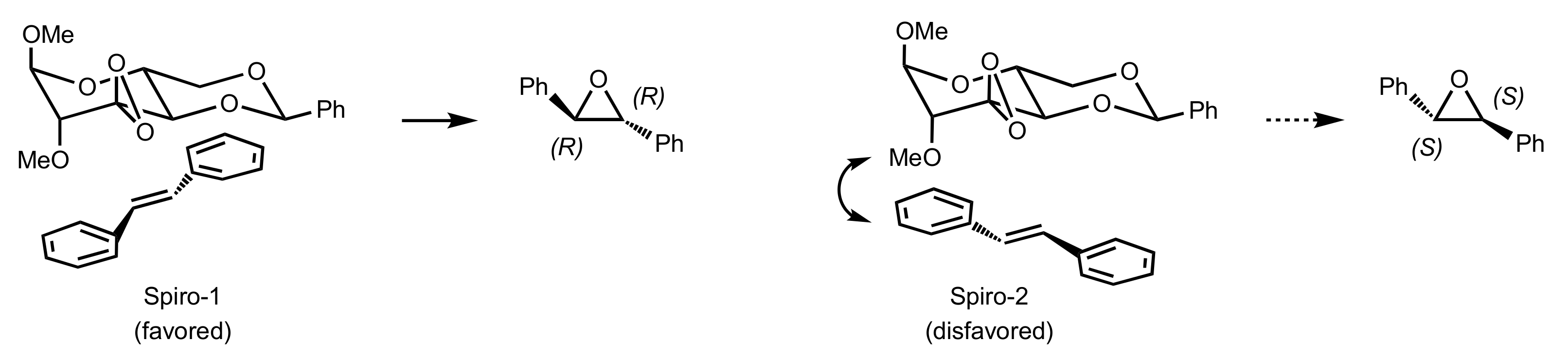
Figure 22.
The proposed transition states for the epoxidation of trans-stilbene catalysed by 261a.
Figure 22.
The proposed transition states for the epoxidation of trans-stilbene catalysed by 261a.
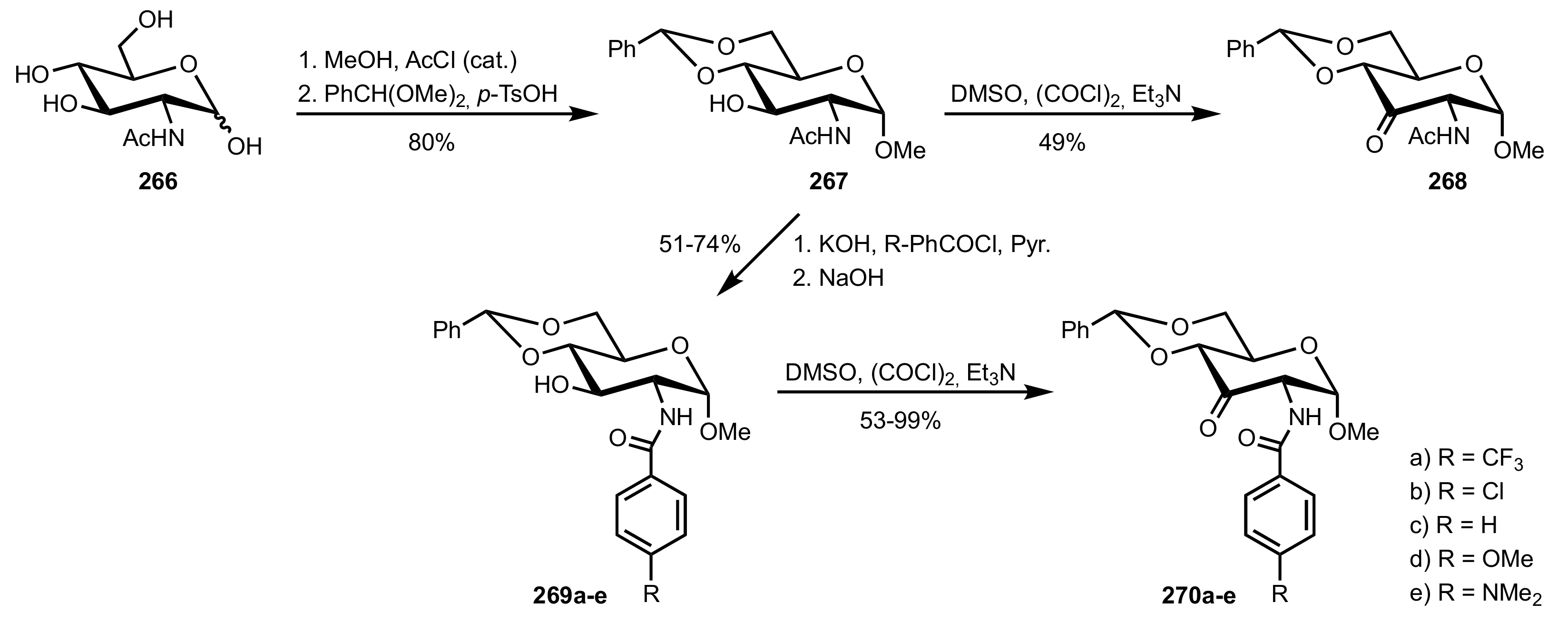
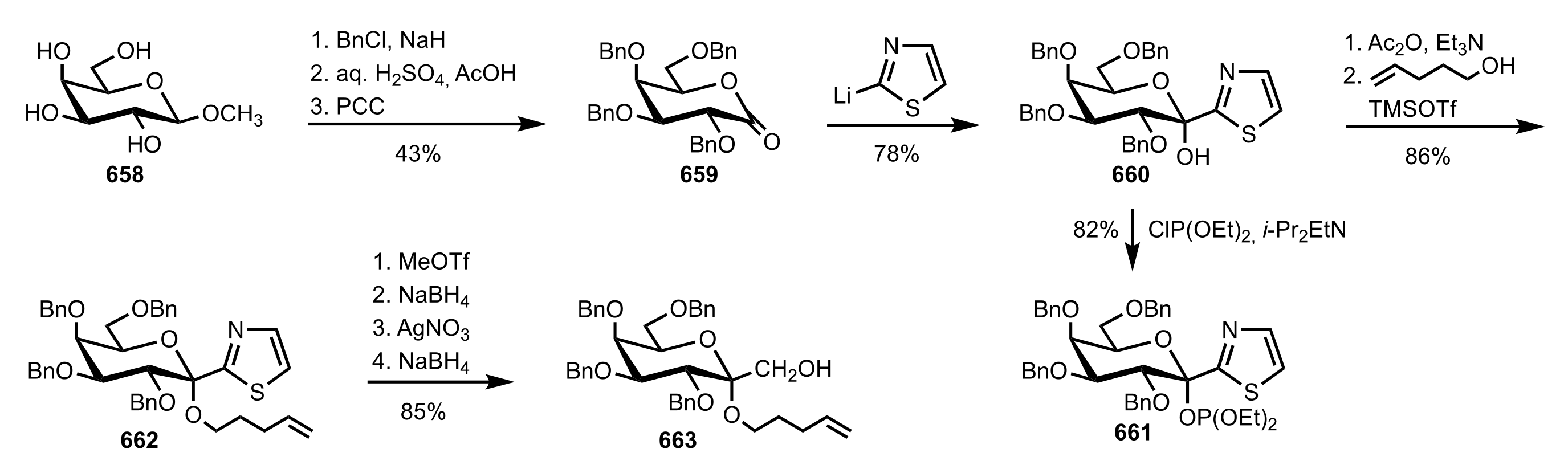
Scheme 72.
The synthesis of d-glucosamine-derived organocatalysts 268 and 270a-e.
Scheme 72.
The synthesis of d-glucosamine-derived organocatalysts 268 and 270a-e.
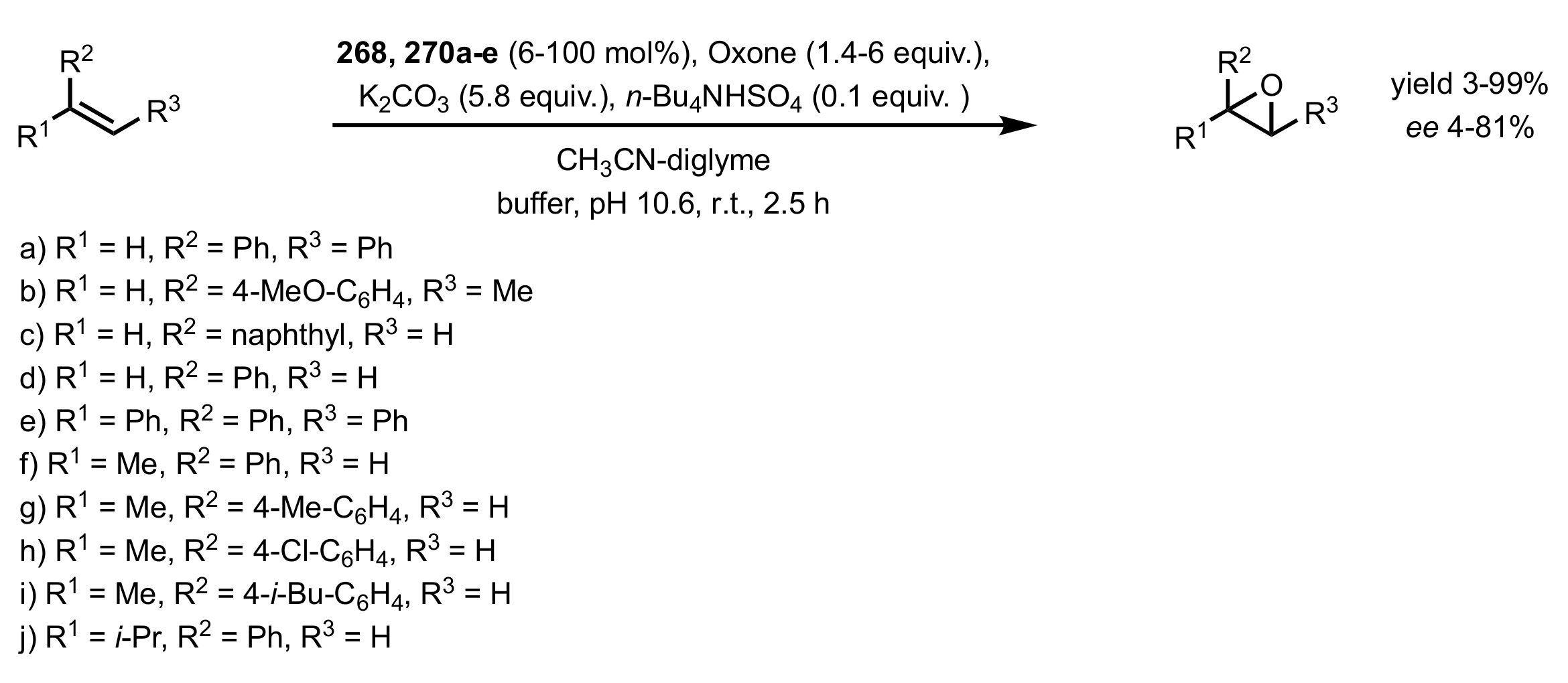
Scheme 73.
The epoxidation of 2,2-disubstituted and terminal alkenes catalysed by 268 and 270a-e.
Scheme 73.
The epoxidation of 2,2-disubstituted and terminal alkenes catalysed by 268 and 270a-e.
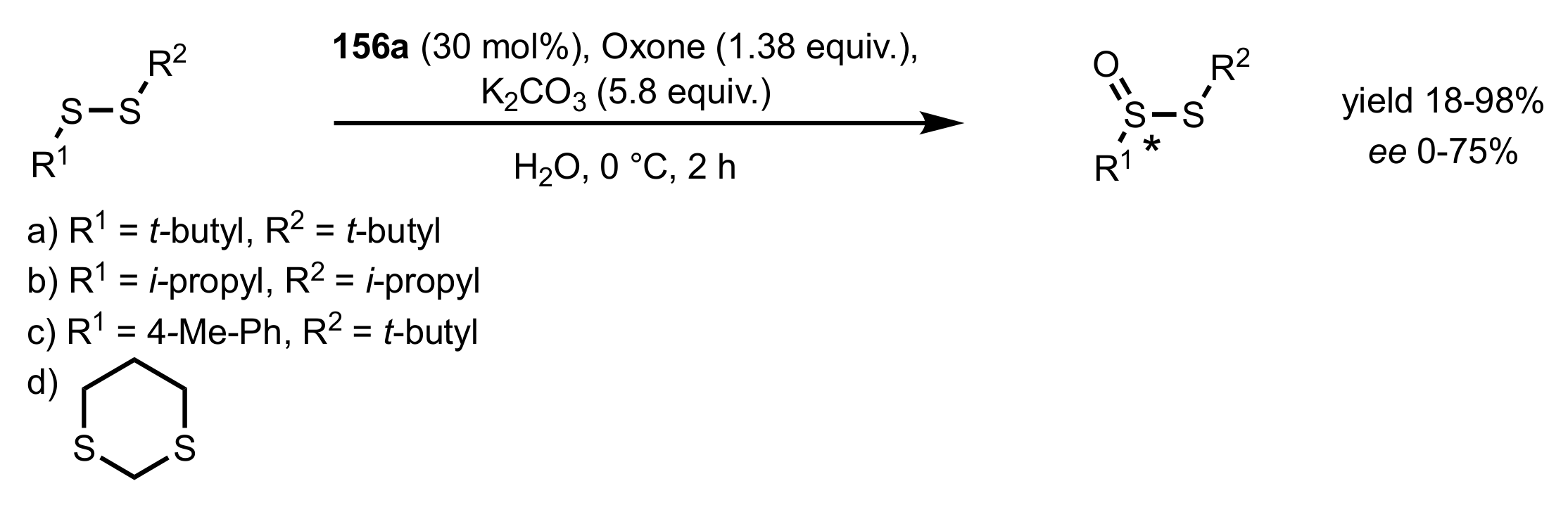
Scheme 74.
Enantioselective synthesis of thiosulfinates catalysed by 156a.
Scheme 74.
Enantioselective synthesis of thiosulfinates catalysed by 156a.
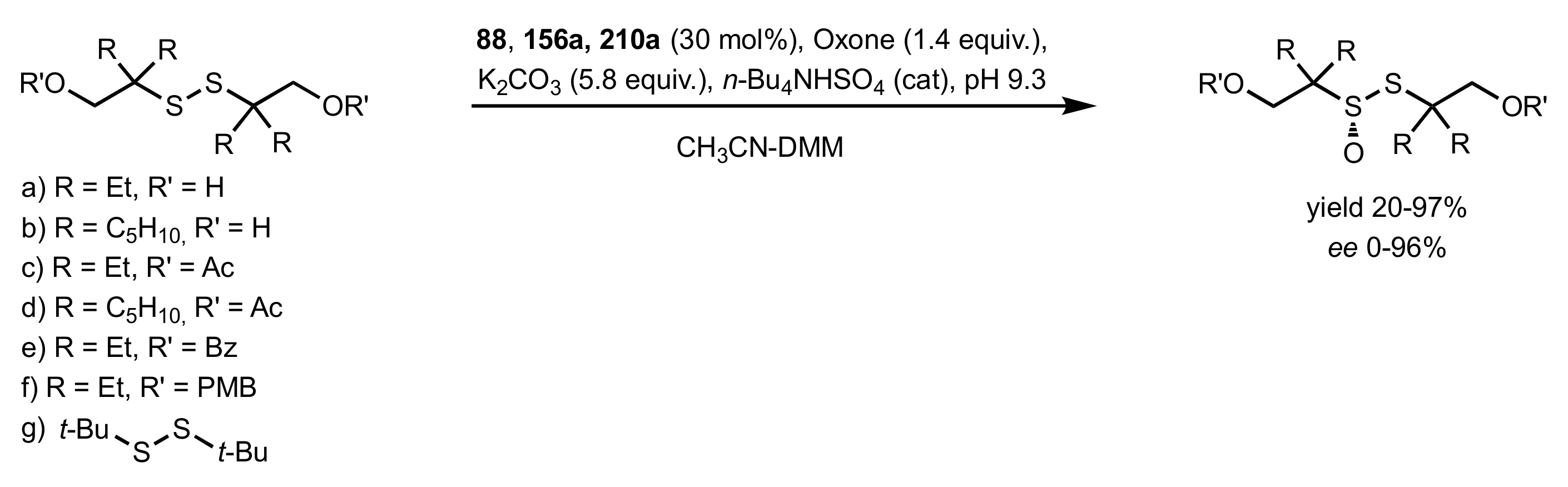
Scheme 75.
The synthesis of chiral sulfinyl derivatives catalysed by 88, 156a and 210a.
Scheme 75.
The synthesis of chiral sulfinyl derivatives catalysed by 88, 156a and 210a.
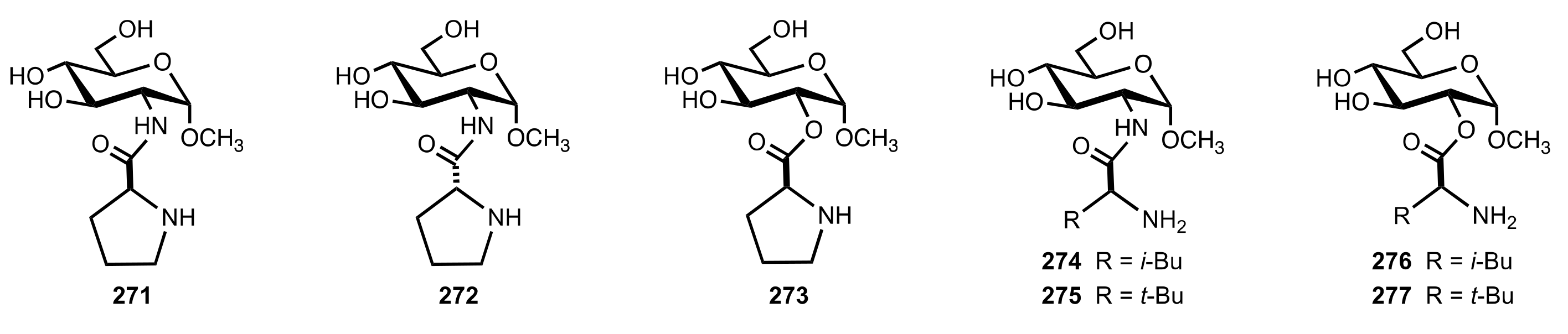
Figure 23.
Sugar-proline and sugar-leucine derivatives prepared by Machinami and co-workers.
Figure 23.
Sugar-proline and sugar-leucine derivatives prepared by Machinami and co-workers.
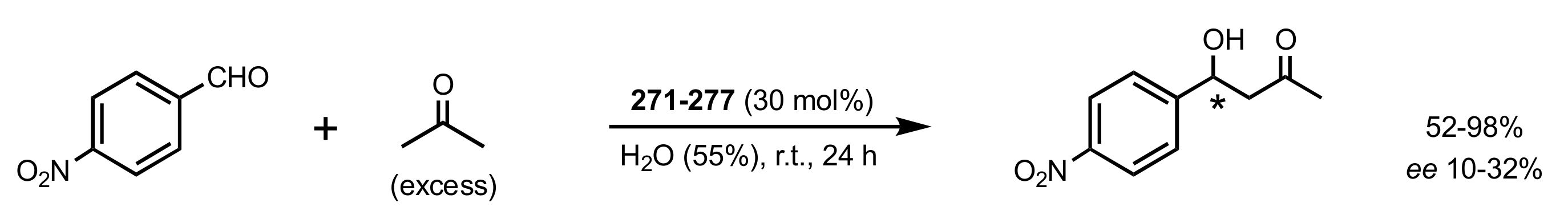
Scheme 76.
Aldol reaction catalysed by 271–277.
Scheme 76.
Aldol reaction catalysed by 271–277.
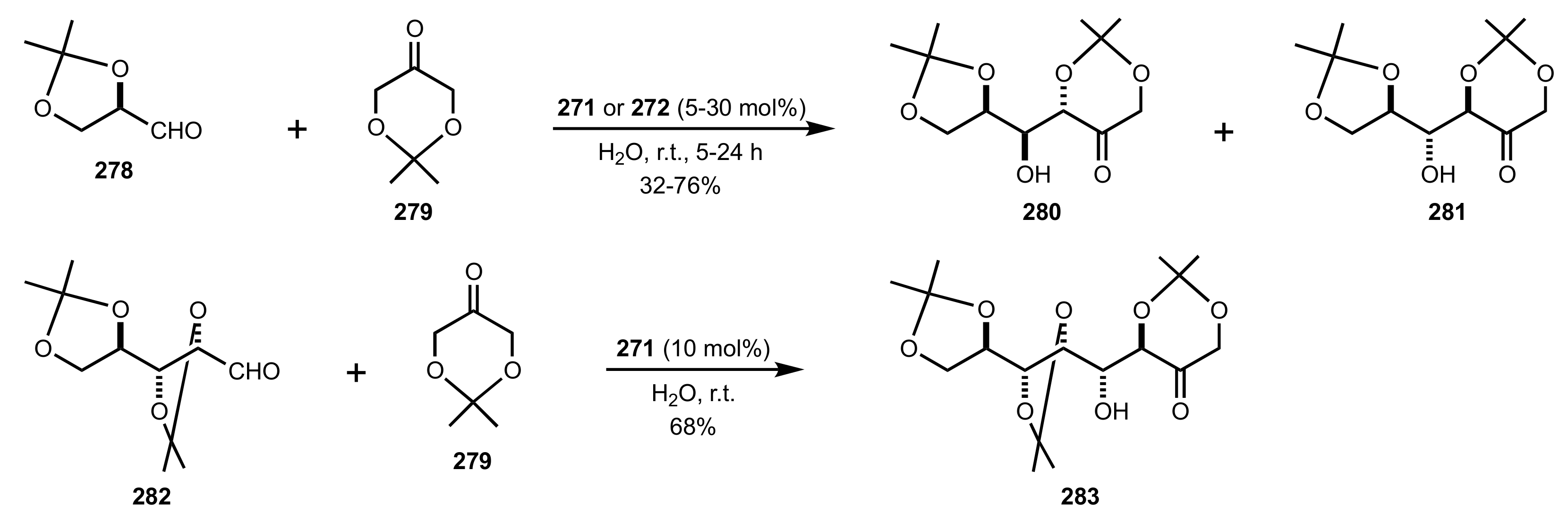
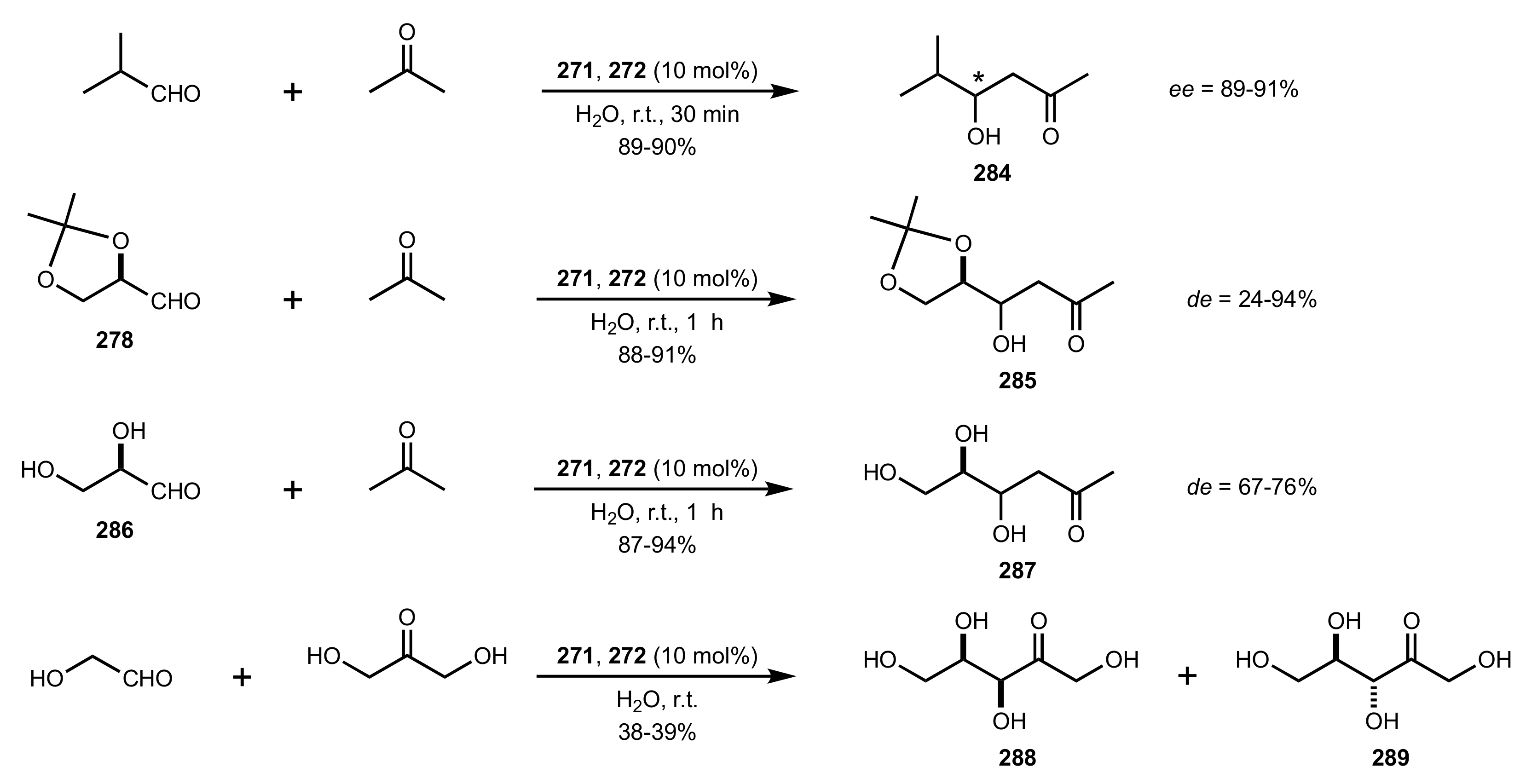
Scheme 77.
Aldol reactions of aldehydo-sugars catalysed by 271 or 272.
Scheme 77.
Aldol reactions of aldehydo-sugars catalysed by 271 or 272.
Scheme 78.
Aldol reactions catalysed by 271 and 272.
Scheme 78.
Aldol reactions catalysed by 271 and 272.
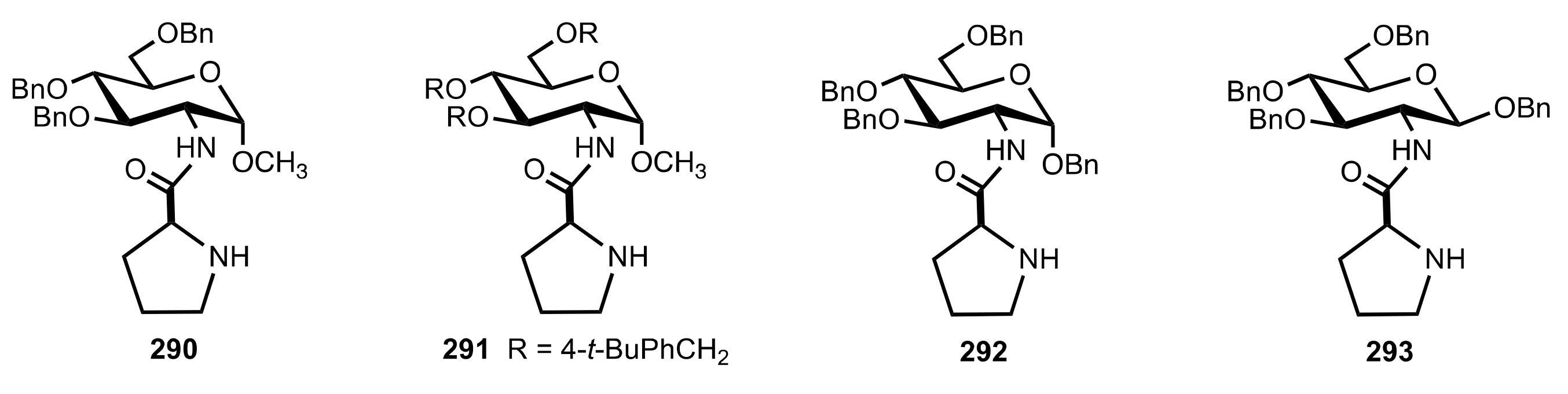
Figure 24.
Sugar prolinamide organocatalysts prepared by Agarwal and Peddinti.
Figure 24.
Sugar prolinamide organocatalysts prepared by Agarwal and Peddinti.
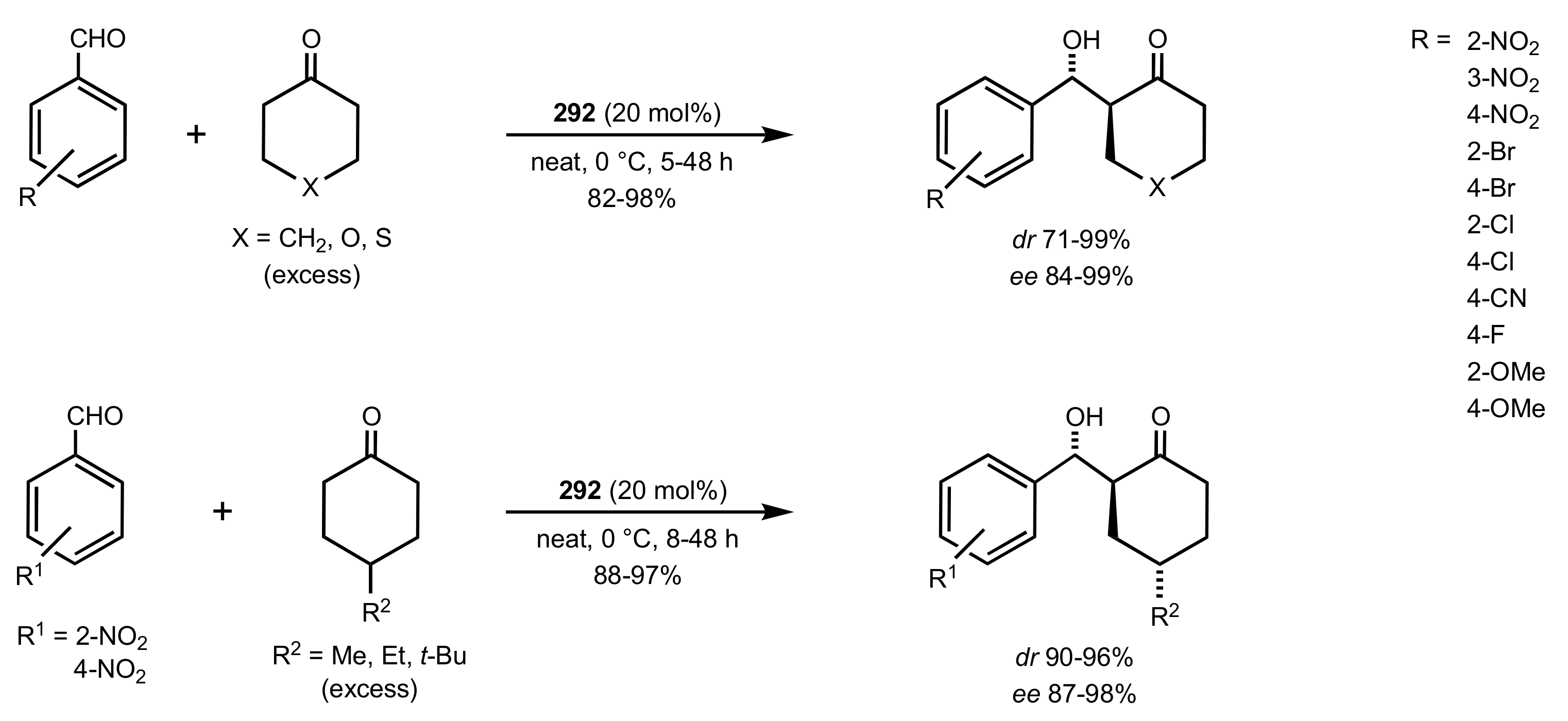
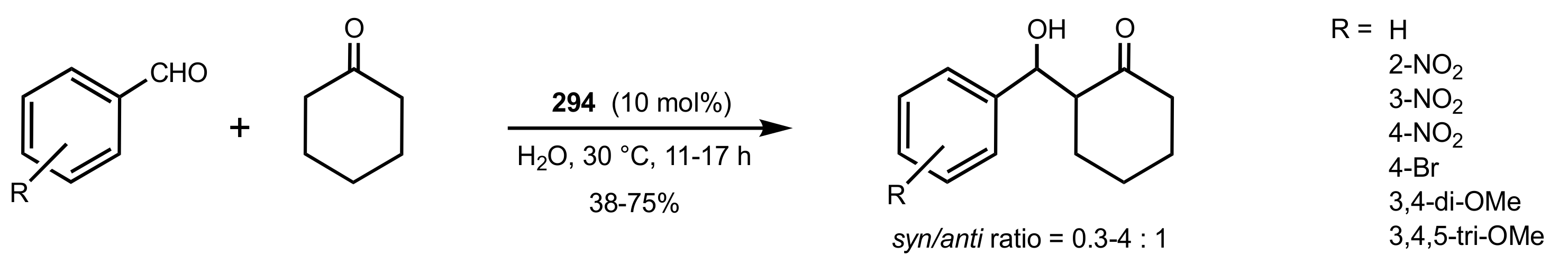
Scheme 79.
Aldol reactions catalysed by 292.
Scheme 79.
Aldol reactions catalysed by 292.
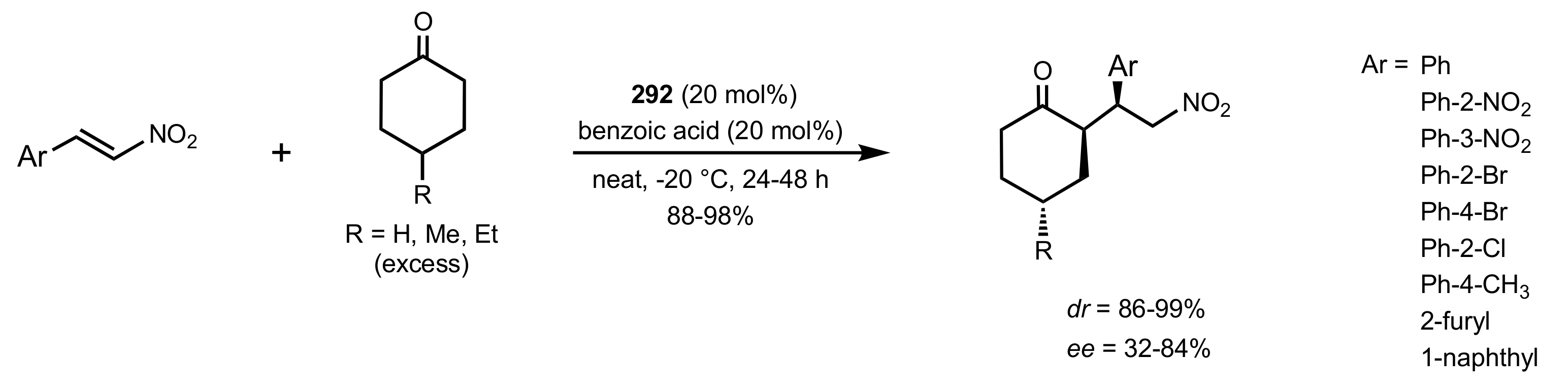
Scheme 80.
The Michael additions catalysed by 292.
Scheme 80.
The Michael additions catalysed by 292.
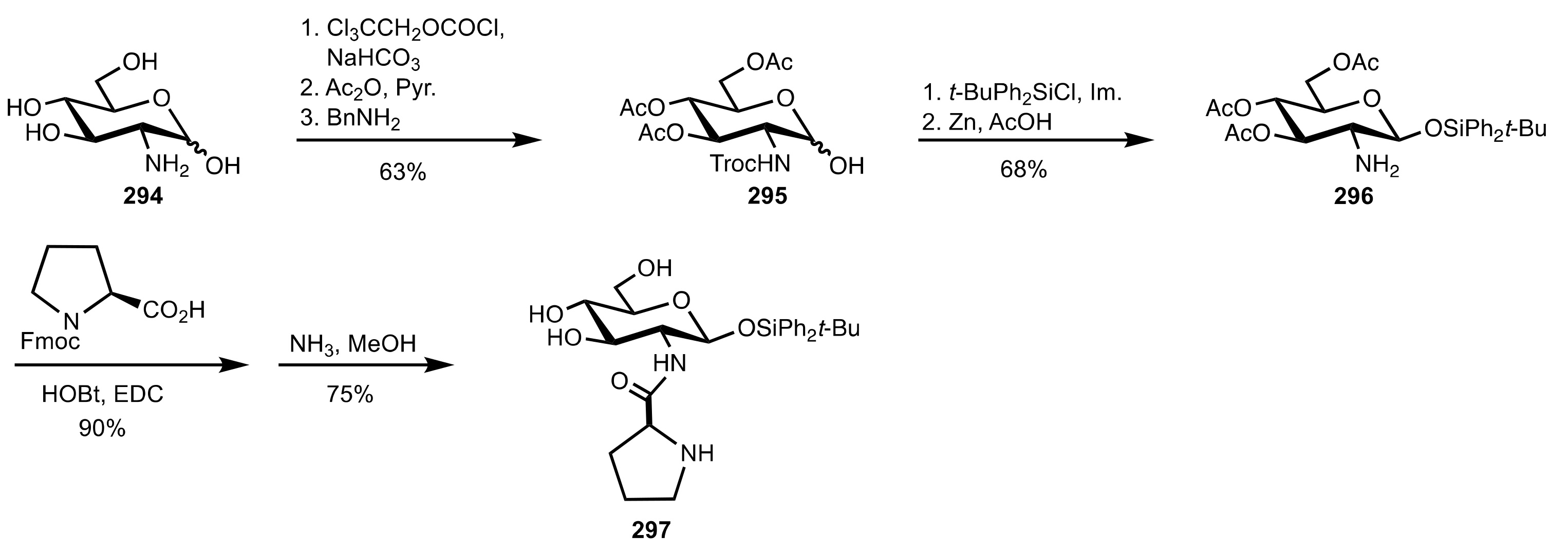
Scheme 81.
The synthesis of the sugar prolinamide organocatalyst 297.
Scheme 81.
The synthesis of the sugar prolinamide organocatalyst 297.
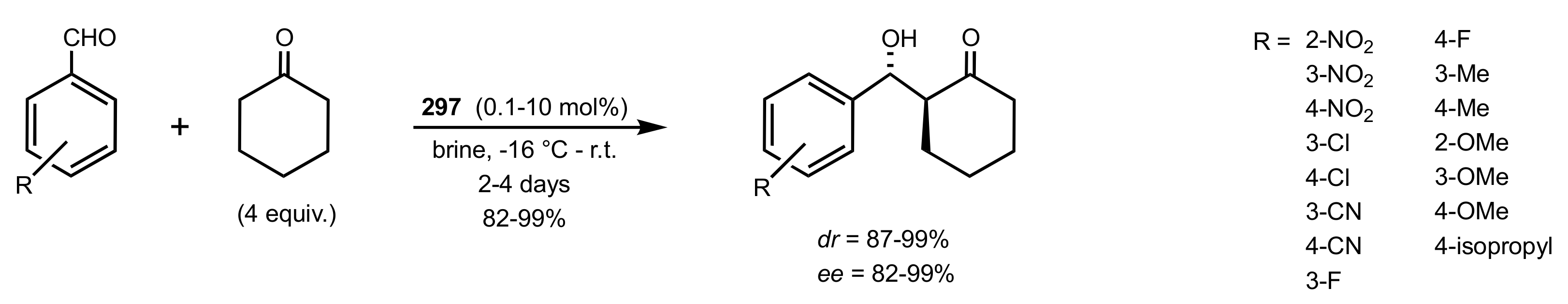
Scheme 82.
Aldol reactions catalysed by 297.
Scheme 82.
Aldol reactions catalysed by 297.
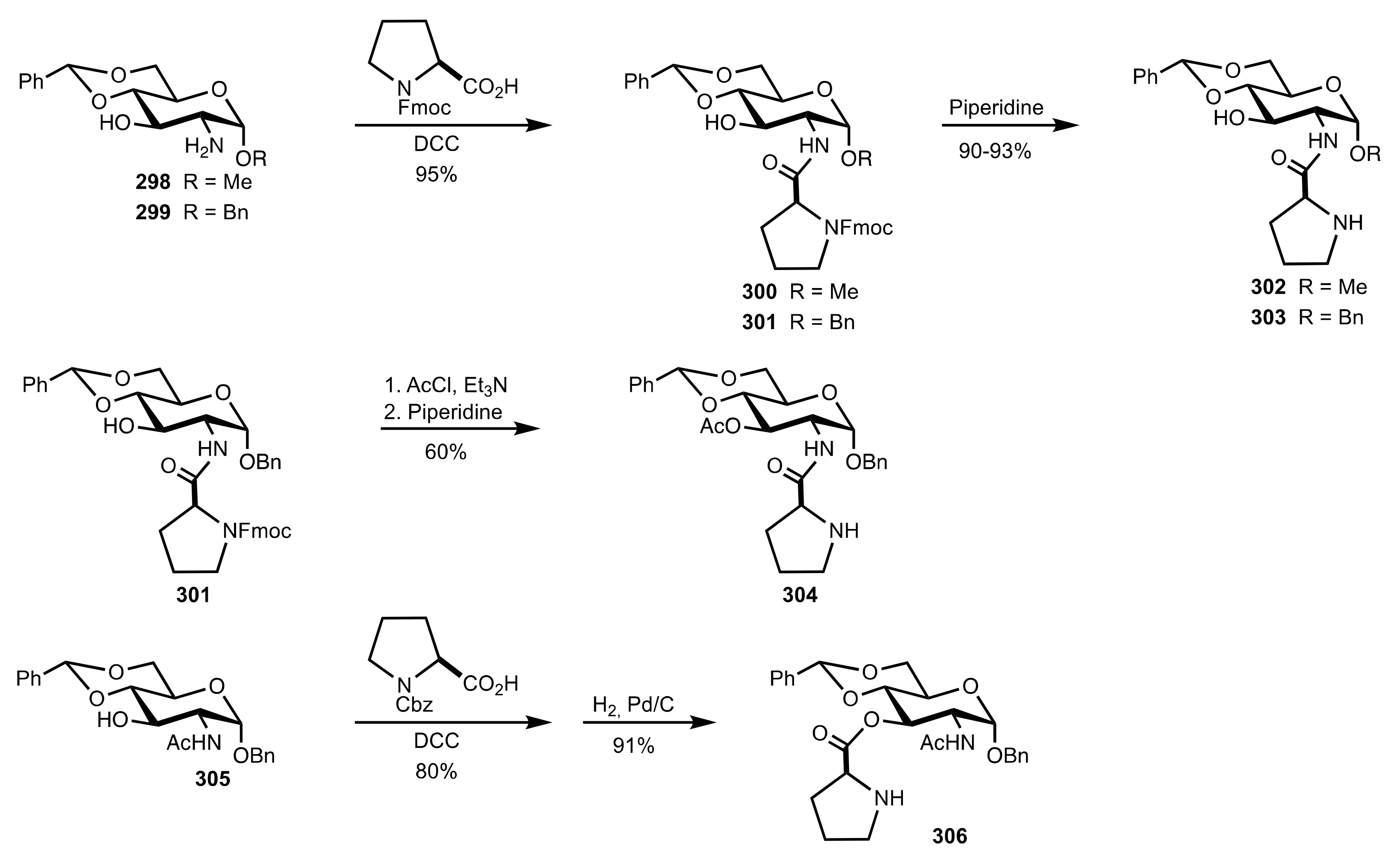
Scheme 83.
The synthesis of the sugar prolinamide organocatalysts 302–304 and 306.
Scheme 83.
The synthesis of the sugar prolinamide organocatalysts 302–304 and 306.
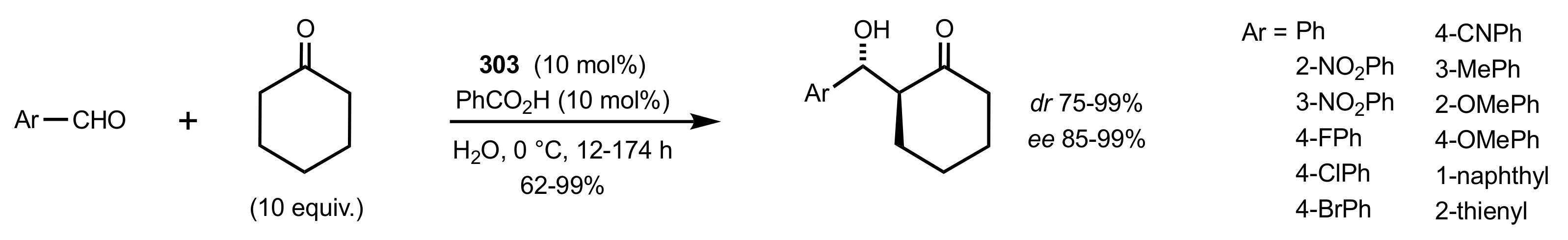
Scheme 84.
Aldol reactions catalysed by 303.
Scheme 84.
Aldol reactions catalysed by 303.
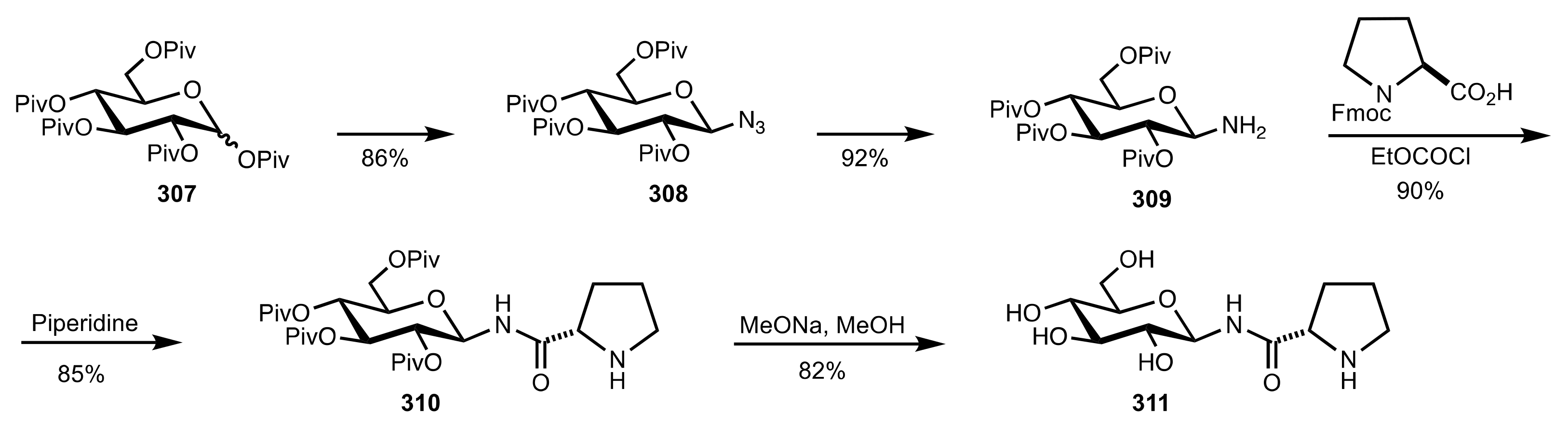
Scheme 85.
The synthesis of the sugar prolinamide organocatalysts 310 and 311.
Scheme 85.
The synthesis of the sugar prolinamide organocatalysts 310 and 311.
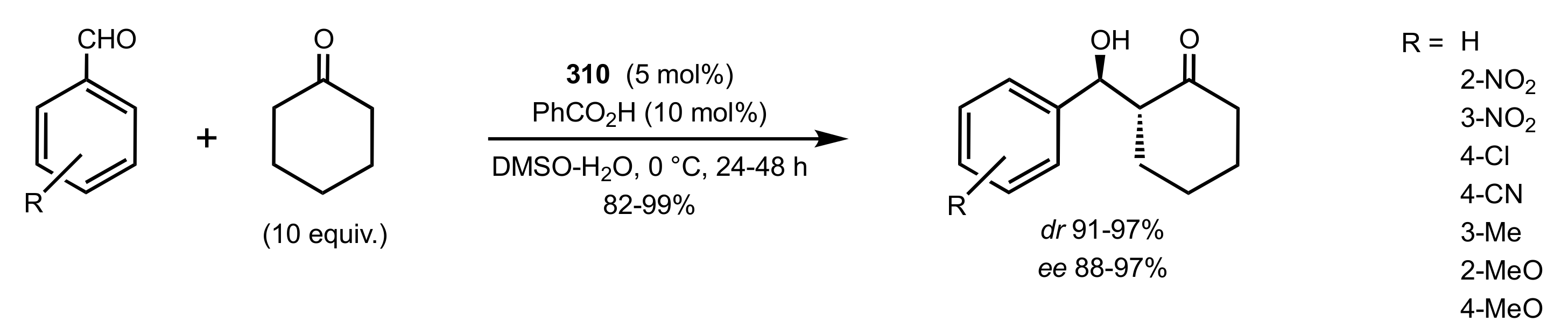
Scheme 86.
Aldol reactions catalysed by 310.
Scheme 86.
Aldol reactions catalysed by 310.
Scheme 87.
The synthesis of the sugar prolinamide organocatalysts 318 and 322.
Scheme 87.
The synthesis of the sugar prolinamide organocatalysts 318 and 322.
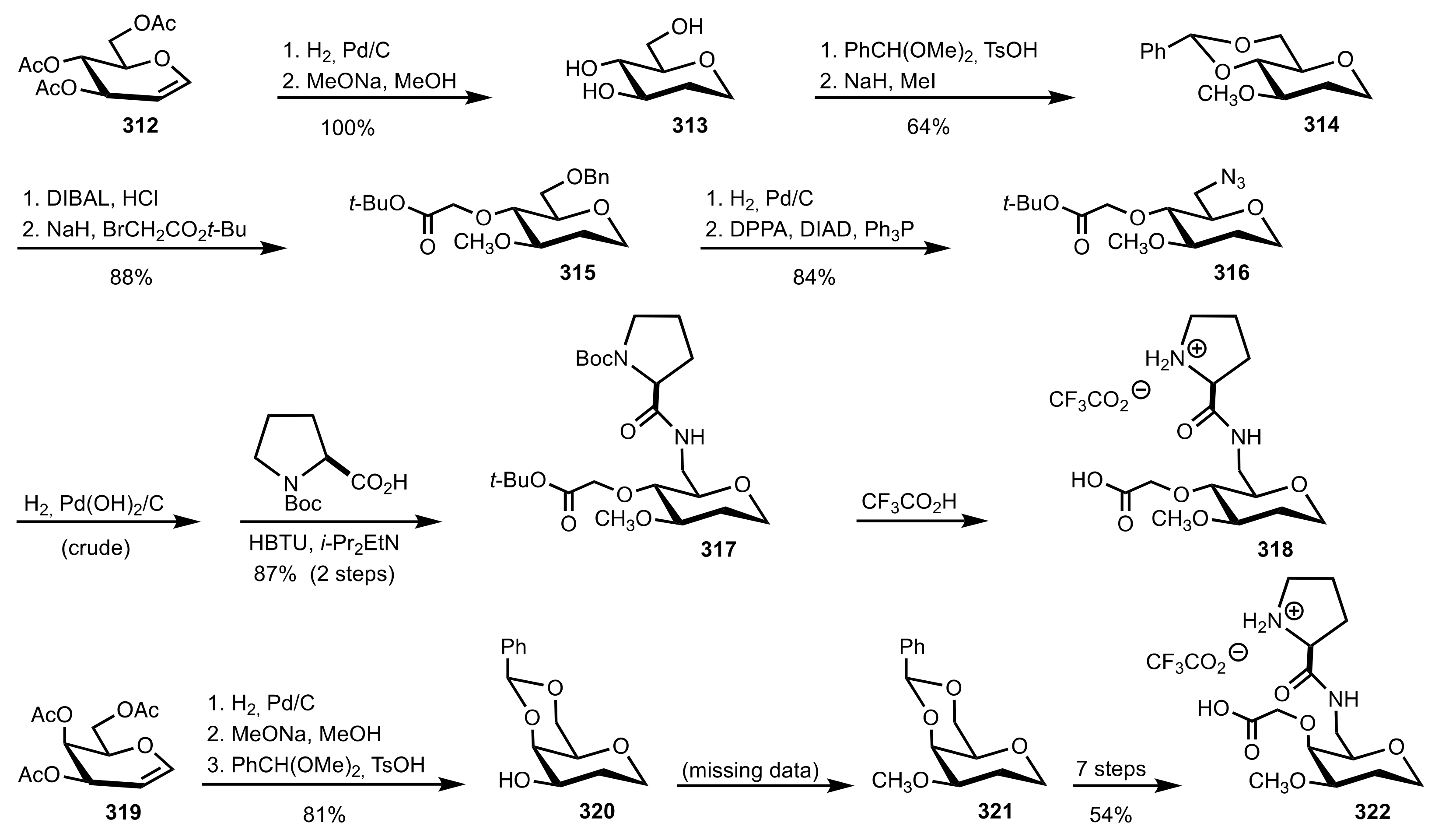
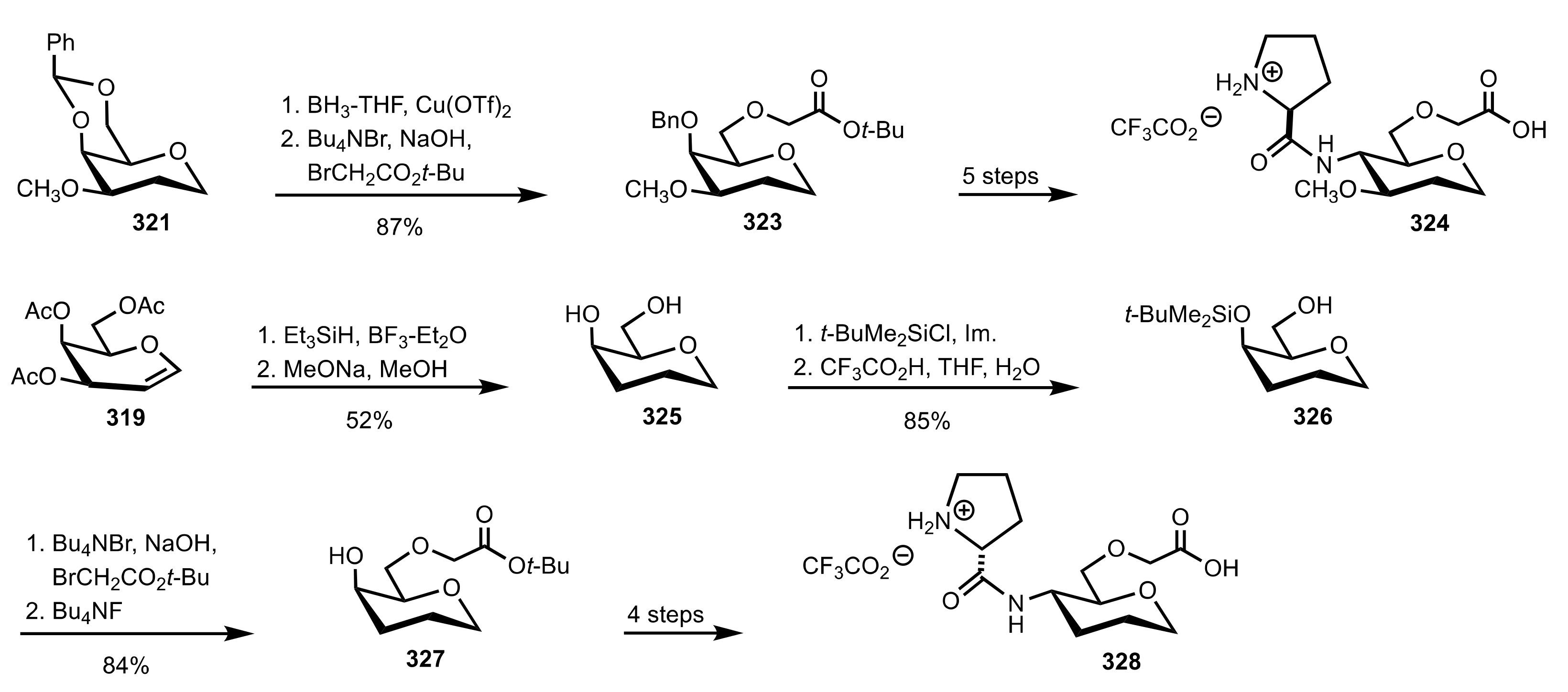
Scheme 88.
The synthesis of the sugar prolinamide organocatalysts 324 and 328.
Scheme 88.
The synthesis of the sugar prolinamide organocatalysts 324 and 328.
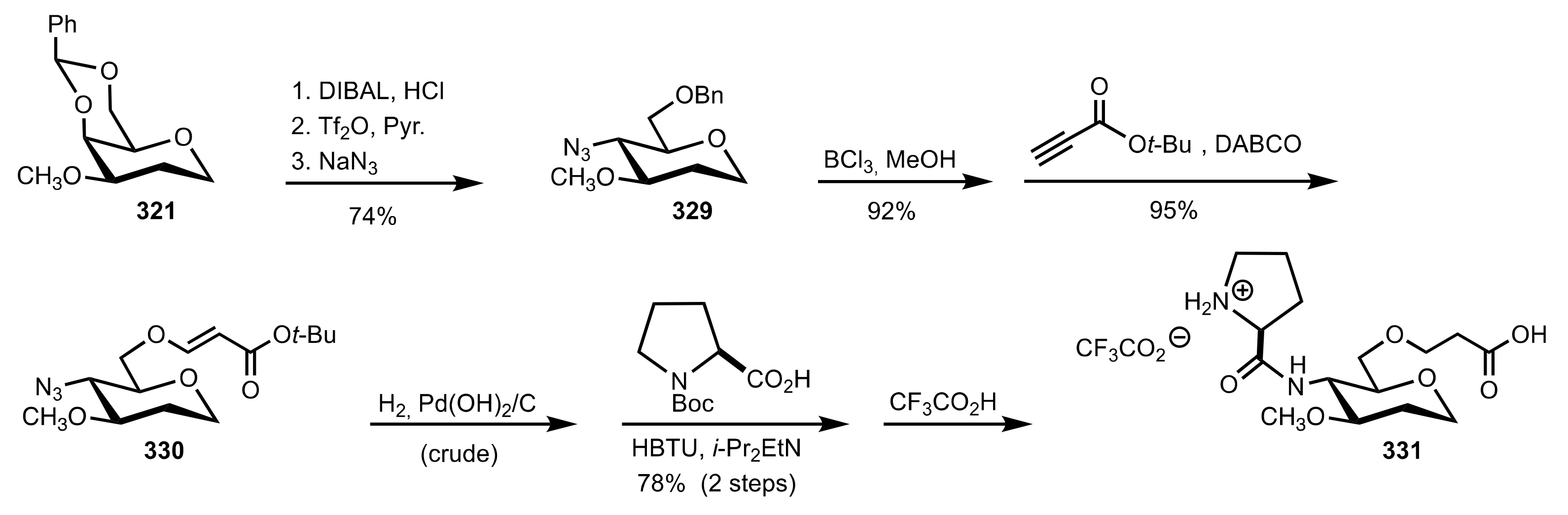
Scheme 89.
The synthesis of the sugar prolinamide organocatalyst 331.
Scheme 89.
The synthesis of the sugar prolinamide organocatalyst 331.
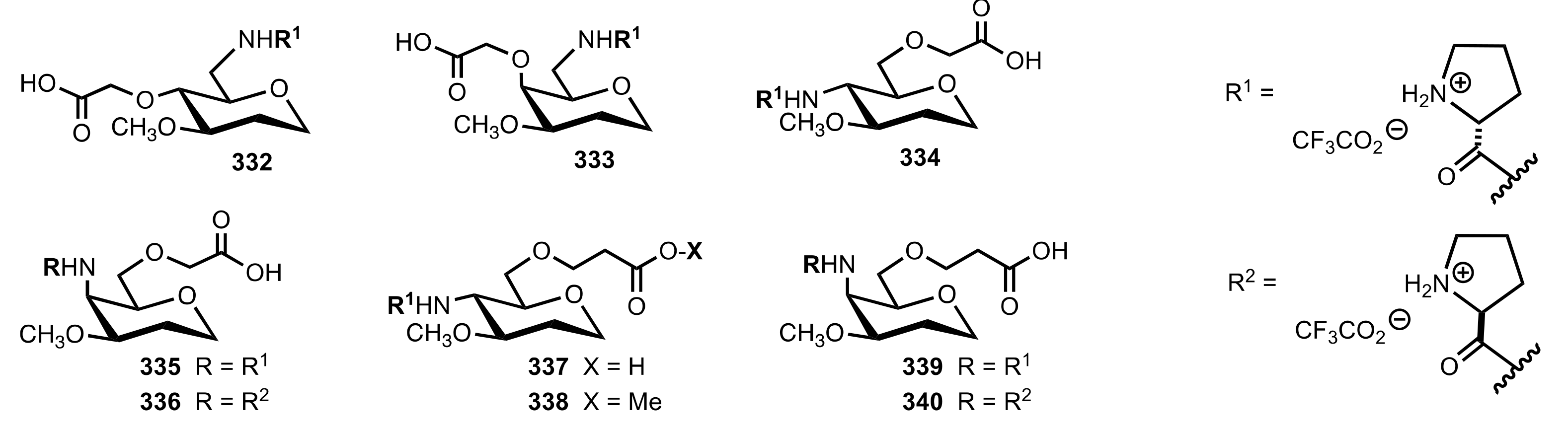
Figure 25.
Other sugar d- and l-prolinamide organocatalysts prepared by Martín and co-workers.
Figure 25.
Other sugar d- and l-prolinamide organocatalysts prepared by Martín and co-workers.
Scheme 90.
The Michael additions catalysed by 337.
Scheme 90.
The Michael additions catalysed by 337.
Scheme 91.
The synthesis of the sugar prolinamide organocatalysts 344 and 345.
Scheme 91.
The synthesis of the sugar prolinamide organocatalysts 344 and 345.
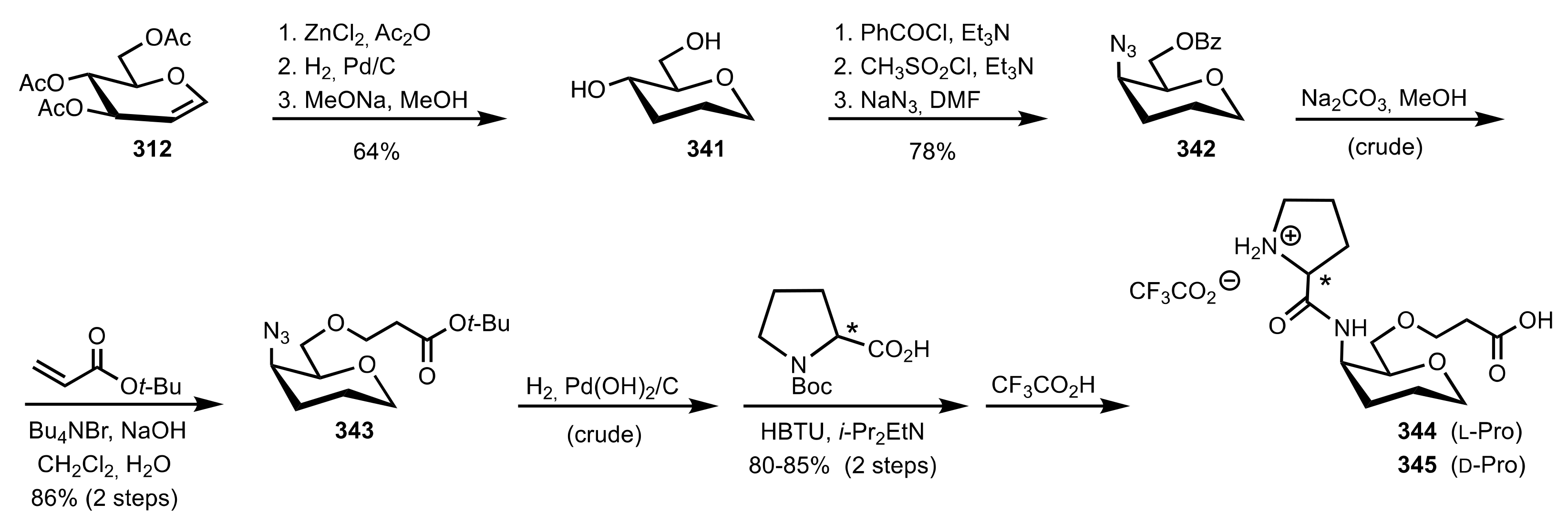
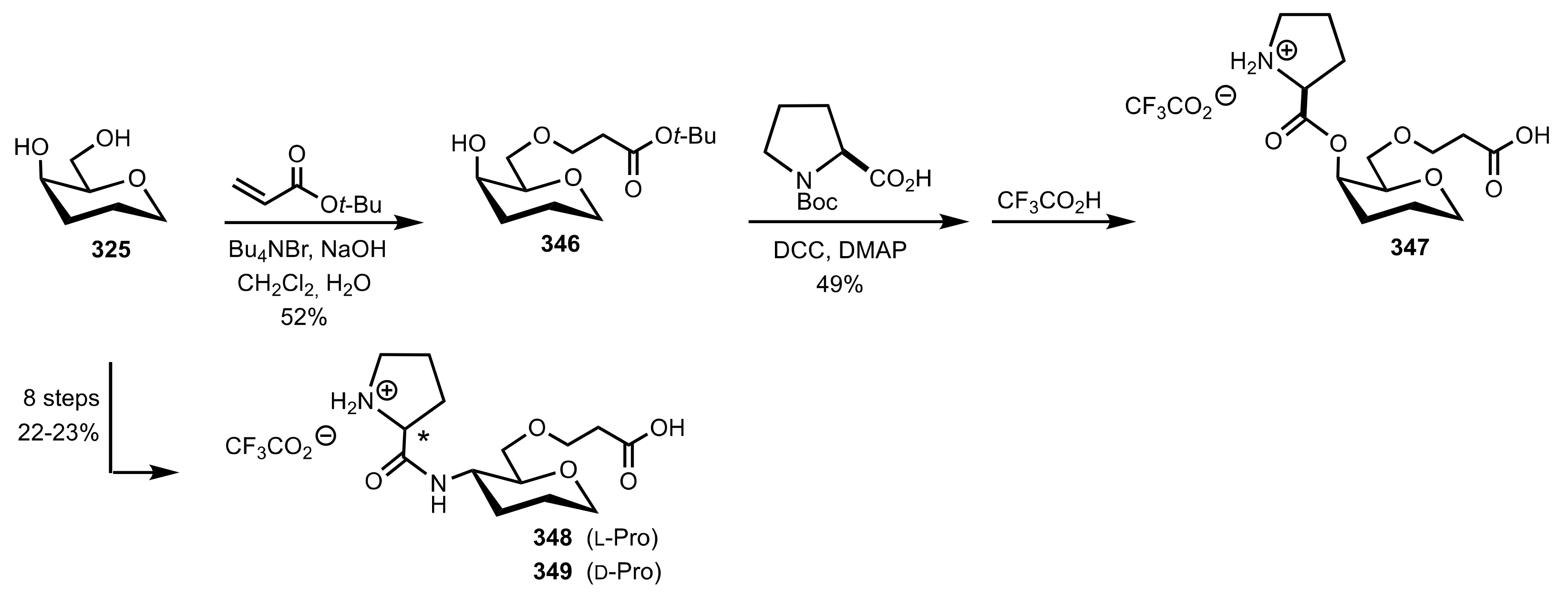
Scheme 92.
Syntheses of the organocatalysts 347–349.
Scheme 92.
Syntheses of the organocatalysts 347–349.
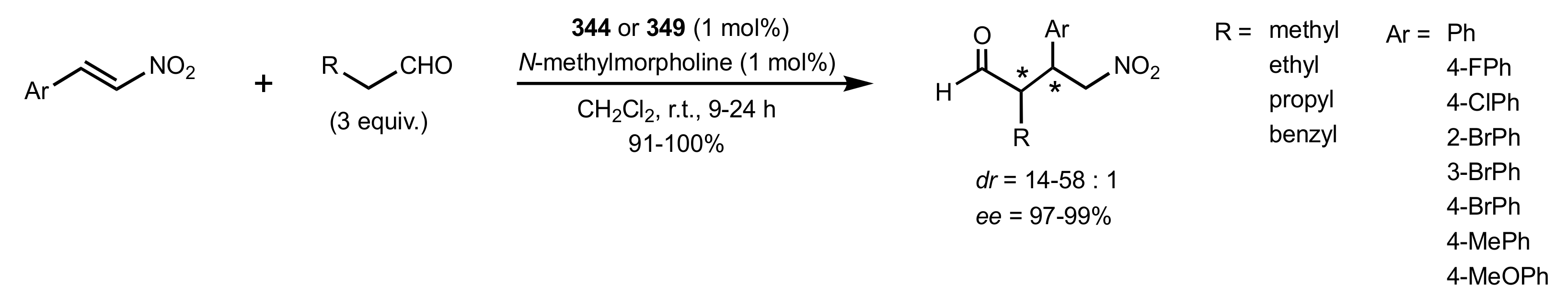
Scheme 93.
The Michael additions catalysed by 344 or 349.
Scheme 93.
The Michael additions catalysed by 344 or 349.
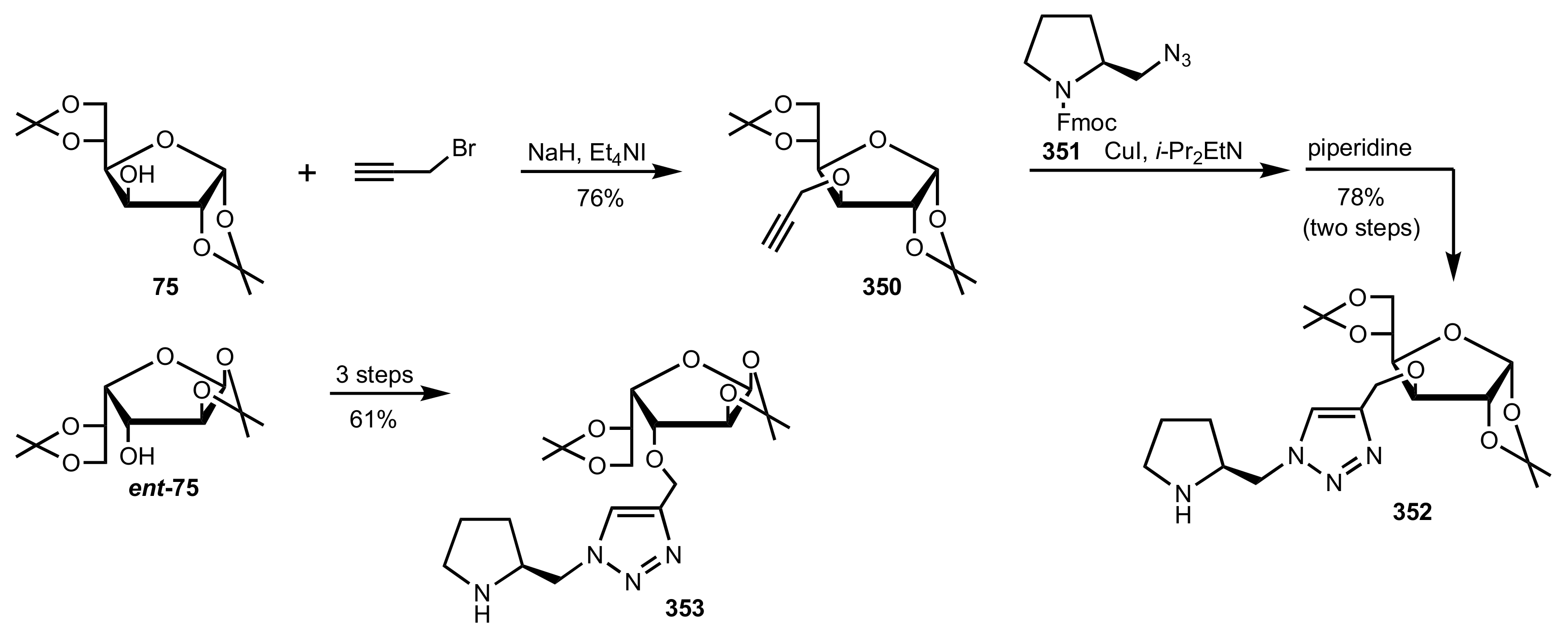
Scheme 94.
The synthesis of the sugar-pyrrolidine organocatalysts 352 and 353.
Scheme 94.
The synthesis of the sugar-pyrrolidine organocatalysts 352 and 353.
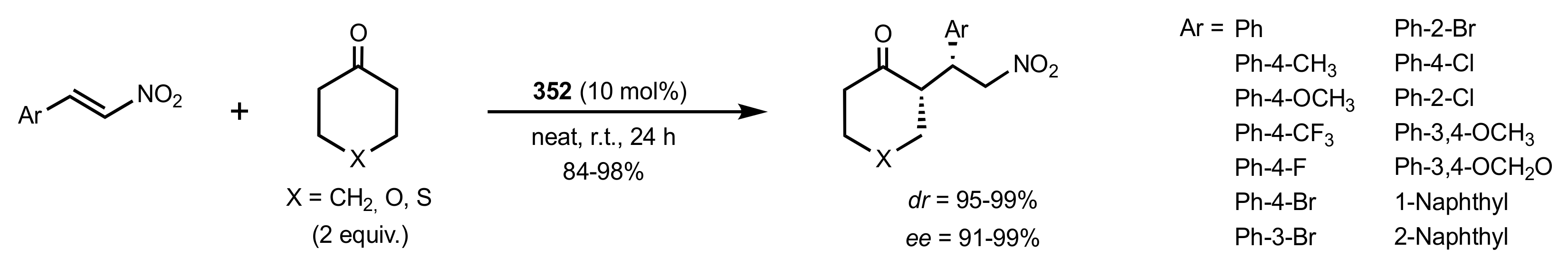
Scheme 95.
The Michael additions catalysed by 352.
Scheme 95.
The Michael additions catalysed by 352.
Scheme 96.
The synthesis of the sugar-pyrrolidine organocatalysts 357 and 359.
Scheme 96.
The synthesis of the sugar-pyrrolidine organocatalysts 357 and 359.
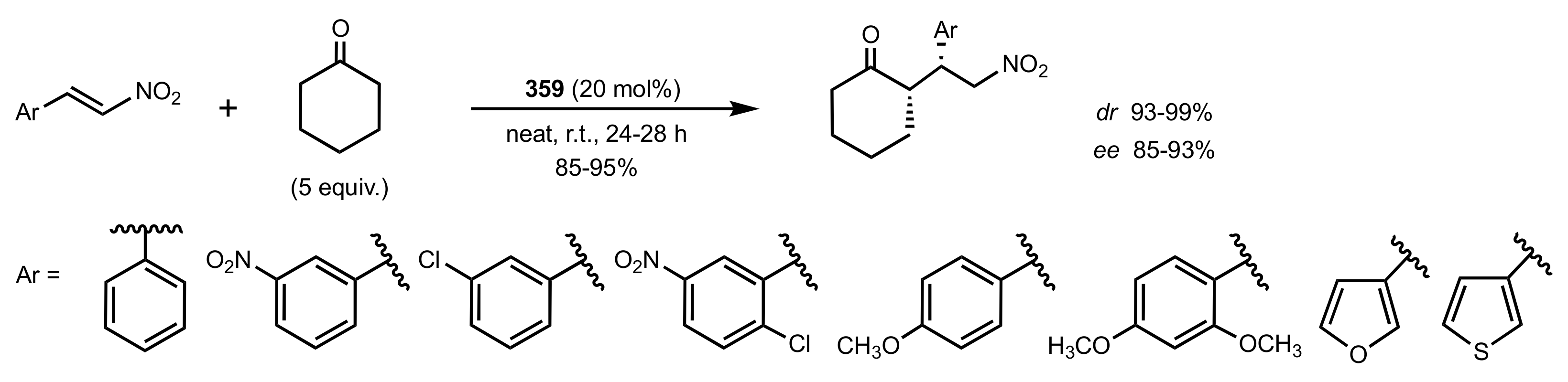
Scheme 97.
The Michael additions catalysed by 359.
Scheme 97.
The Michael additions catalysed by 359.
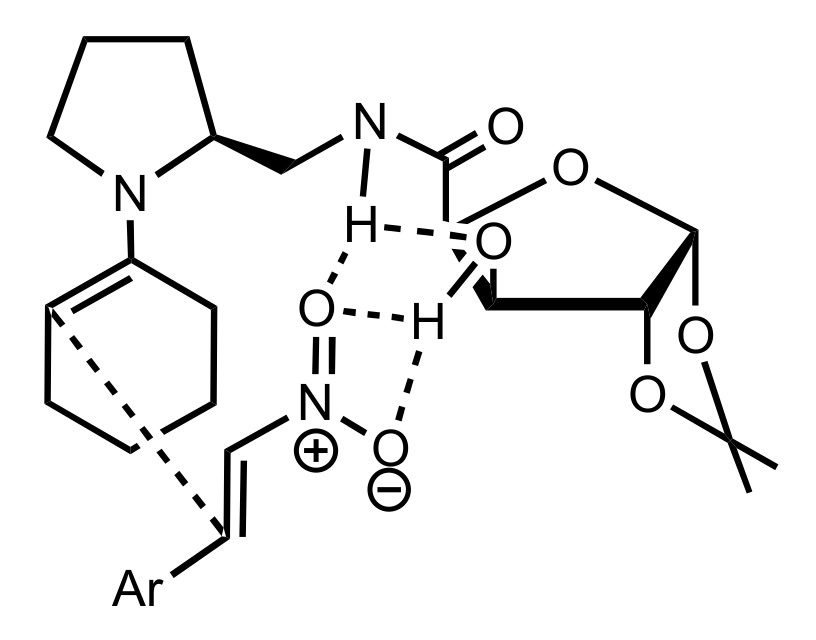
Figure 26.
The proposed transition state for the Michael additions catalysed by 359.
Figure 26.
The proposed transition state for the Michael additions catalysed by 359.
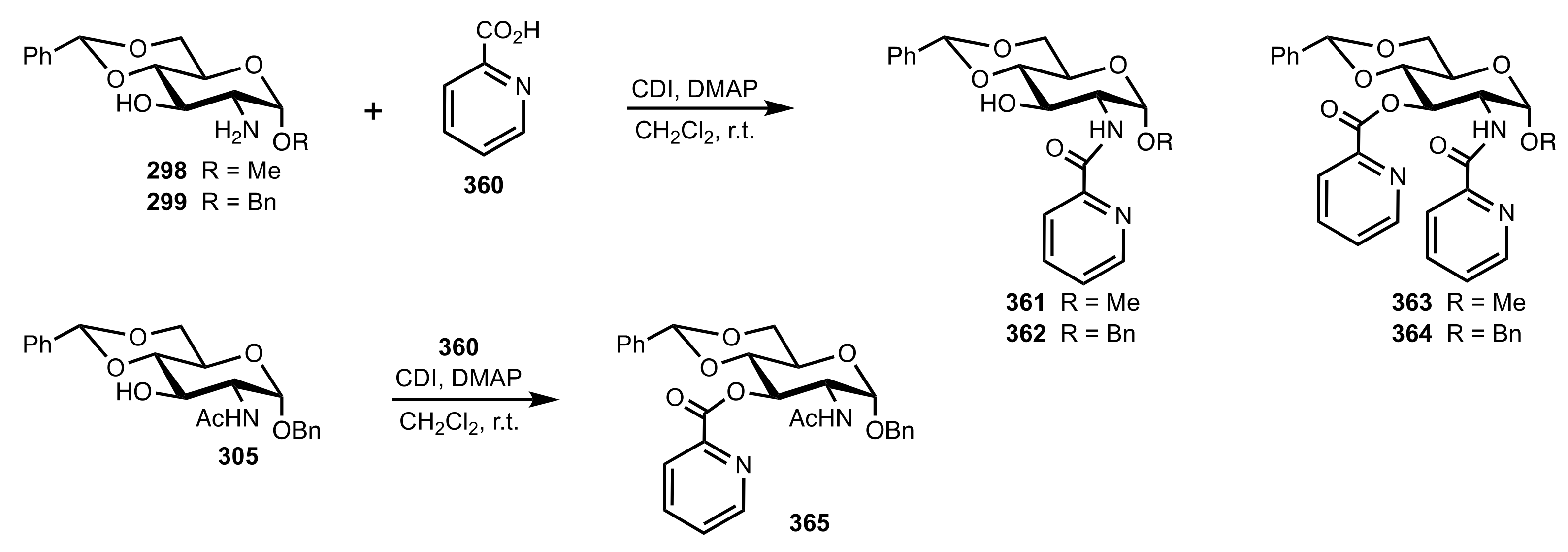
Scheme 98.
The synthesis of the sugar-pyridine organocatalysts 361–365.
Scheme 98.
The synthesis of the sugar-pyridine organocatalysts 361–365.
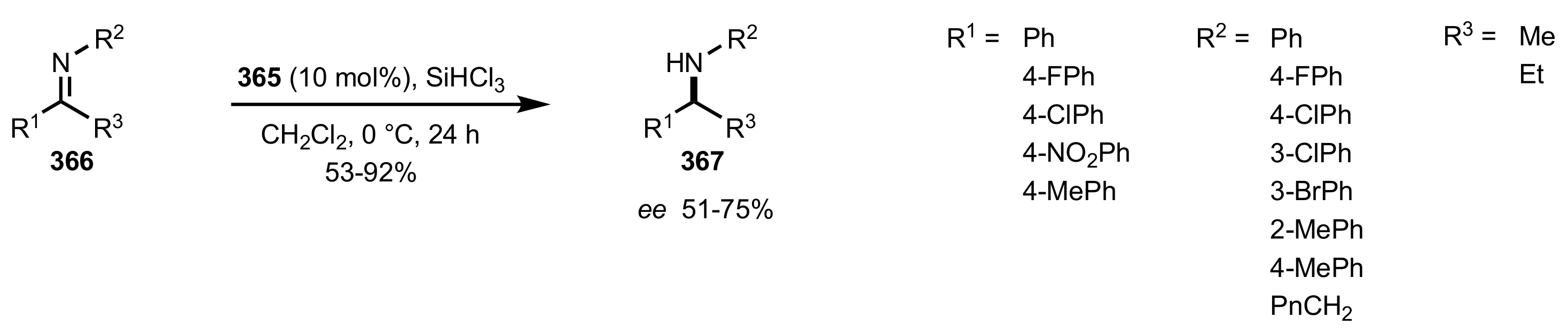
Scheme 99.
The reduction of imines catalysed by 365.
Scheme 99.
The reduction of imines catalysed by 365.
Figure 27.
The proposed mechanism for the imines reduction catalysed by 365.
Figure 27.
The proposed mechanism for the imines reduction catalysed by 365.
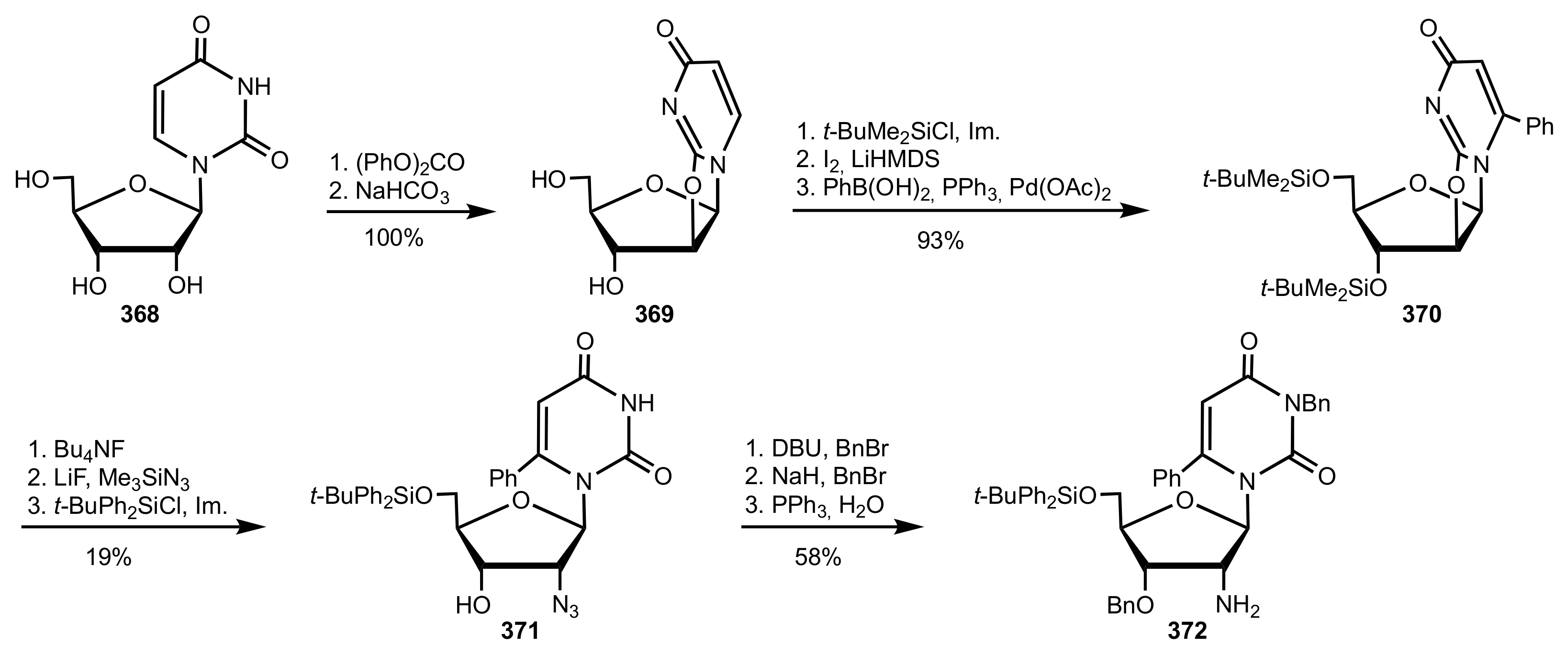
Scheme 100.
The synthesis of the 2′-aminouridine organocatalyst 372.
Scheme 100.
The synthesis of the 2′-aminouridine organocatalyst 372.
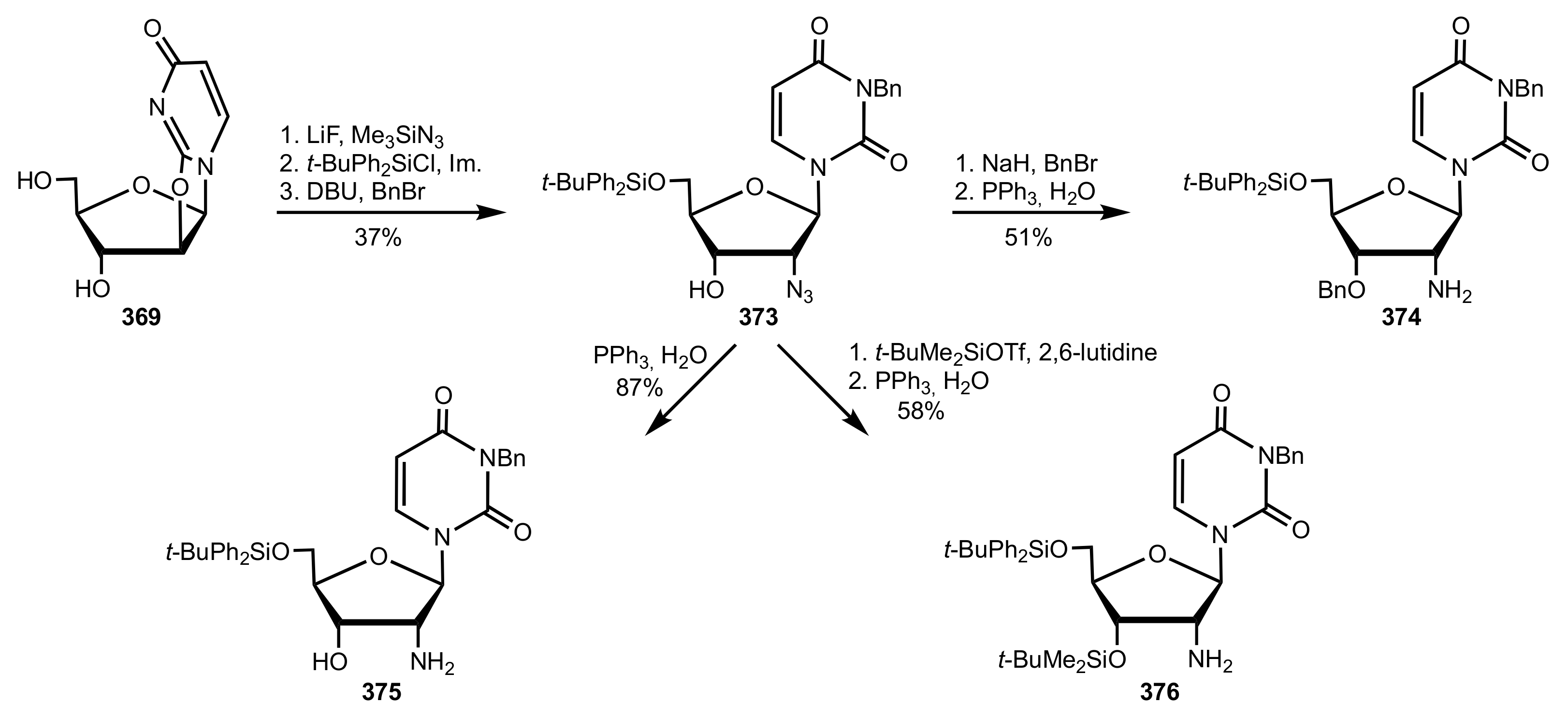
Scheme 101.
The synthesis of the 2′-aminouridine organocatalysts 374–376.
Scheme 101.
The synthesis of the 2′-aminouridine organocatalysts 374–376.
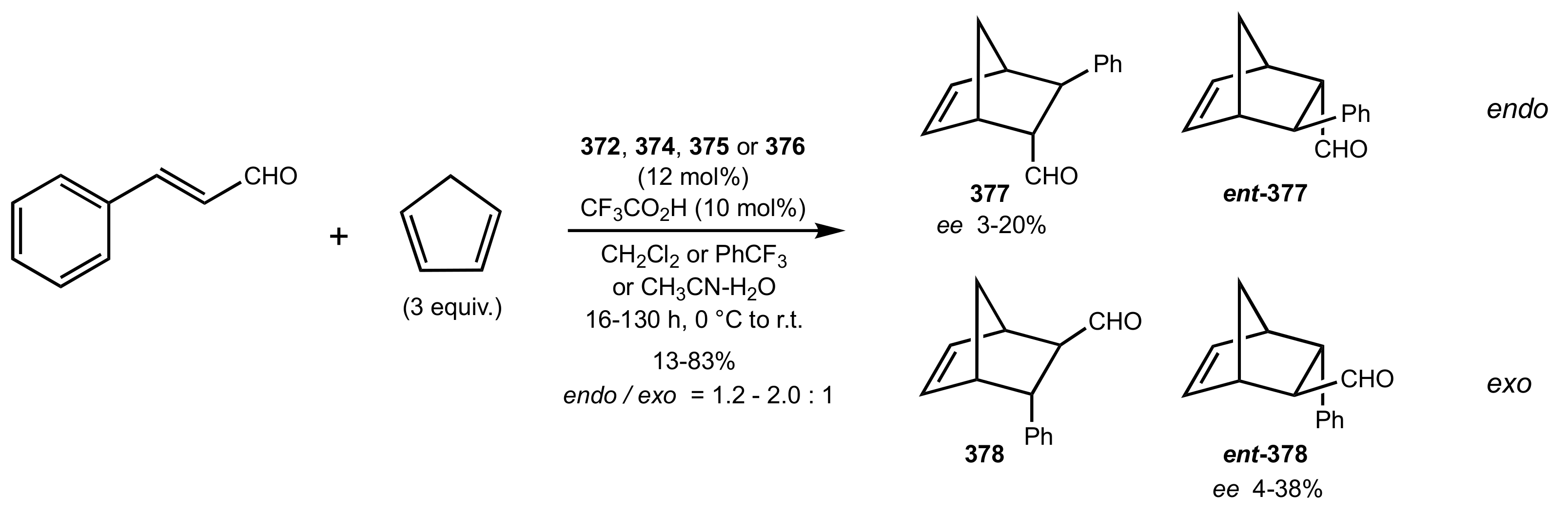
Scheme 102.
Diels–Alder reactions catalysed by 372 or 374–376.
Scheme 102.
Diels–Alder reactions catalysed by 372 or 374–376.
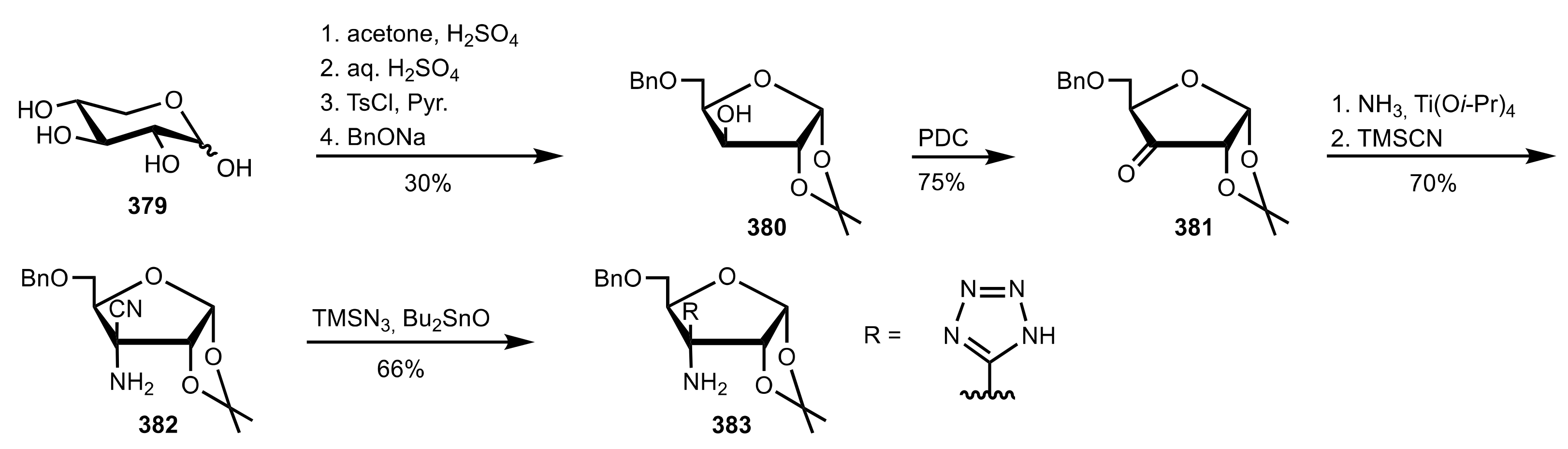
Scheme 103.
The synthesis of the sugar tetrazole organocatalyst 383.
Scheme 103.
The synthesis of the sugar tetrazole organocatalyst 383.
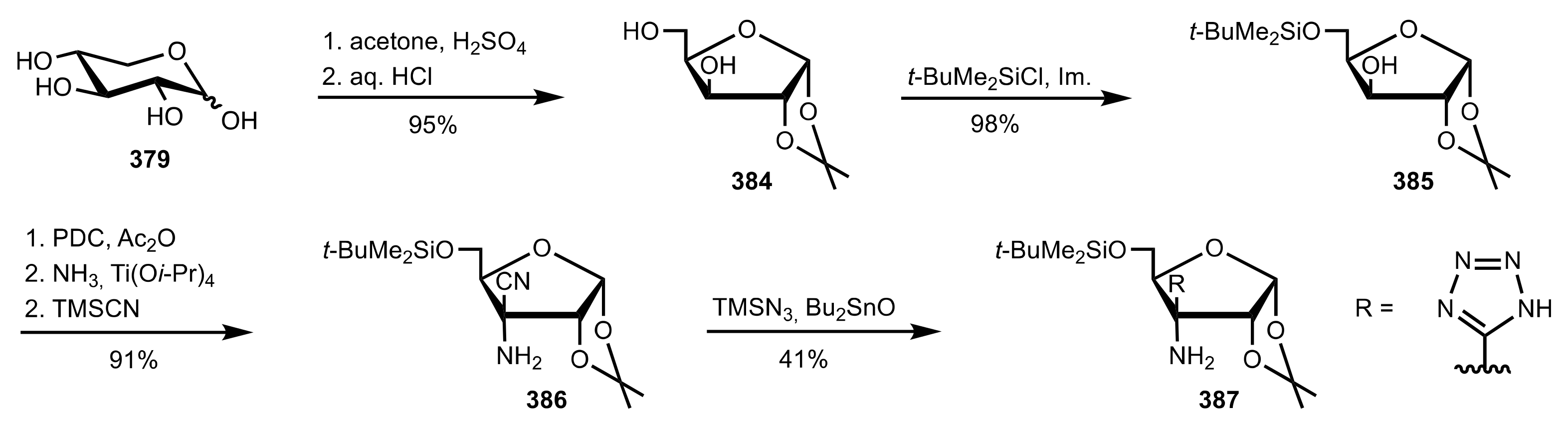
Scheme 104.
The synthesis of the sugar tetrazole organocatalyst 387.
Scheme 104.
The synthesis of the sugar tetrazole organocatalyst 387.
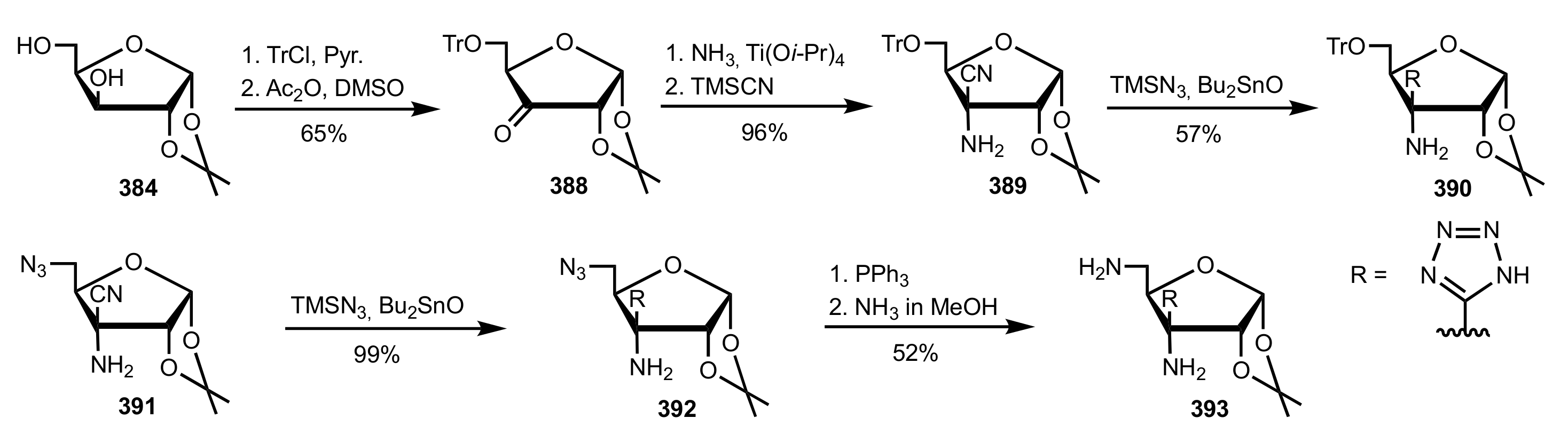
Scheme 105.
The synthesis of the sugar tetrazole organocatalysts 390, 392 and 393.
Scheme 105.
The synthesis of the sugar tetrazole organocatalysts 390, 392 and 393.
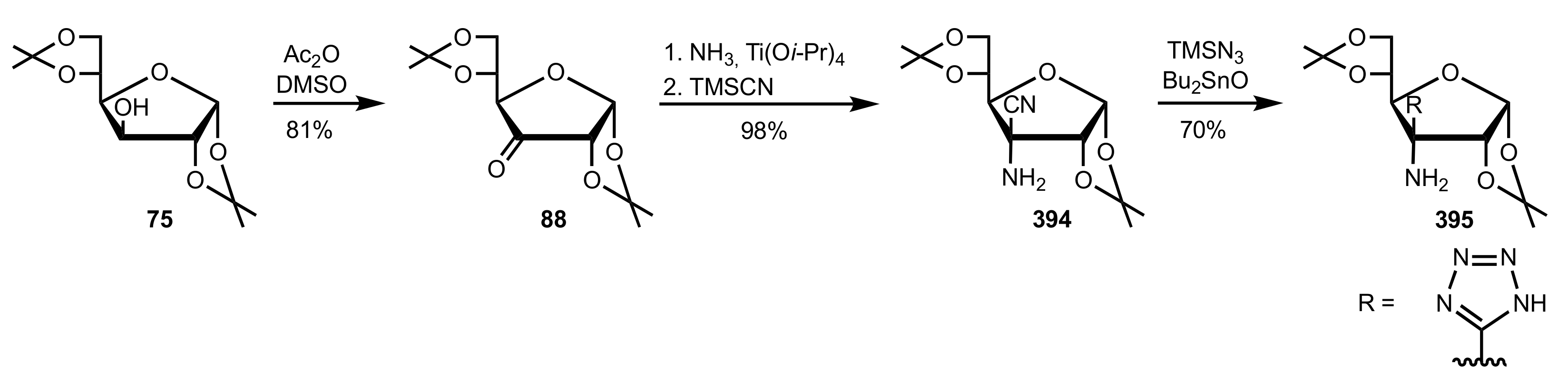
Scheme 106.
The synthesis of the sugar tetrazole organocatalyst 395.
Scheme 106.
The synthesis of the sugar tetrazole organocatalyst 395.
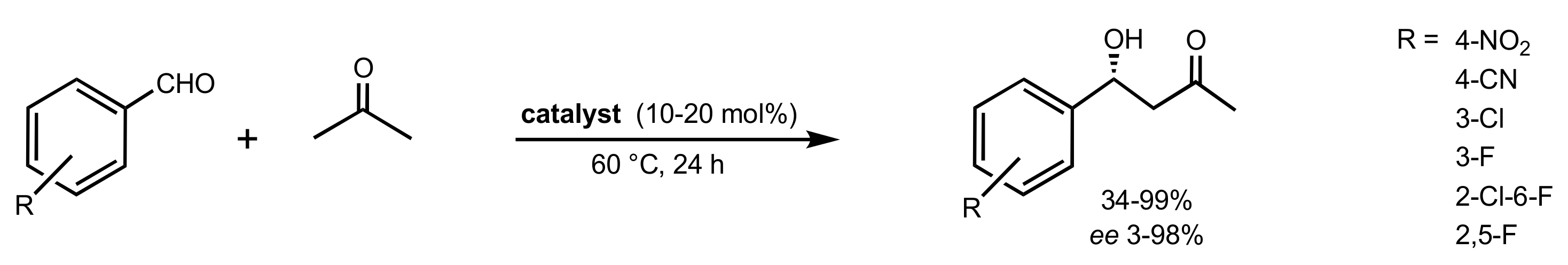
Scheme 107.
Aldol reaction catalysed by 383, 387, 390, 392, 393 or 395.
Scheme 107.
Aldol reaction catalysed by 383, 387, 390, 392, 393 or 395.
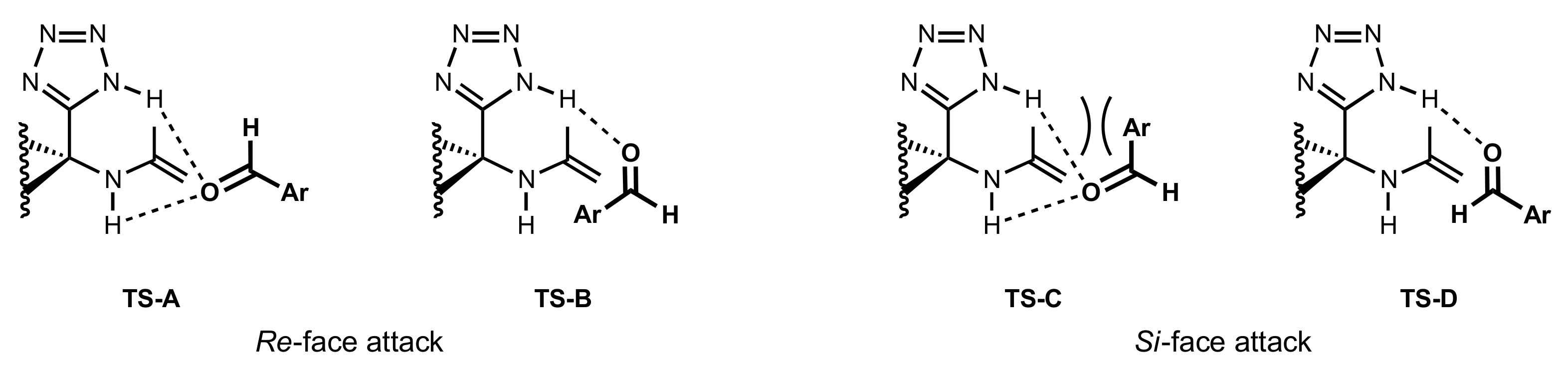
Figure 28.
Transition states of the aldol reaction catalysed by the sugar tetrazole catalysts.
Figure 28.
Transition states of the aldol reaction catalysed by the sugar tetrazole catalysts.
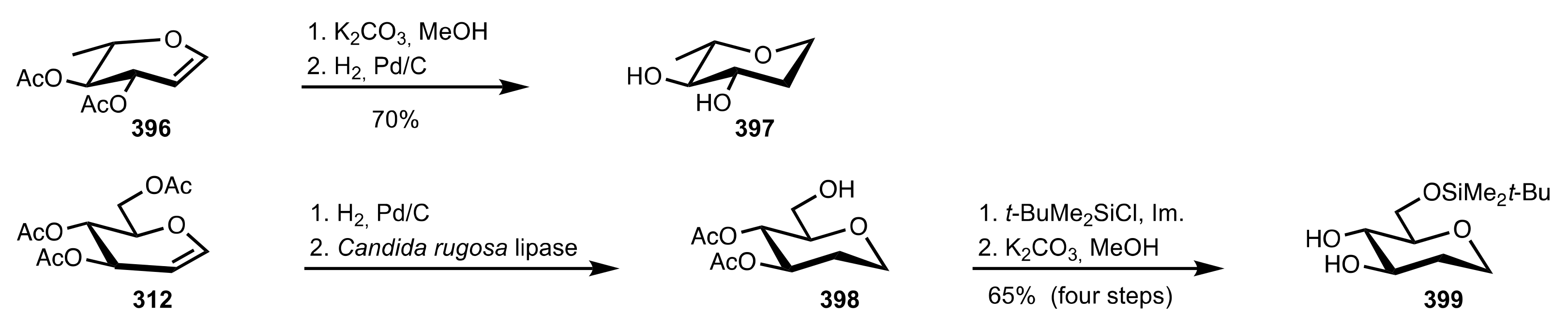
Scheme 108.
The synthesis of the sugar diol organocatalysts 397 and 399.
Scheme 108.
The synthesis of the sugar diol organocatalysts 397 and 399.
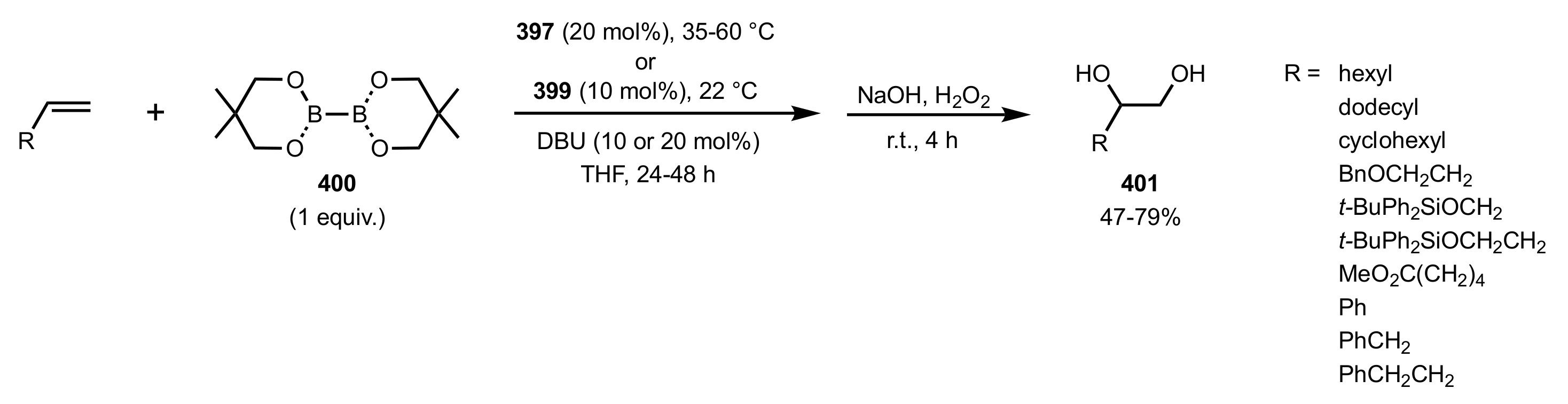
Scheme 109.
The diboration–oxidation of alkenes organocatalysed by sugar diols 397 and 399.
Scheme 109.
The diboration–oxidation of alkenes organocatalysed by sugar diols 397 and 399.
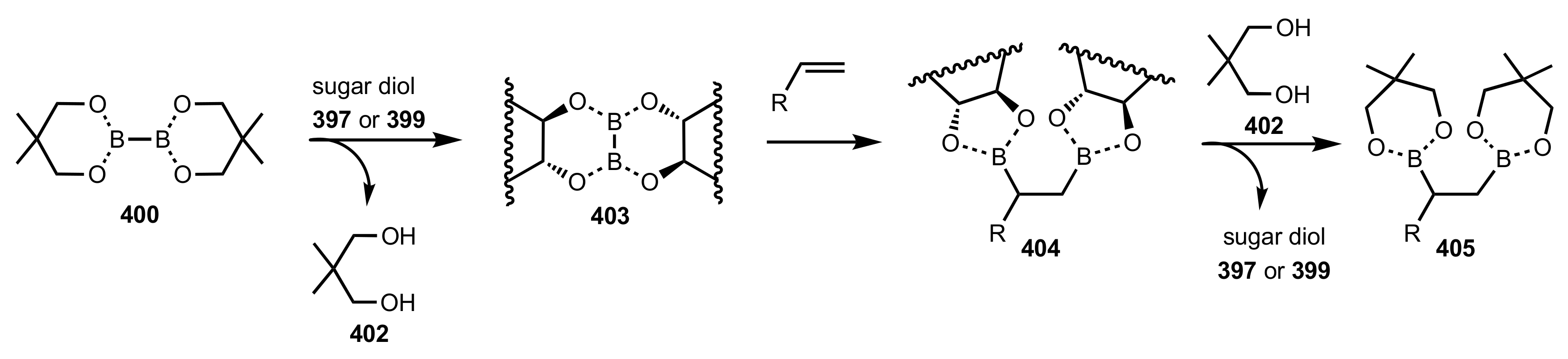
Scheme 110.
Mechanism of the diboration–oxidation of alkenes catalysed by 397 or 399.
Scheme 110.
Mechanism of the diboration–oxidation of alkenes catalysed by 397 or 399.
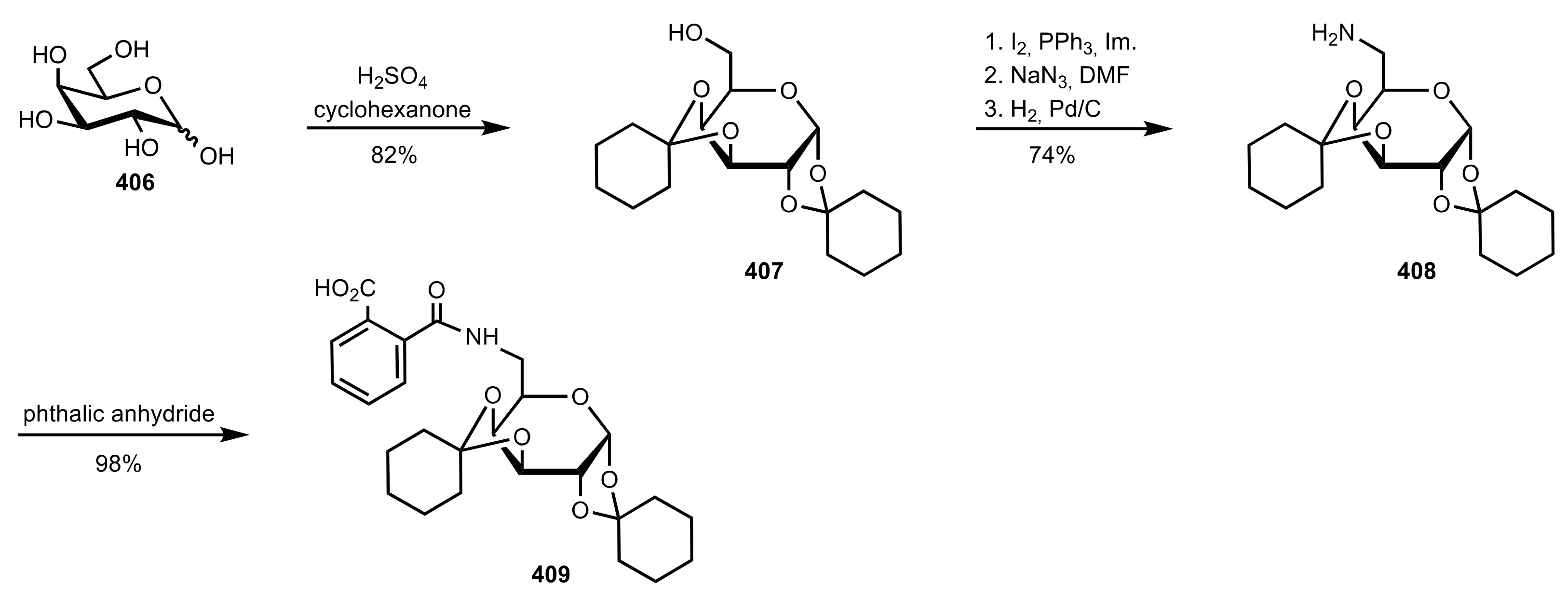
Scheme 111.
The synthesis of the sugar carboxylic acid organocatalyst 409.
Scheme 111.
The synthesis of the sugar carboxylic acid organocatalyst 409.
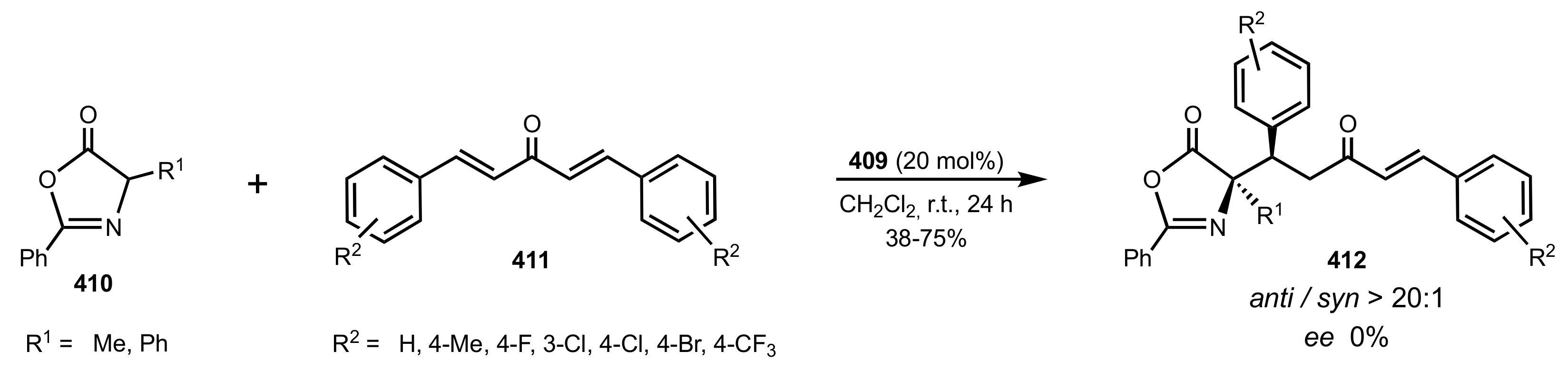
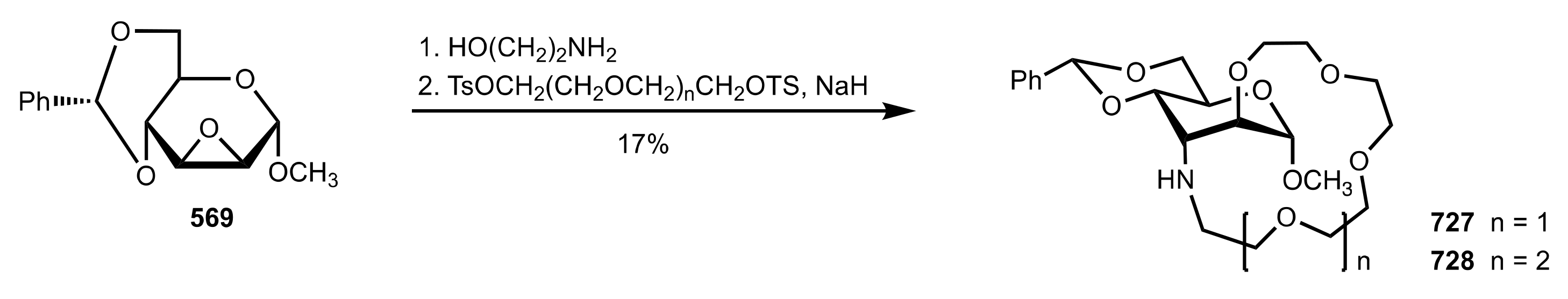
Scheme 112.
The Michael addition catalysed by 409.
Scheme 112.
The Michael addition catalysed by 409.
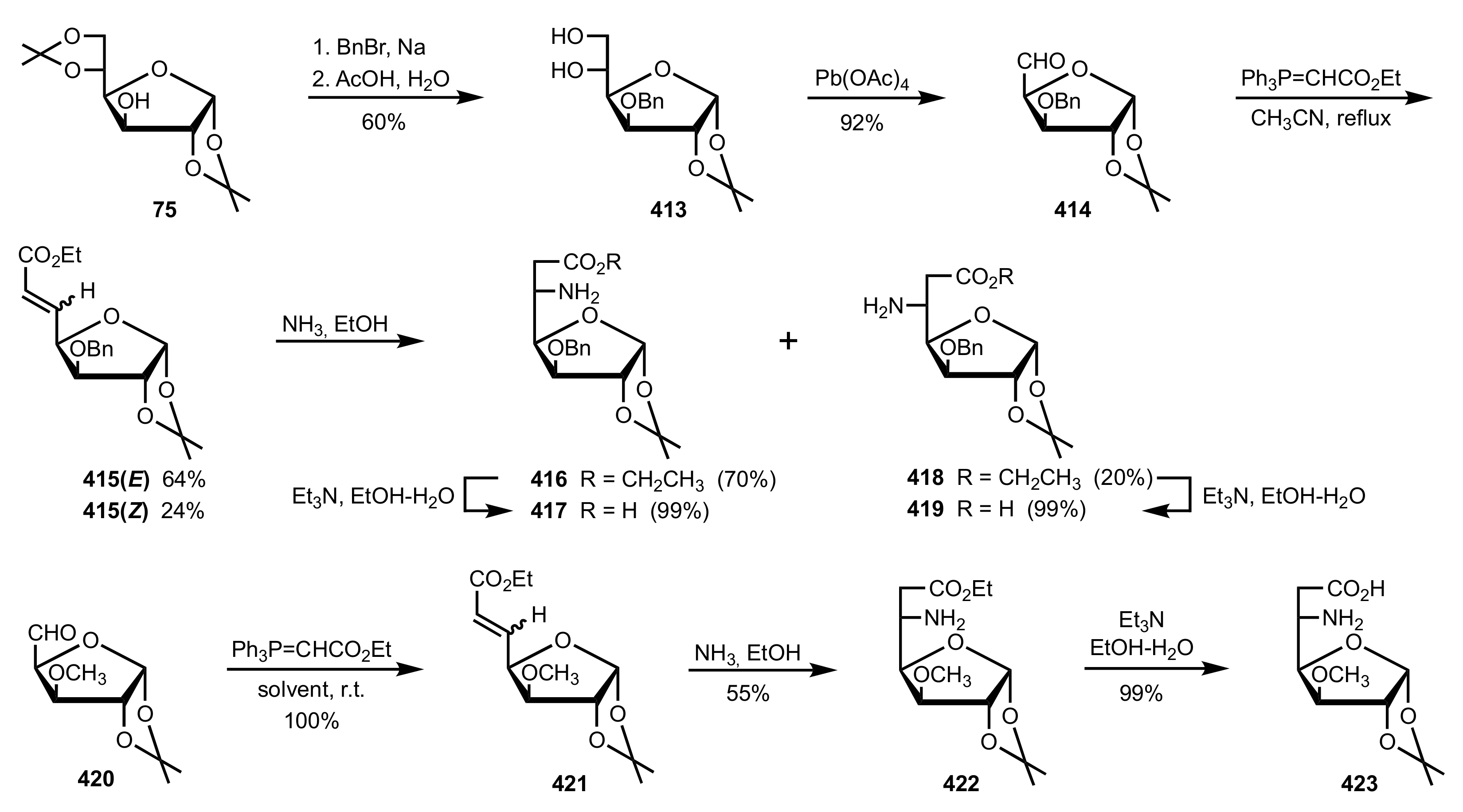
Scheme 113.
The synthesis of the sugar aminoacid organocatalysts 416, 417, 419 and 423.
Scheme 113.
The synthesis of the sugar aminoacid organocatalysts 416, 417, 419 and 423.
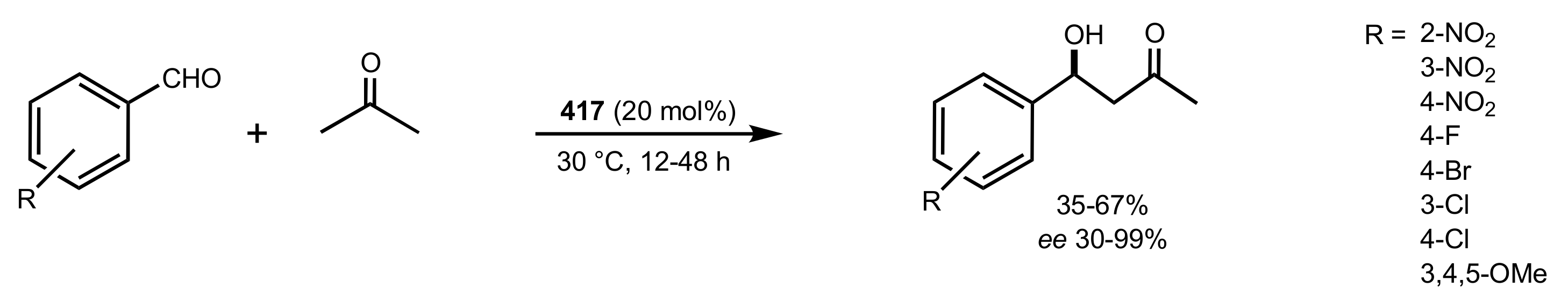
Scheme 114.
Aldol reactions catalysed by 417.
Scheme 114.
Aldol reactions catalysed by 417.
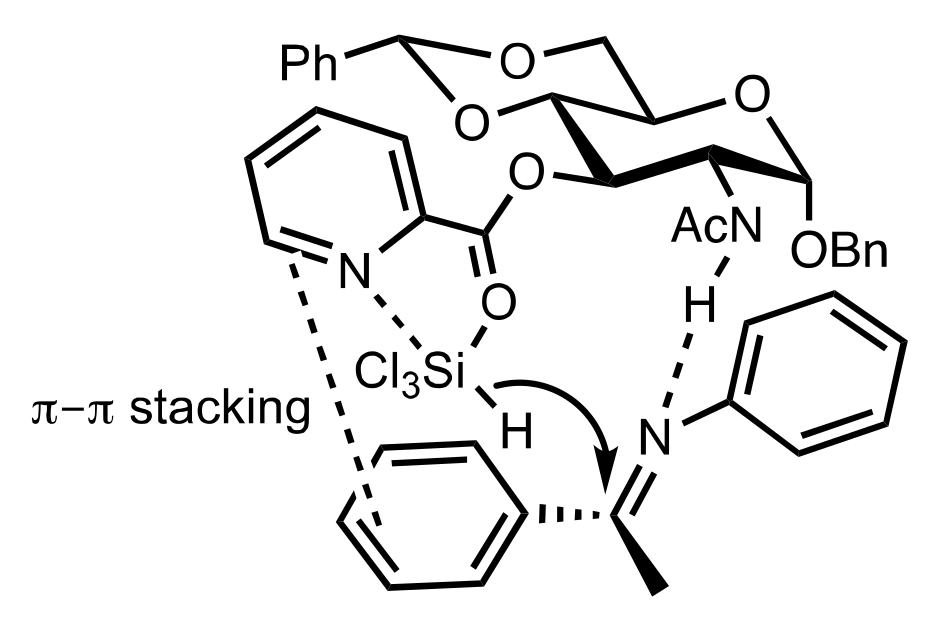
Figure 29.
Favoured transition state leading to the (S)-configured aldol.
Figure 29.
Favoured transition state leading to the (S)-configured aldol.
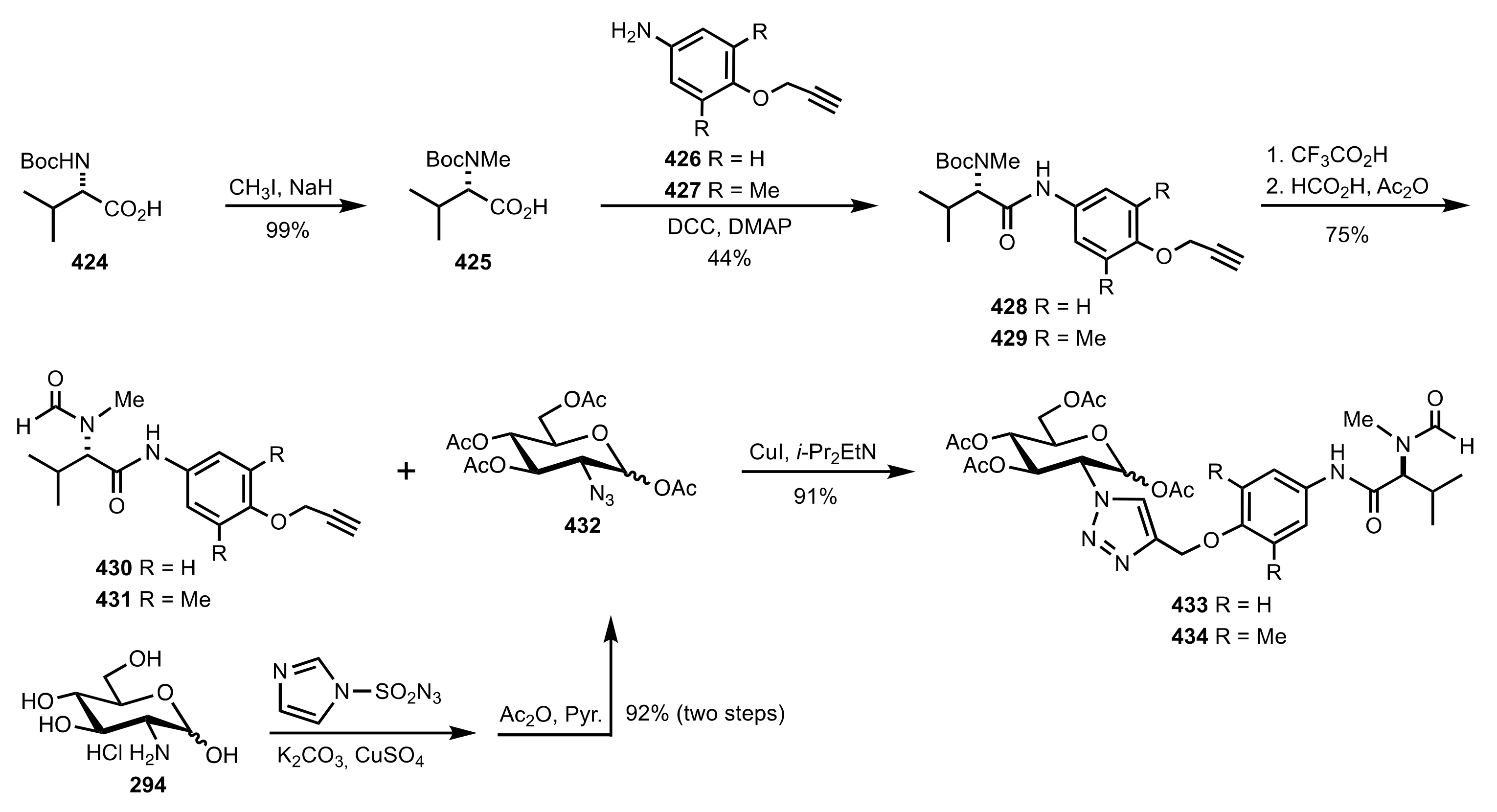
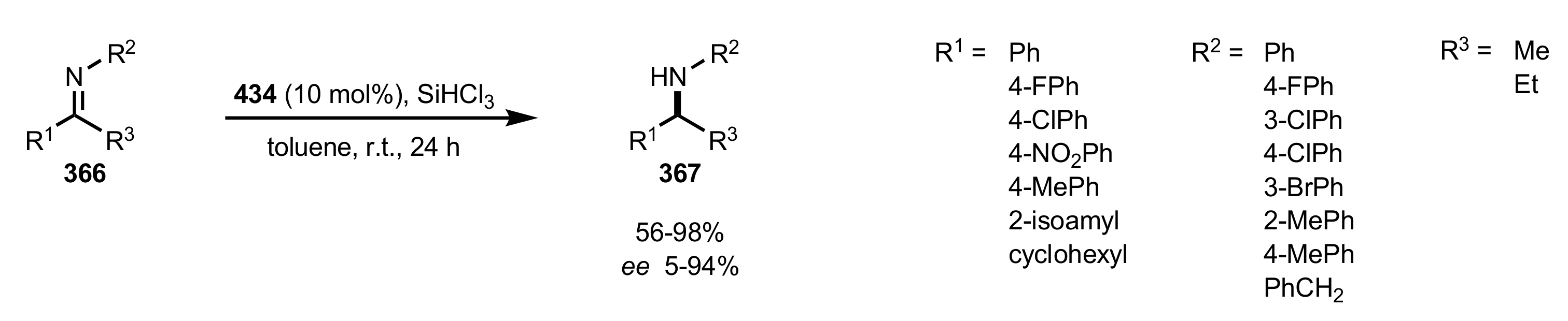
Scheme 115.
The synthesis of the sugar aminoacid organocatalysts 433 and 434.
Scheme 115.
The synthesis of the sugar aminoacid organocatalysts 433 and 434.
Scheme 116.
The reduction of imines catalysed by 434.
Scheme 116.
The reduction of imines catalysed by 434.
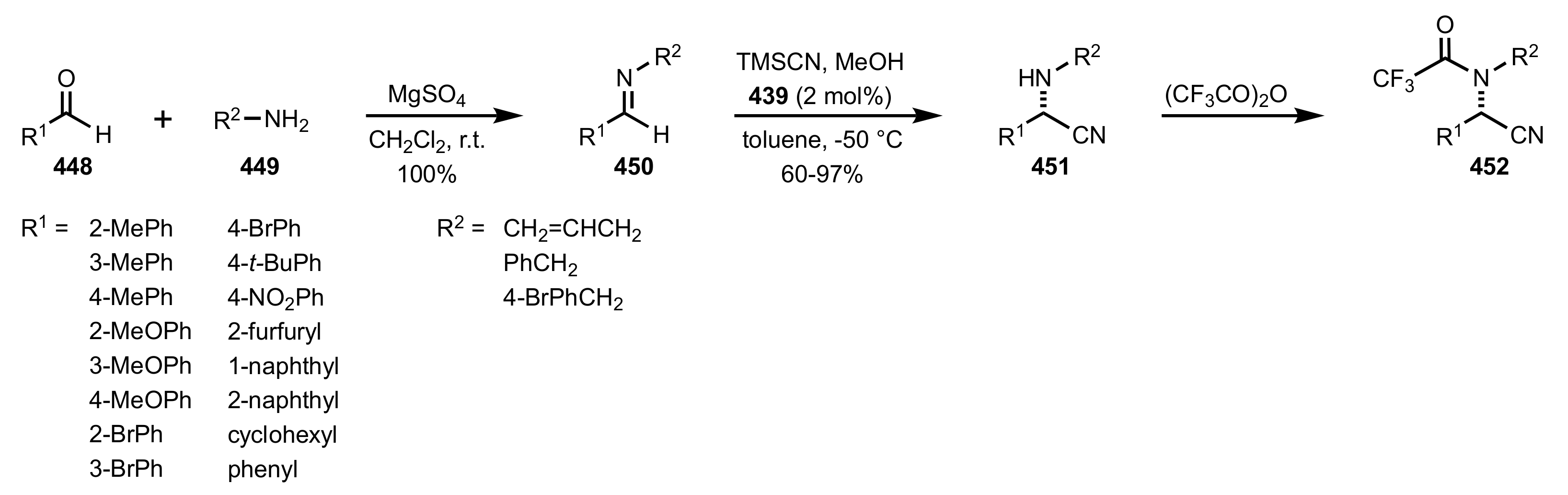
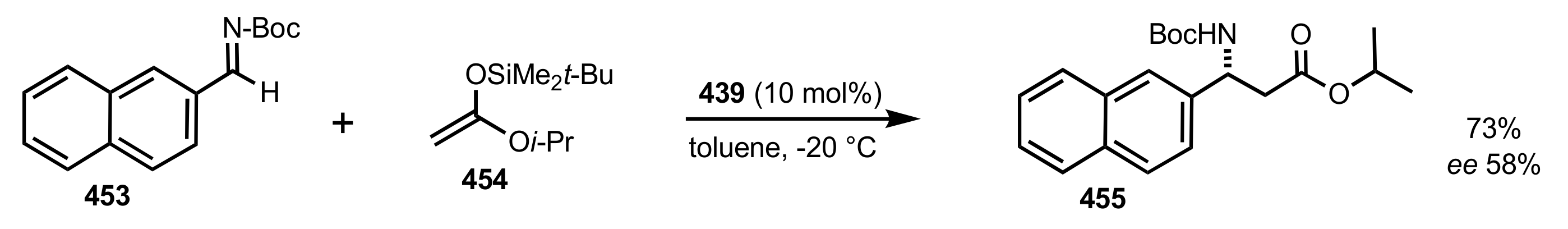
Scheme 117.
The synthesis of the sugar imine organocatalyst 439.
Scheme 117.
The synthesis of the sugar imine organocatalyst 439.
Figure 30.
Other sugar imine organocatalysts used by Kunz and co-workers.
Figure 30.
Other sugar imine organocatalysts used by Kunz and co-workers.
Scheme 118.
Strecker reaction catalysed by 439.
Scheme 118.
Strecker reaction catalysed by 439.
Scheme 119.
Mannich reaction catalysed by 439.
Scheme 119.
Mannich reaction catalysed by 439.
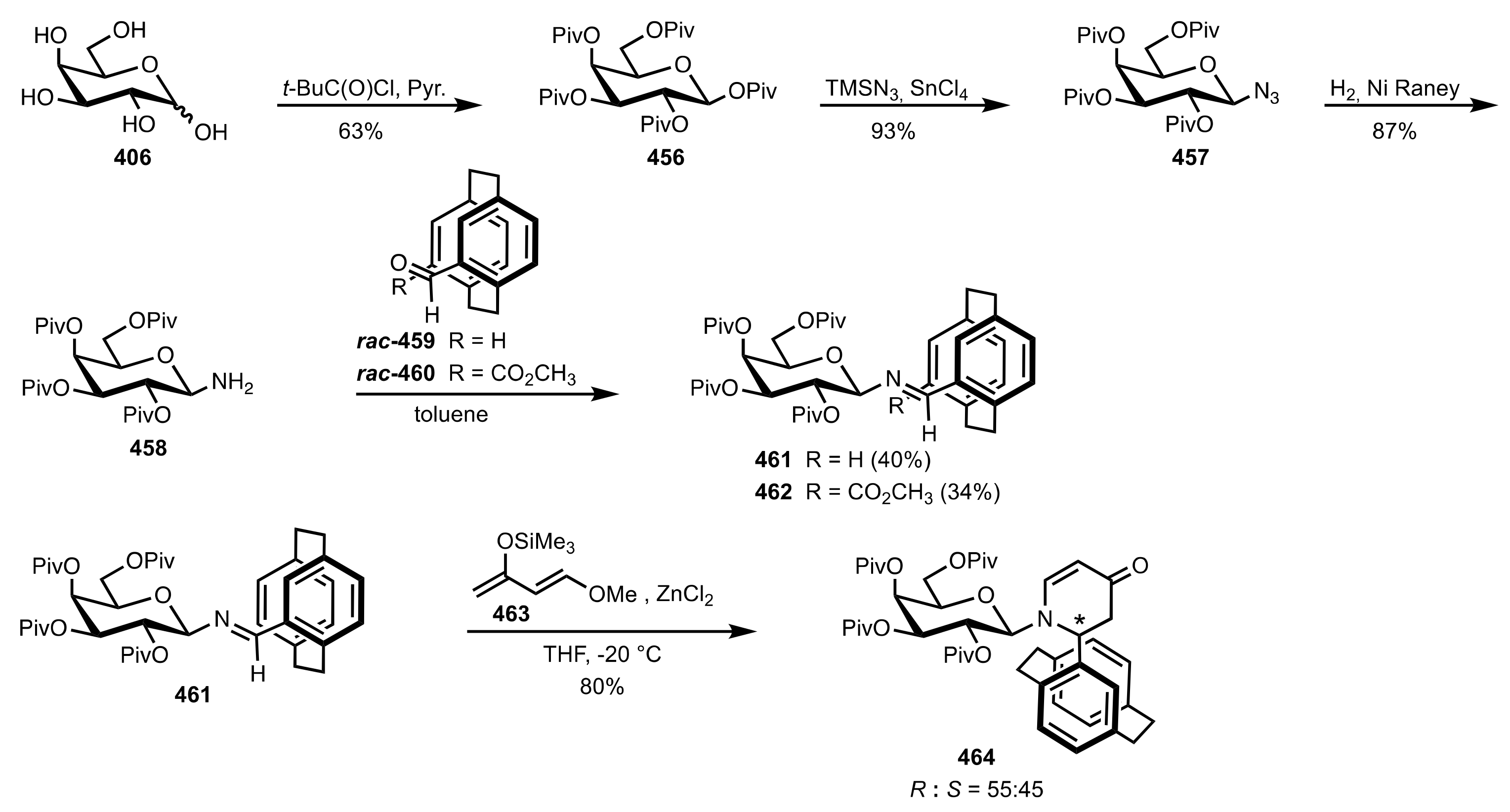
Scheme 120.
The synthesis of the sugar imine organocatalysts 461, 462 and 464.
Scheme 120.
The synthesis of the sugar imine organocatalysts 461, 462 and 464.
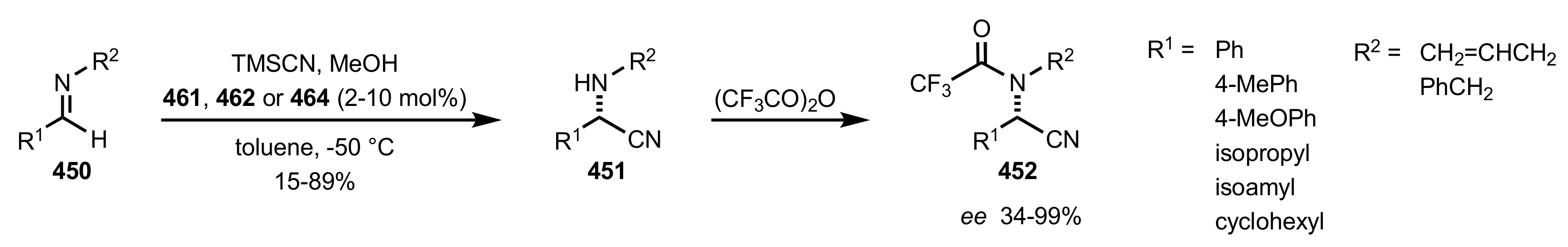
Scheme 121.
Strecker reactions catalysed by 461, 462 and 464.
Scheme 121.
Strecker reactions catalysed by 461, 462 and 464.
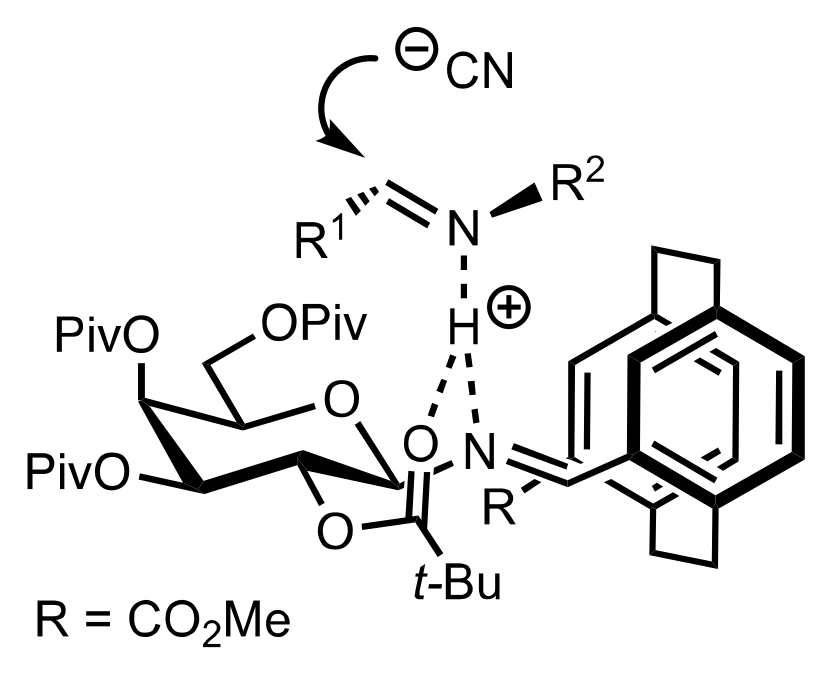
Figure 31.
The proposed mechanism for the (S)-stereoselective formation of α-amino-nitriles.
Figure 31.
The proposed mechanism for the (S)-stereoselective formation of α-amino-nitriles.
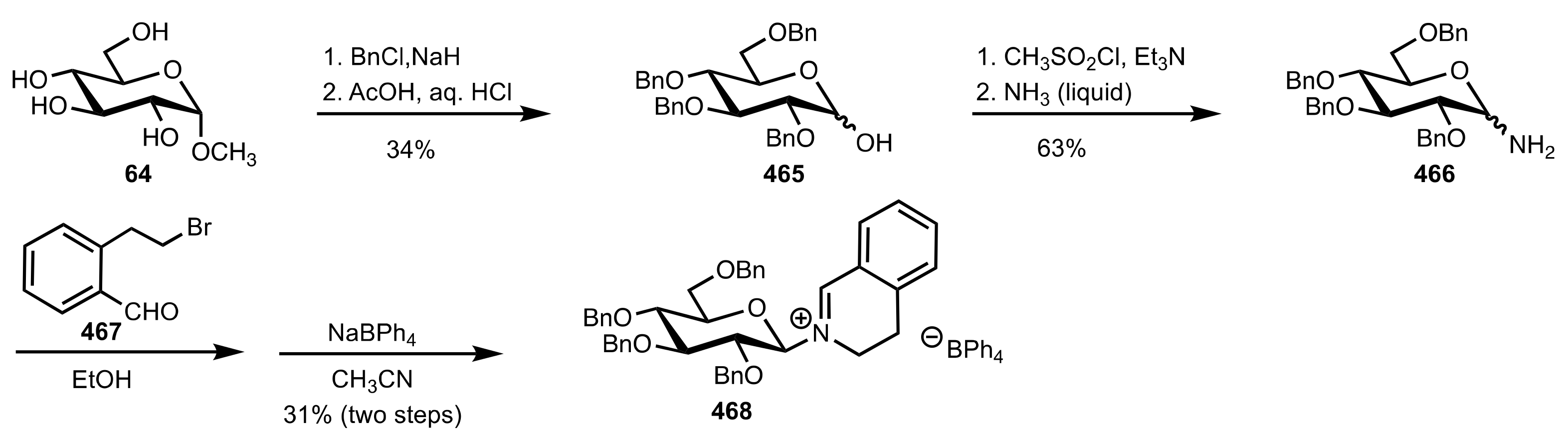
Scheme 122.
The synthesis of the sugar iminium salt organocatalyst 468.
Scheme 122.
The synthesis of the sugar iminium salt organocatalyst 468.
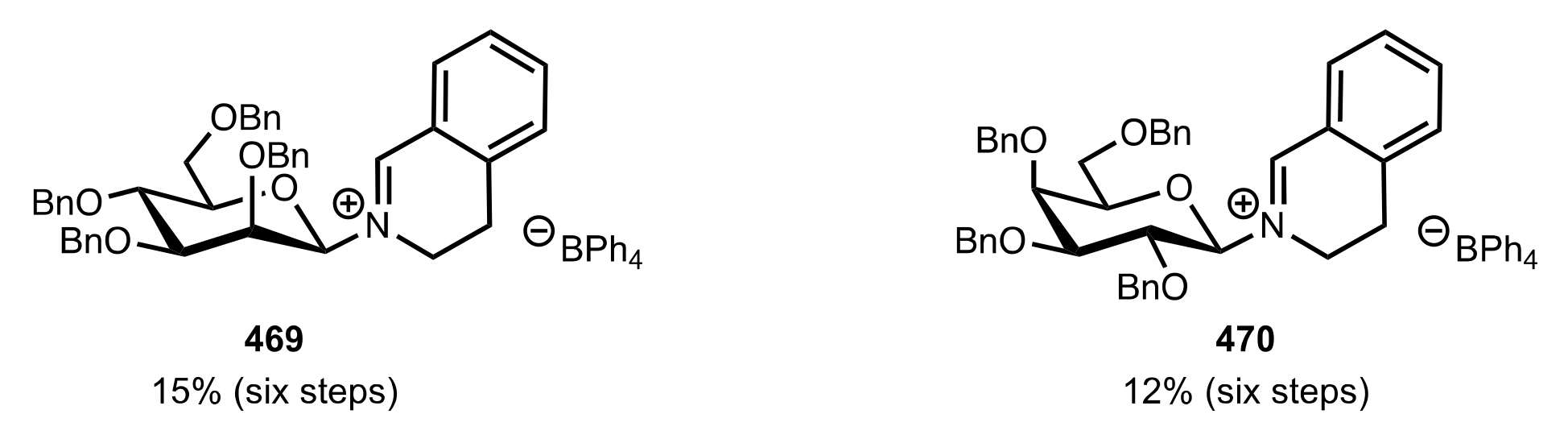
Figure 32.
The sugar iminium organocatalysts 469 and 470.
Figure 32.
The sugar iminium organocatalysts 469 and 470.
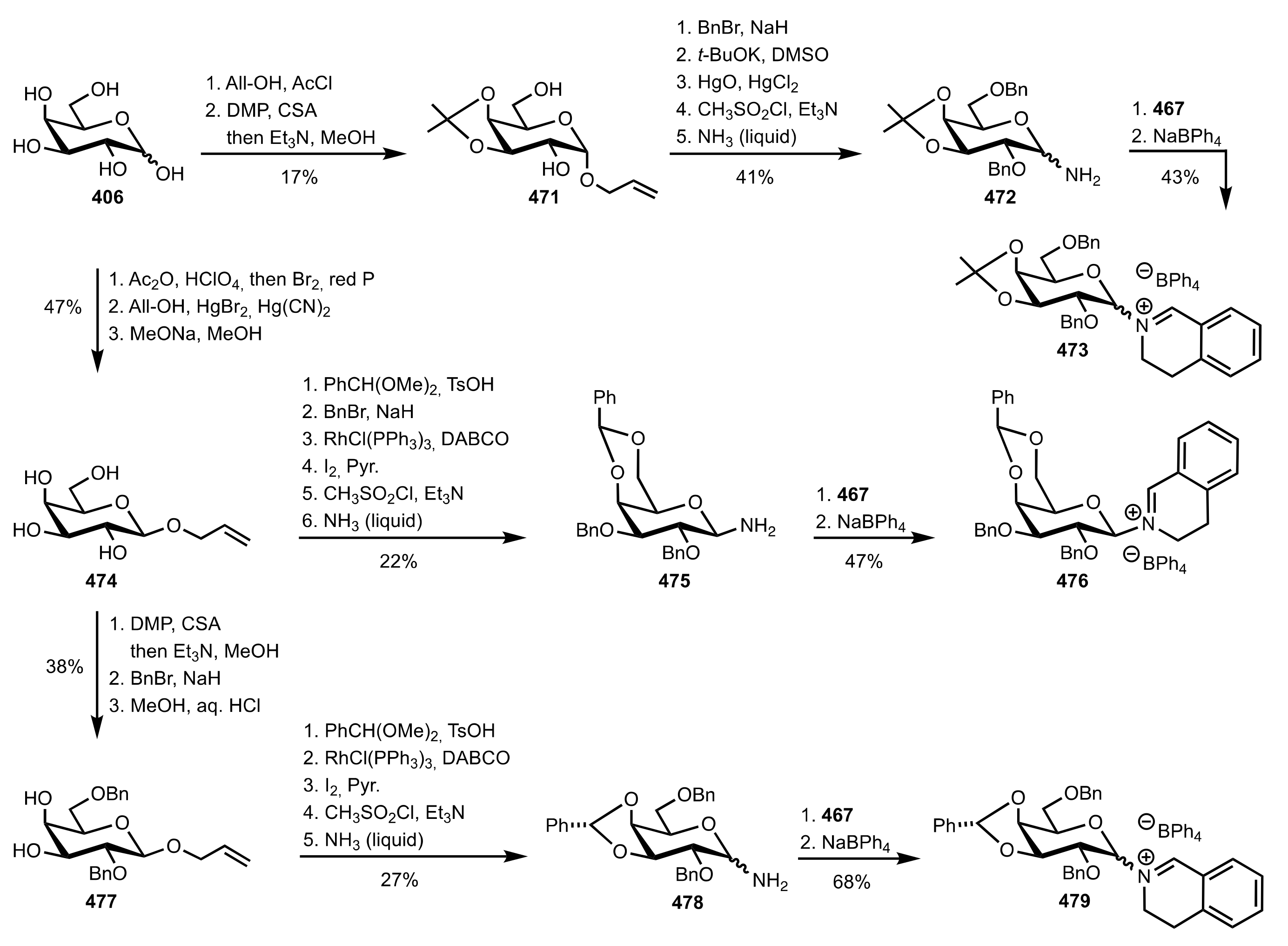
Scheme 123.
The synthesis of the sugar iminium organocatalysts 473, 476 and 479.
Scheme 123.
The synthesis of the sugar iminium organocatalysts 473, 476 and 479.
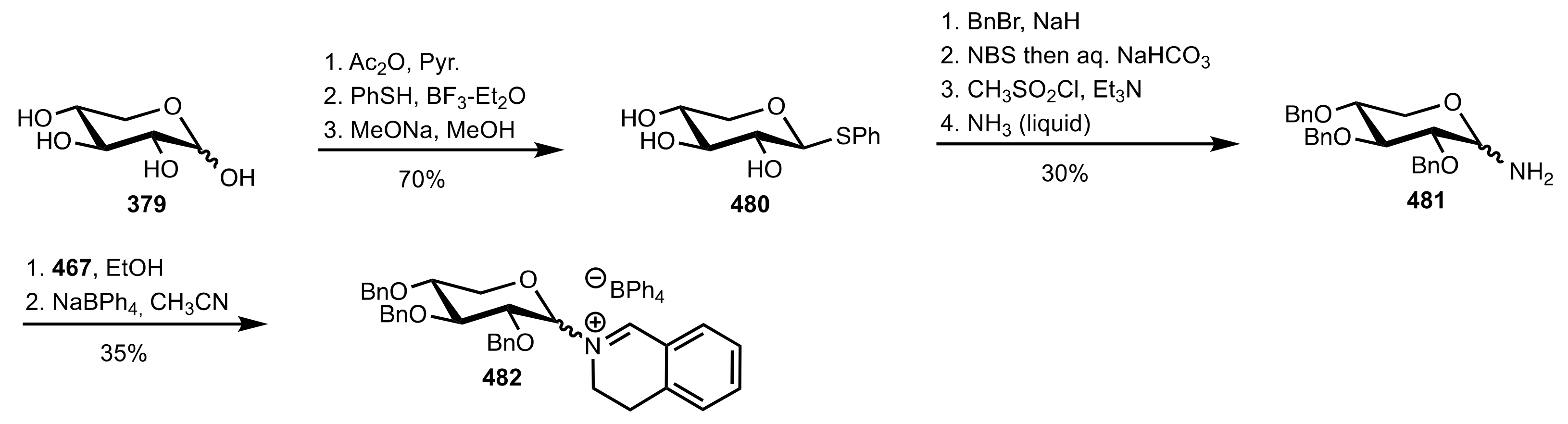
Scheme 124.
The synthesis of the sugar iminium organocatalyst 482.
Scheme 124.
The synthesis of the sugar iminium organocatalyst 482.
Scheme 125.
Asymmetric epoxidations catalysed by 468–470, 473, 476, 479 or 482.
Scheme 125.
Asymmetric epoxidations catalysed by 468–470, 473, 476, 479 or 482.
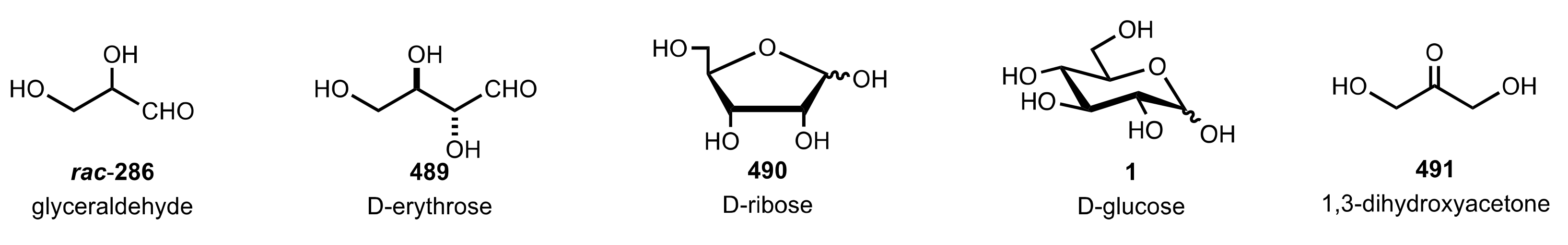
Figure 33.
Aldoses and ketose used as organocatalysts in the hydration of α-amino-nitriles.
Figure 33.
Aldoses and ketose used as organocatalysts in the hydration of α-amino-nitriles.
Scheme 126.
Organocatalysed hydration of α-amino-nitriles.
Scheme 126.
Organocatalysed hydration of α-amino-nitriles.
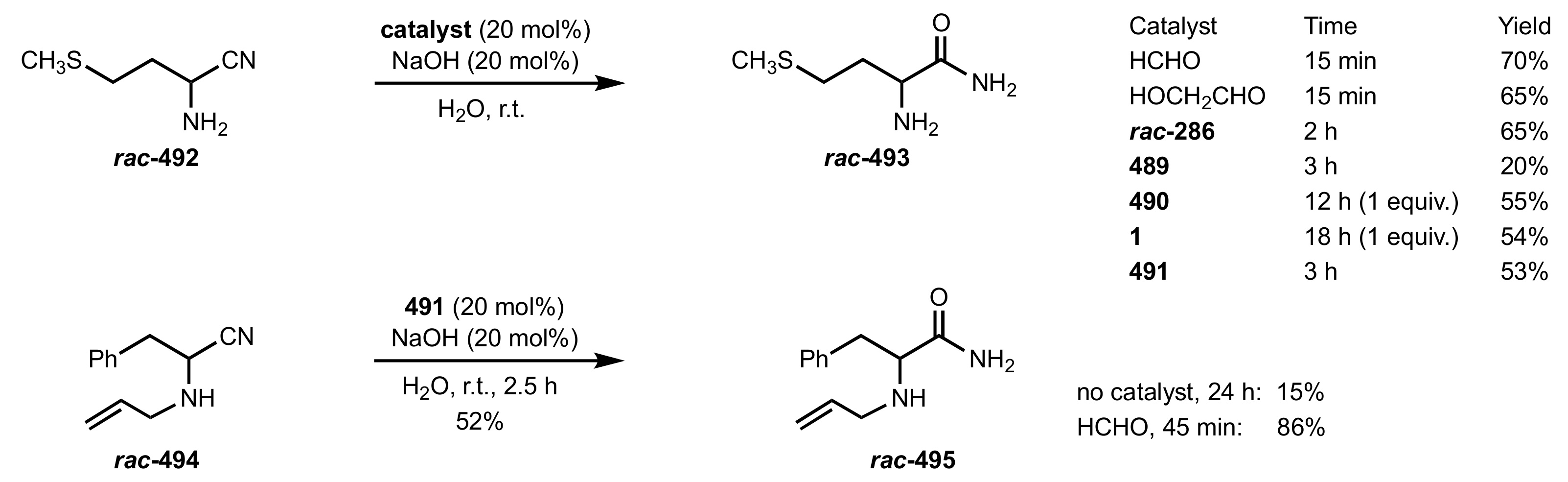
Scheme 127.
Aldol reactions catalysed by d-glucosamine (294).
Scheme 127.
Aldol reactions catalysed by d-glucosamine (294).
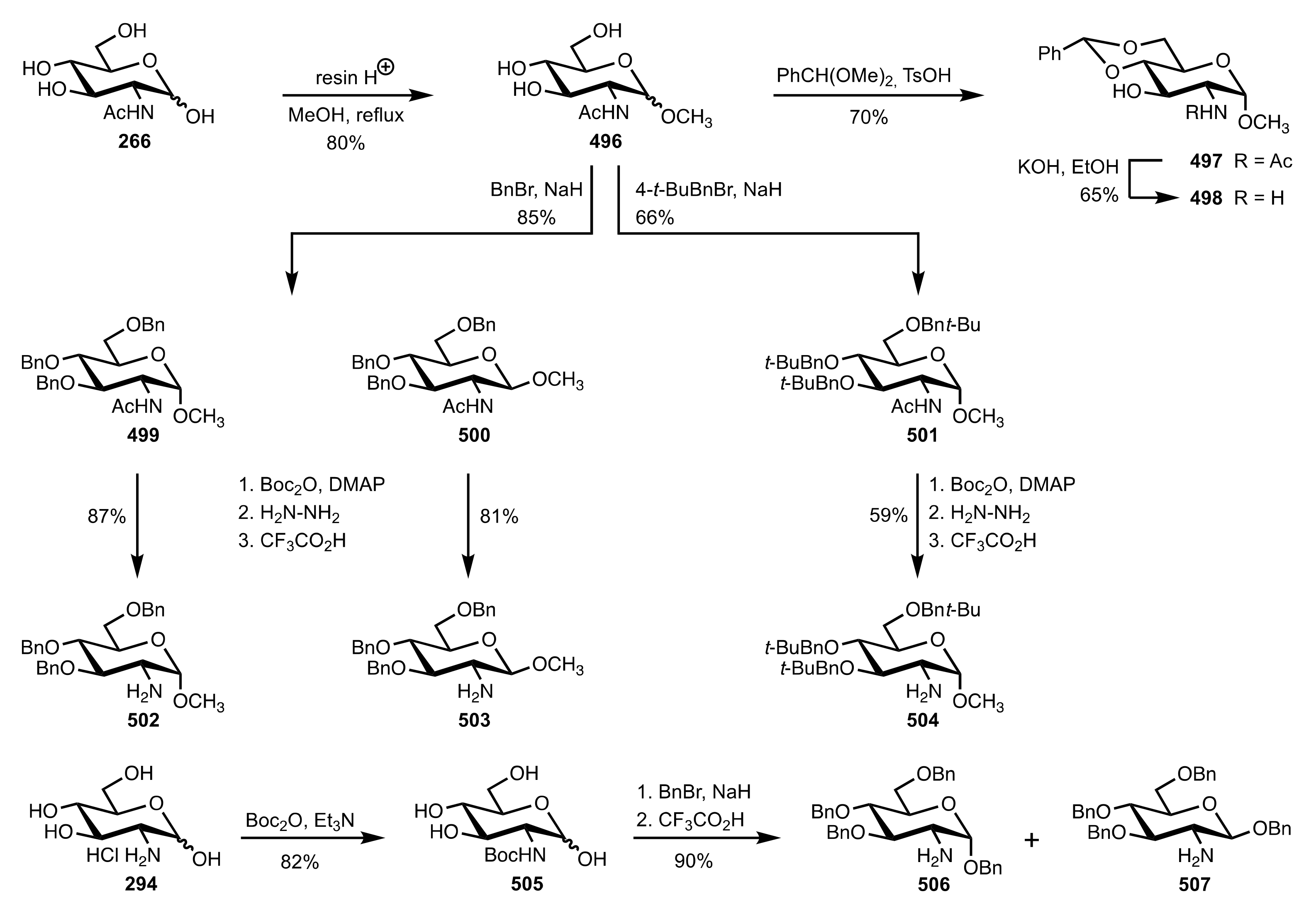
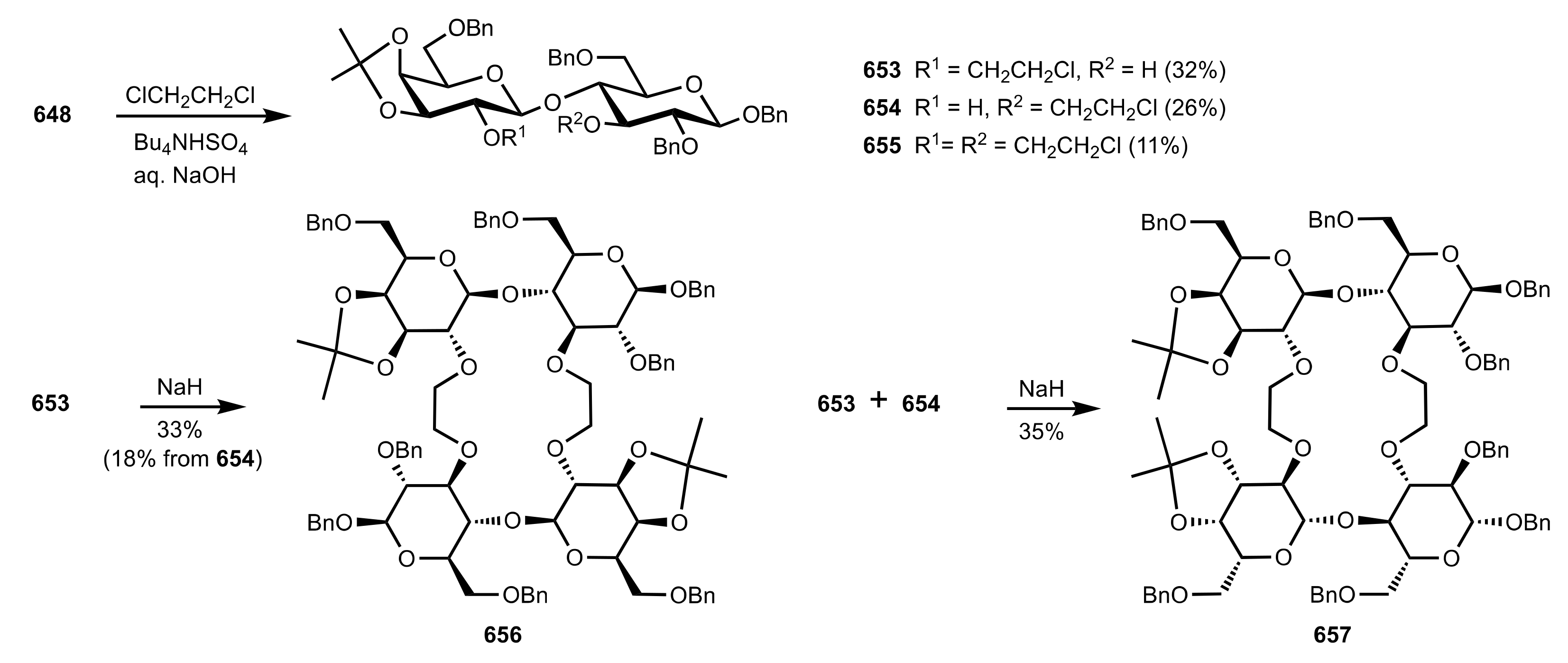
Scheme 128.
The synthesis of the d-glucosamine glycosides organocatalysts.
Scheme 128.
The synthesis of the d-glucosamine glycosides organocatalysts.
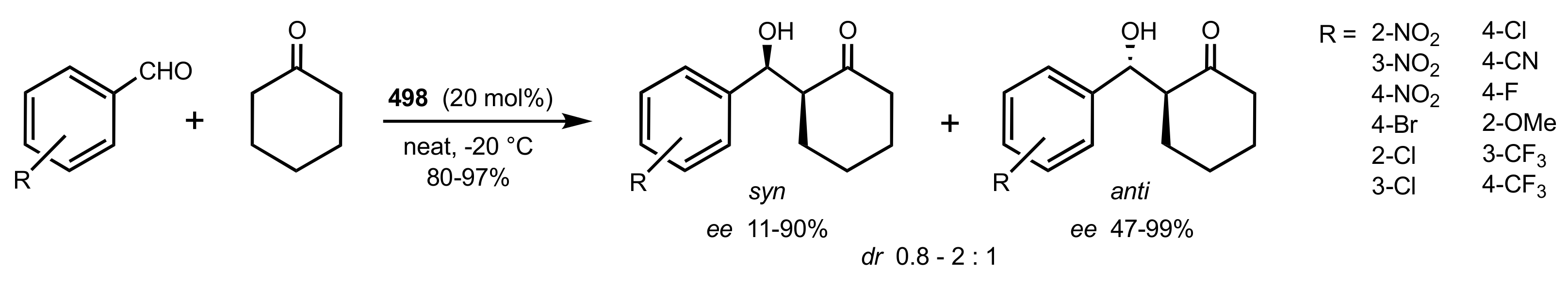
Scheme 129.
Aldol reactions catalysed by the d-glucosamine glycoside 498.
Scheme 129.
Aldol reactions catalysed by the d-glucosamine glycoside 498.
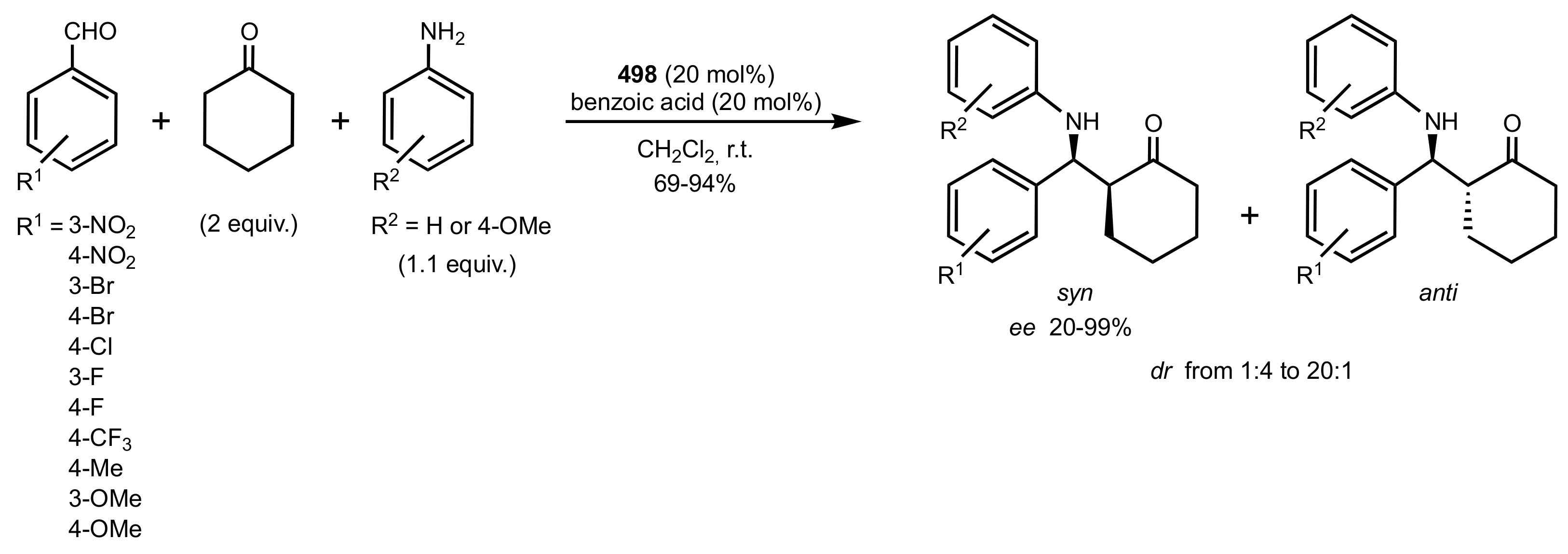
Scheme 130.
Mannich reaction catalysed by the d-glucosamine glycoside 498.
Scheme 130.
Mannich reaction catalysed by the d-glucosamine glycoside 498.
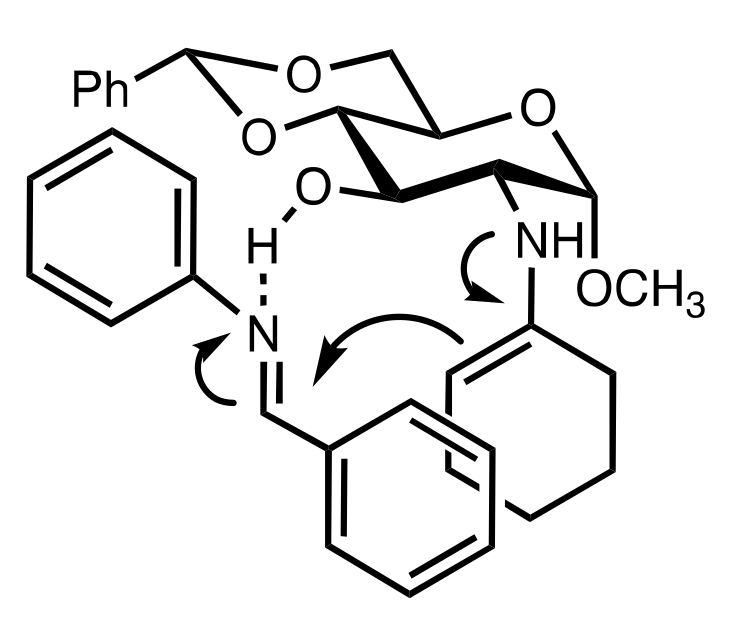
Figure 34.
The proposed transition state of the Mannich reaction leading to the syn adduct.
Figure 34.
The proposed transition state of the Mannich reaction leading to the syn adduct.
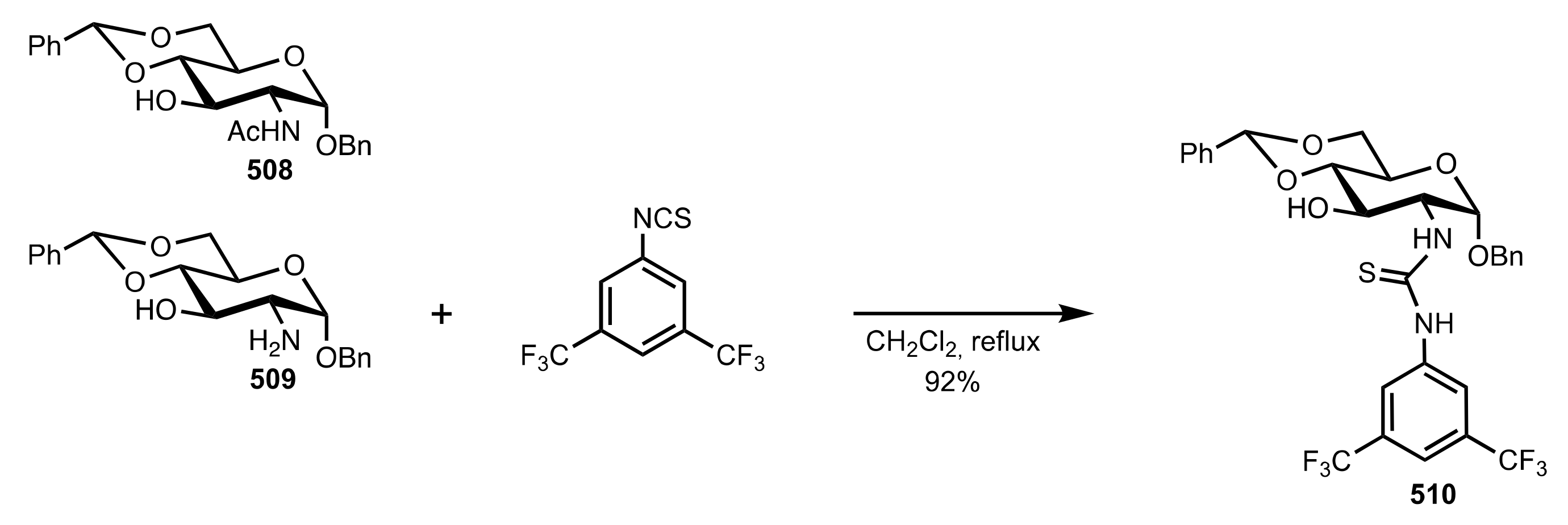
Scheme 131.
d-glucosamine glycosides organocatalysts 508–510 used for the aldol reaction.
Scheme 131.
d-glucosamine glycosides organocatalysts 508–510 used for the aldol reaction.
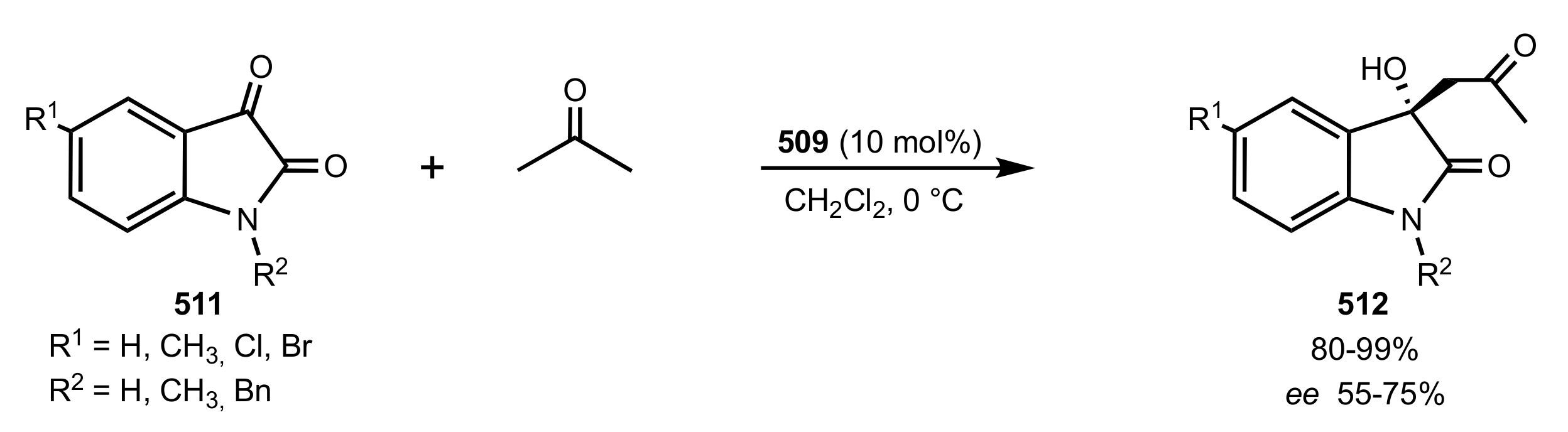
Scheme 132.
Aldol reaction catalysed by the d-glucosamine glycoside 509.
Scheme 132.
Aldol reaction catalysed by the d-glucosamine glycoside 509.
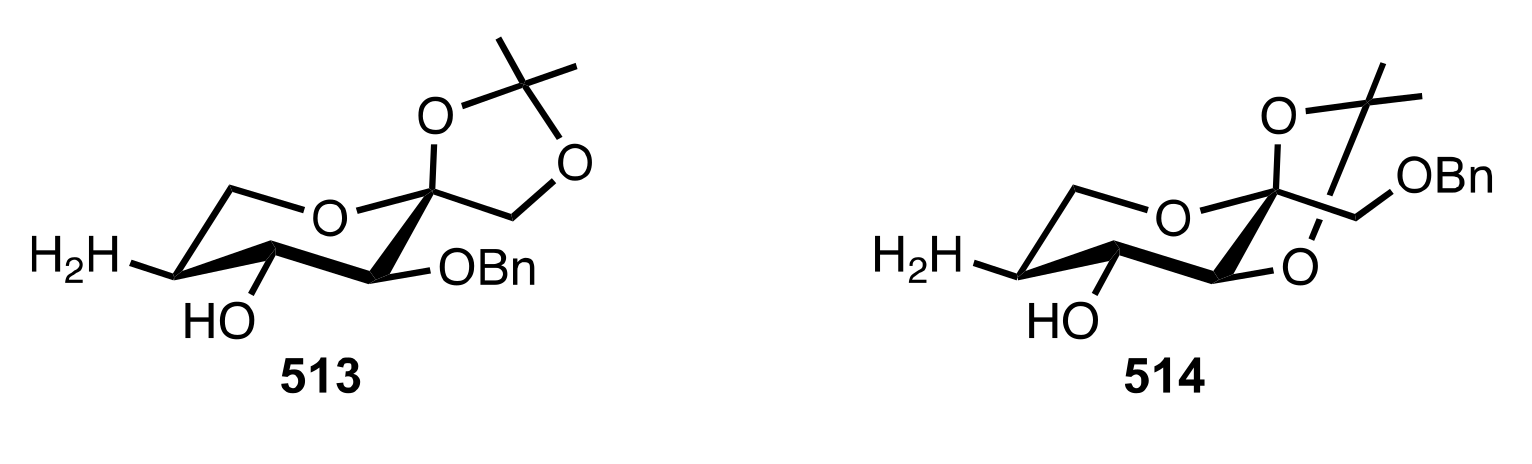
Figure 35.
d-Fructose-derived organocatalysts 513 and 514.
Figure 35.
d-Fructose-derived organocatalysts 513 and 514.
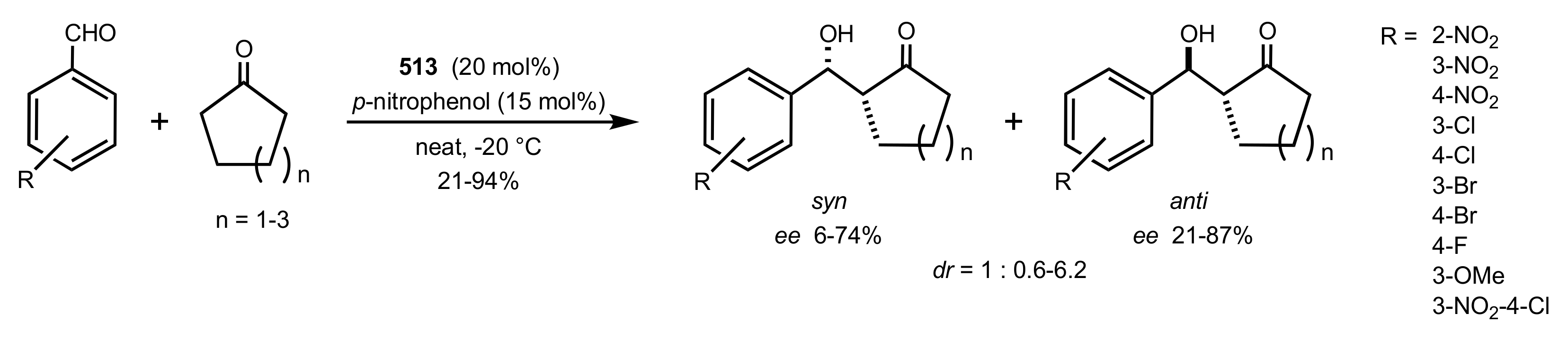
Scheme 133.
Aldol reaction catalysed by the organocatalyst 513.
Scheme 133.
Aldol reaction catalysed by the organocatalyst 513.
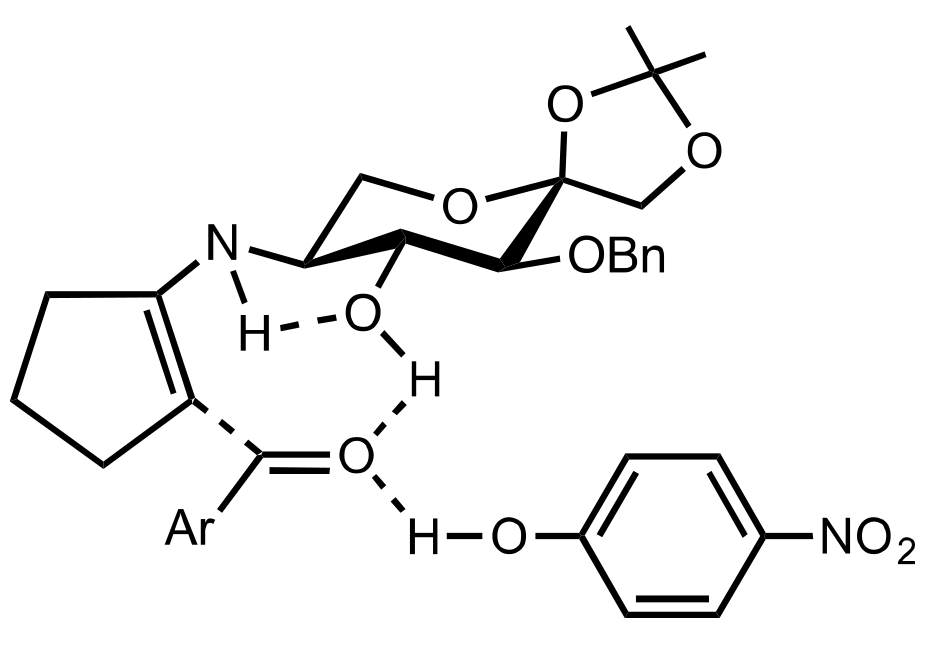
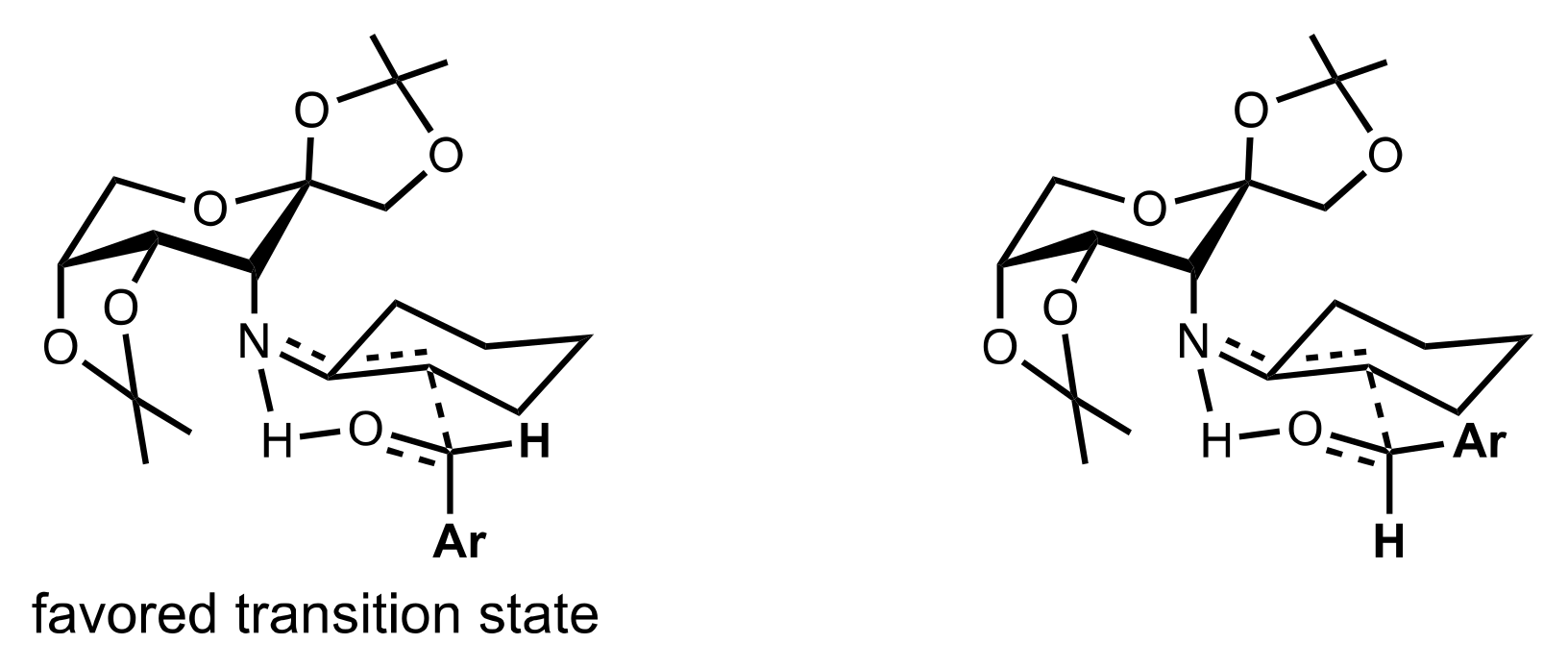
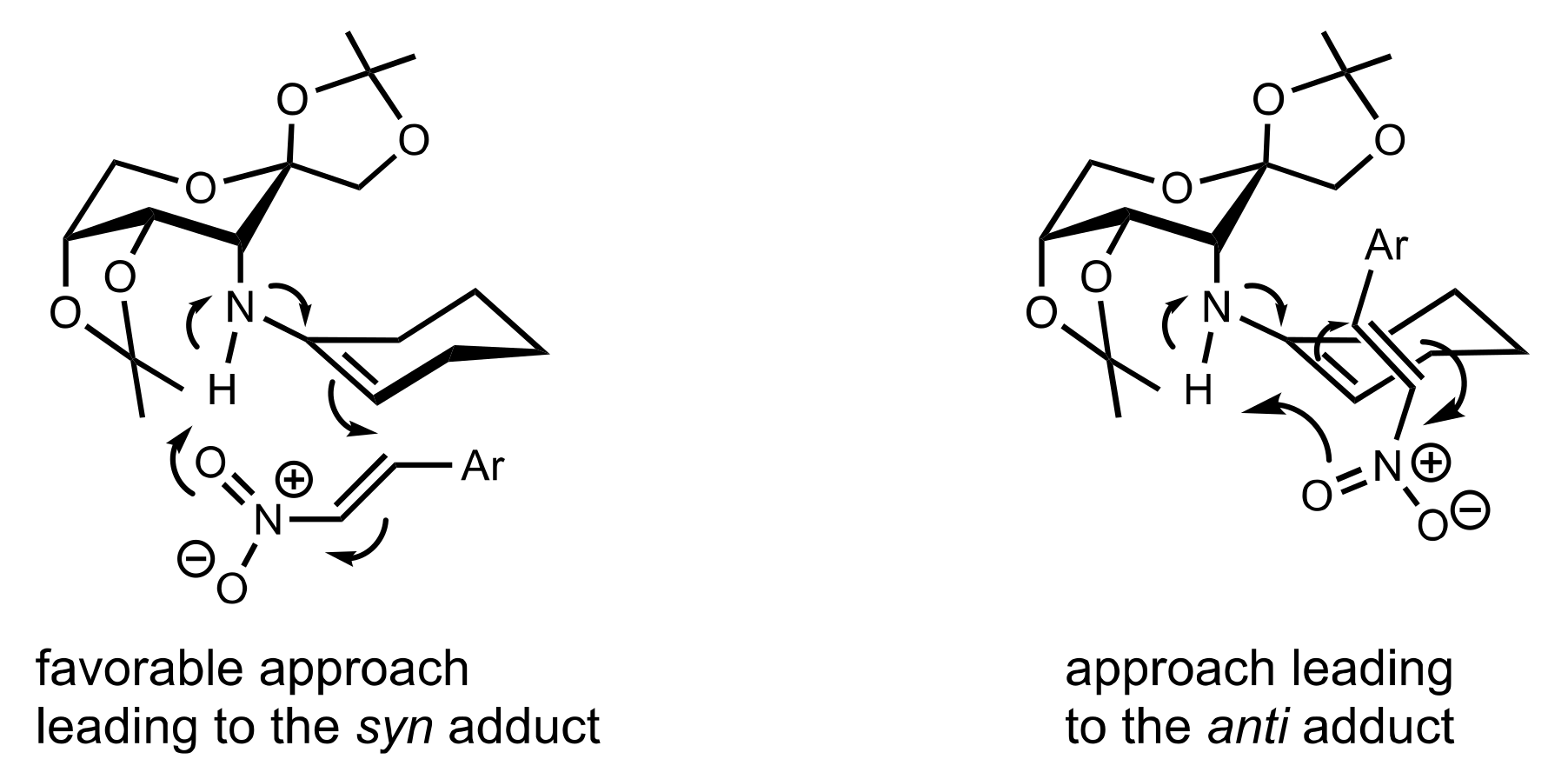
Figure 36.
The proposed transition state of the aldol reaction catalysed by 513 and p-nitrophenol.
Figure 36.
The proposed transition state of the aldol reaction catalysed by 513 and p-nitrophenol.
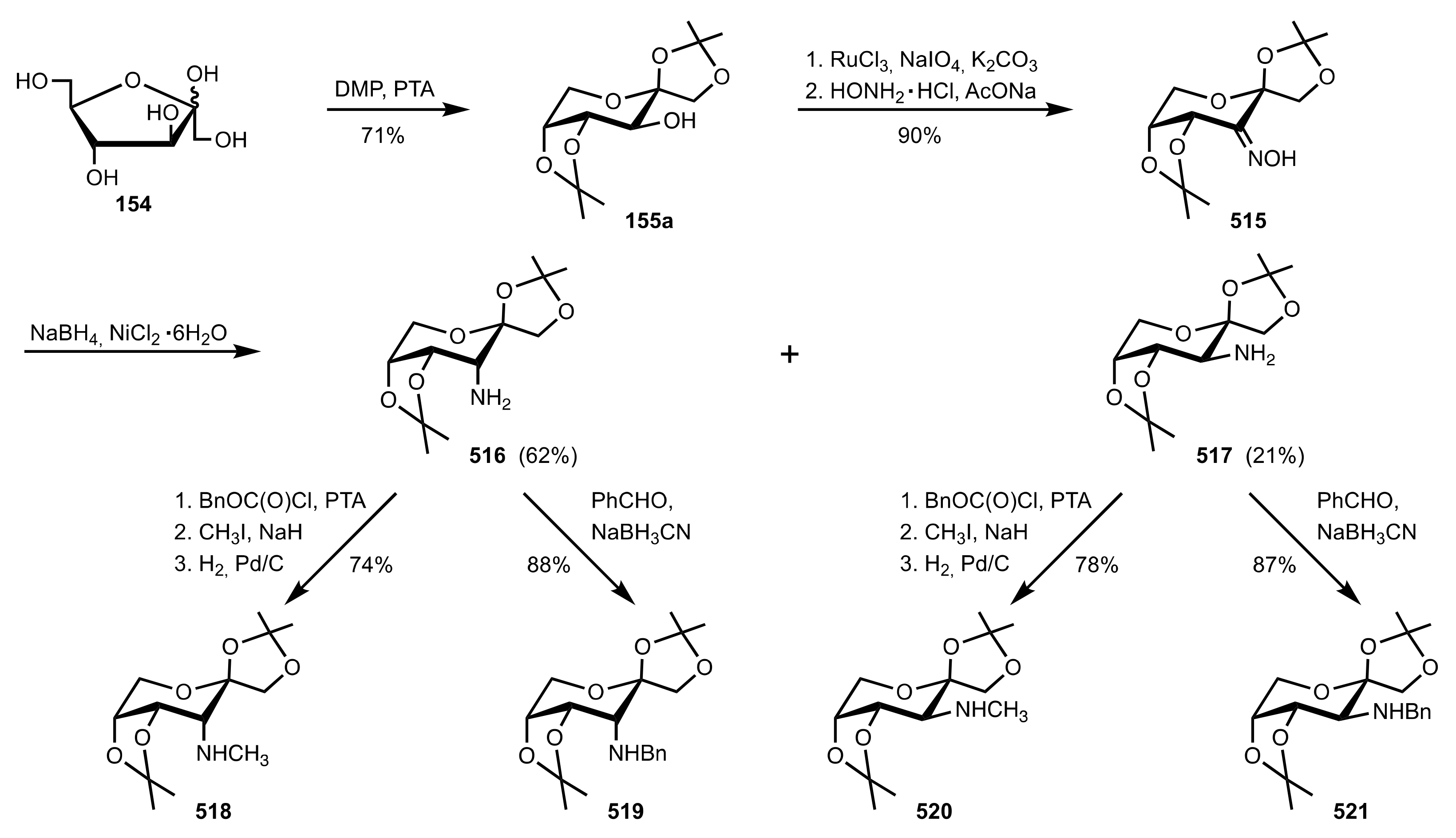
Scheme 134.
The synthesis of the d-fructose derived organocatalysts 516–521.
Scheme 134.
The synthesis of the d-fructose derived organocatalysts 516–521.
Scheme 135.
Aldol reaction catalysed by the organocatalyst 516.
Scheme 135.
Aldol reaction catalysed by the organocatalyst 516.
Figure 37.
The proposed transition state of the aldol reaction catalysed by 516.
Figure 37.
The proposed transition state of the aldol reaction catalysed by 516.
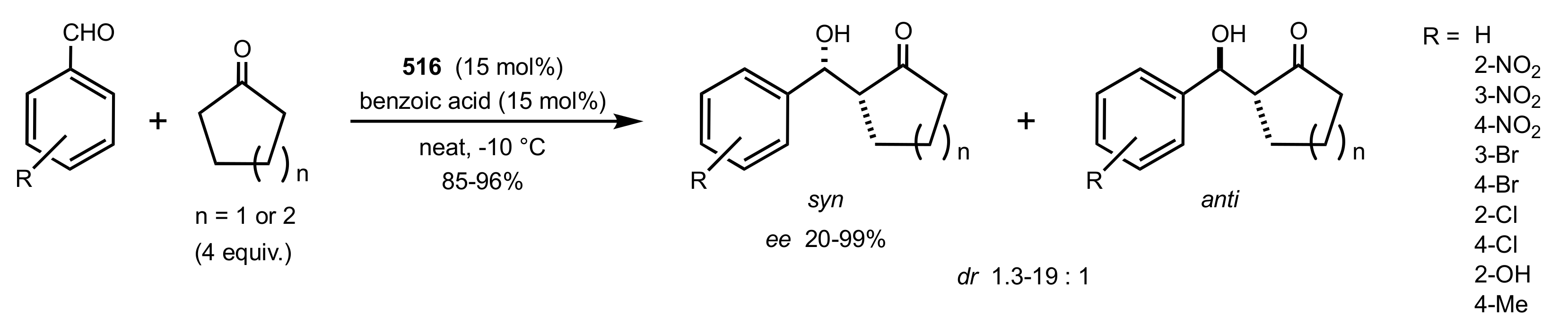
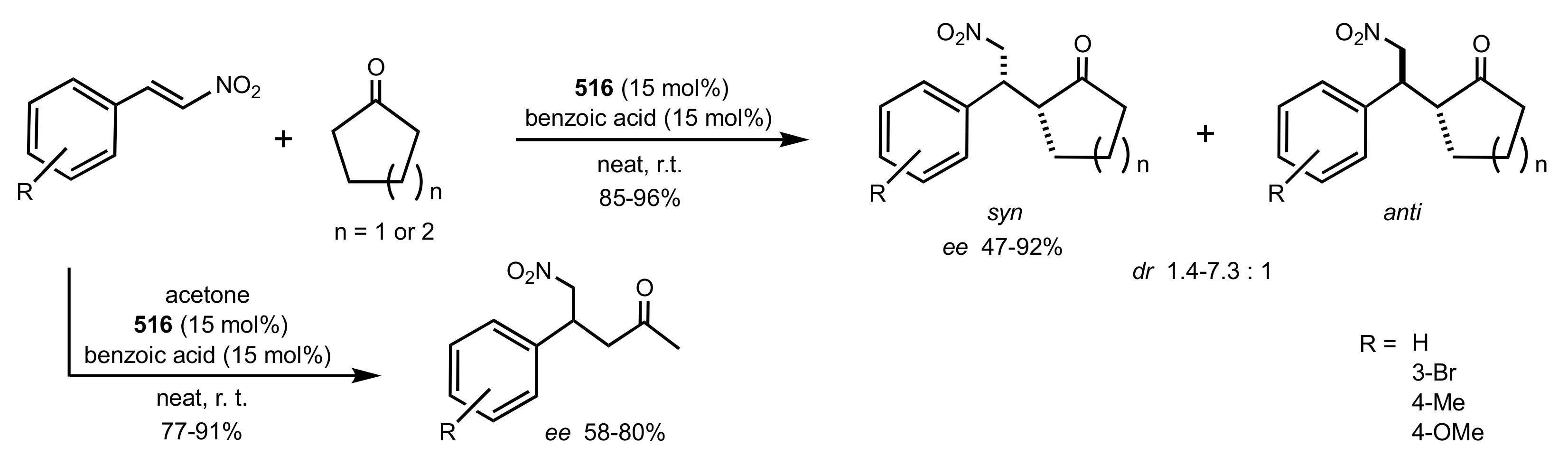
Scheme 136.
The Michael additions catalysed by the organocatalyst 516.
Scheme 136.
The Michael additions catalysed by the organocatalyst 516.
Figure 38.
The proposed mechanism for the Michael additions catalysed by 516.
Figure 38.
The proposed mechanism for the Michael additions catalysed by 516.
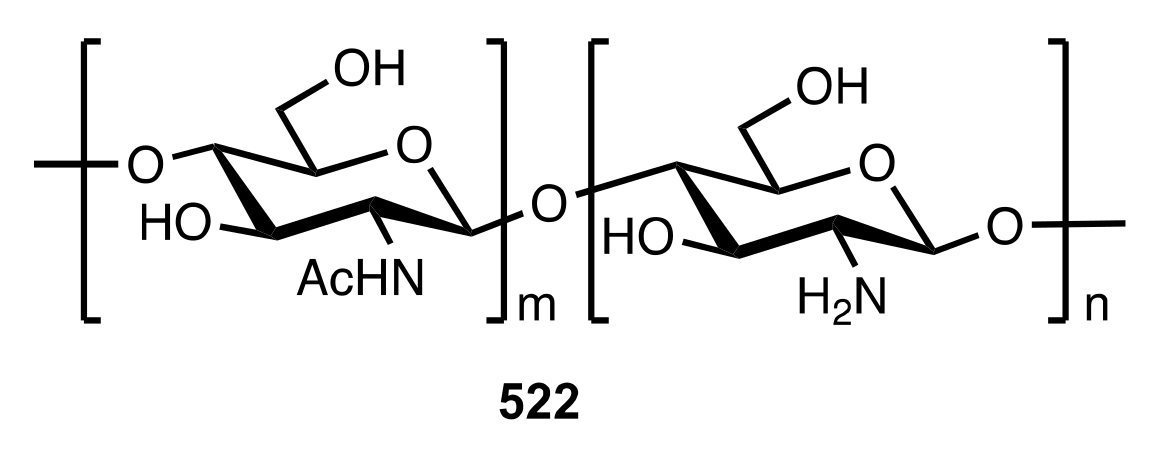
Figure 39.
The structure of the chitosan.
Figure 39.
The structure of the chitosan.
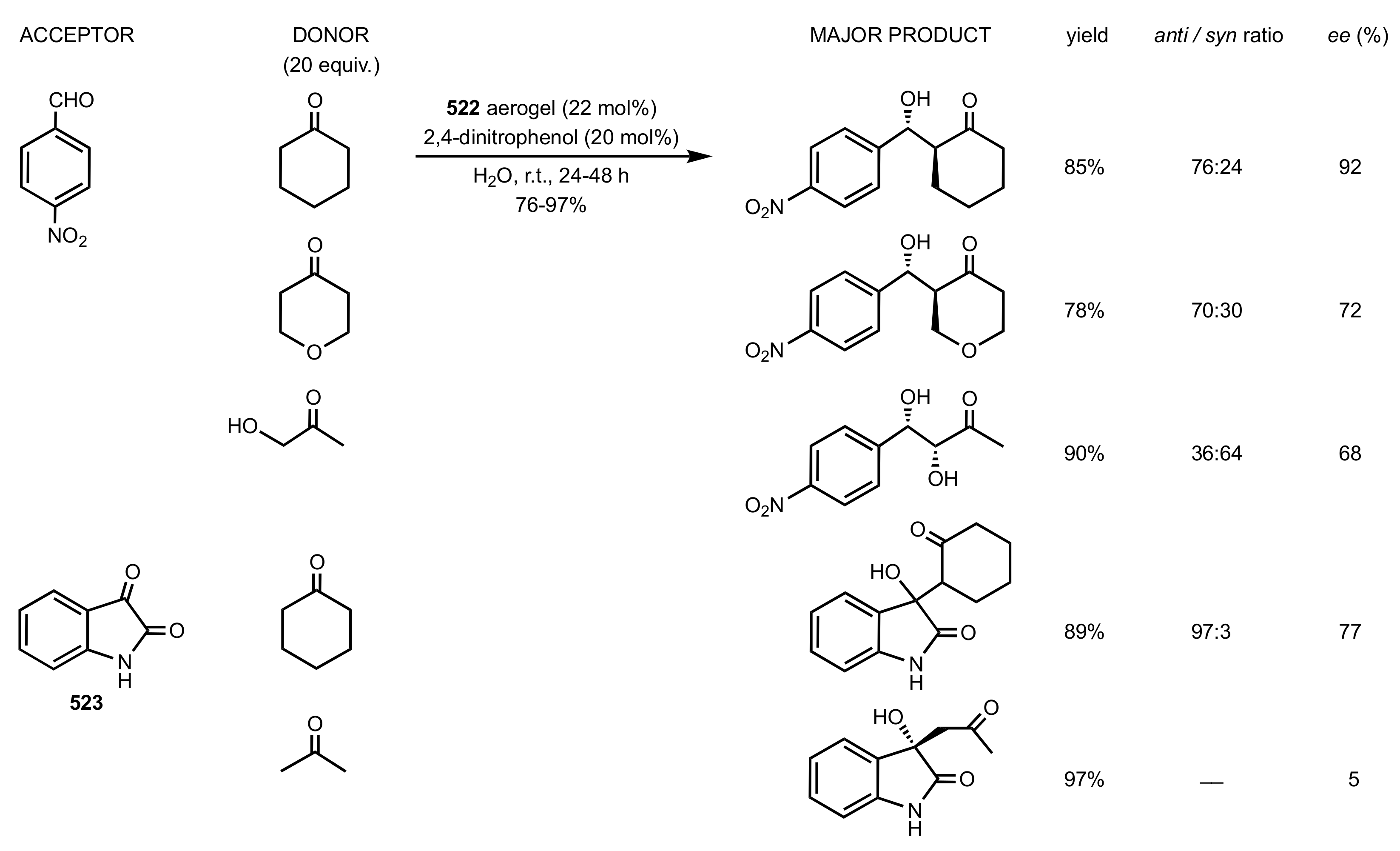
Scheme 137.
Aldol reactions catalysed by chitosan 522 aerogel.
Scheme 137.
Aldol reactions catalysed by chitosan 522 aerogel.
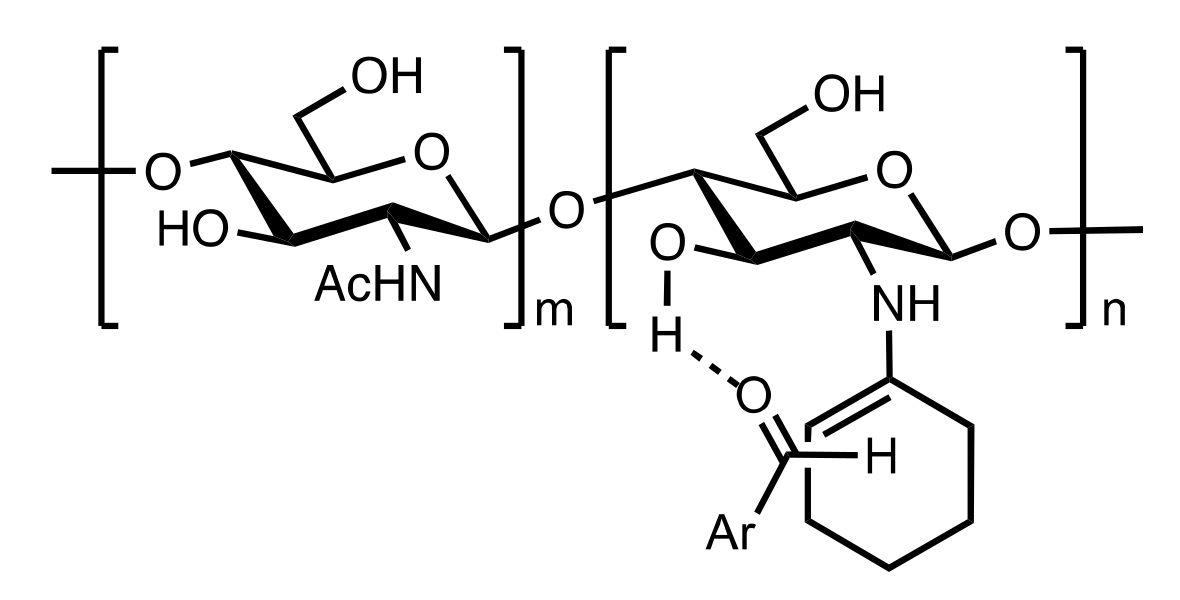
Figure 40.
The proposed transition state for the addition of cyclohexanone to 4-nitrobenzaldehyde to give the anti aldol.
Figure 40.
The proposed transition state for the addition of cyclohexanone to 4-nitrobenzaldehyde to give the anti aldol.
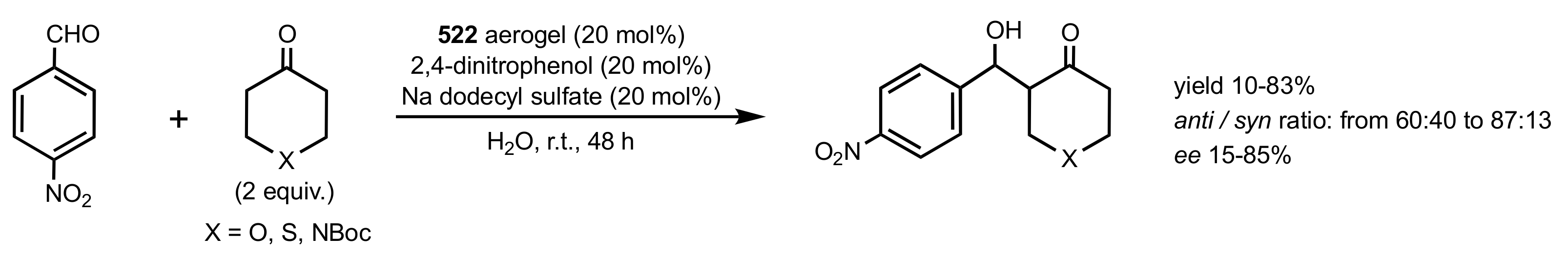
Scheme 138.
Aldol reactions catalysed by chitosan 522 aerogel.
Scheme 138.
Aldol reactions catalysed by chitosan 522 aerogel.
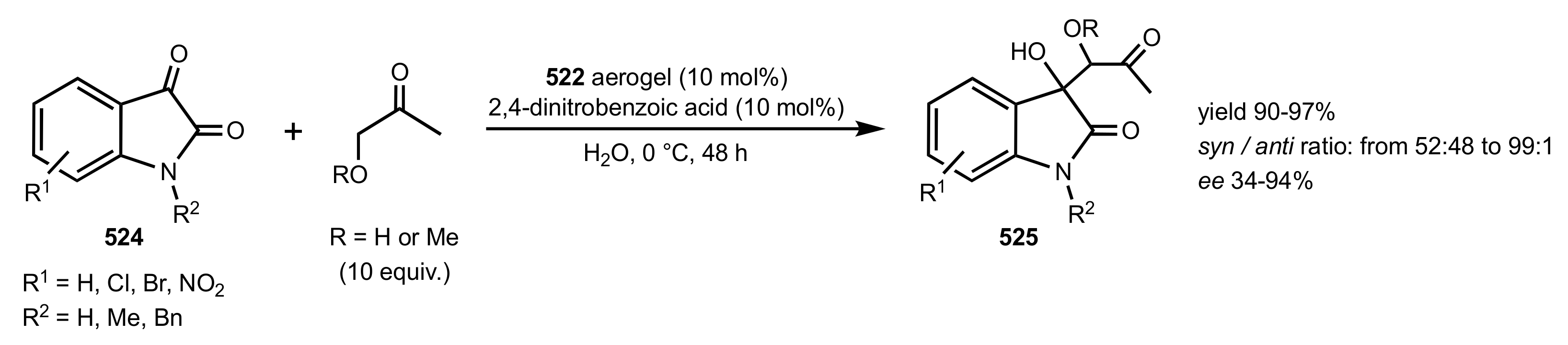
Scheme 139.
Optimized conditions for the aldol reactions catalysed by chitosan 522 aerogel.
Scheme 139.
Optimized conditions for the aldol reactions catalysed by chitosan 522 aerogel.
Scheme 140.
Aldol reactions catalysed by chitosan 522 aerogel.
Scheme 140.
Aldol reactions catalysed by chitosan 522 aerogel.
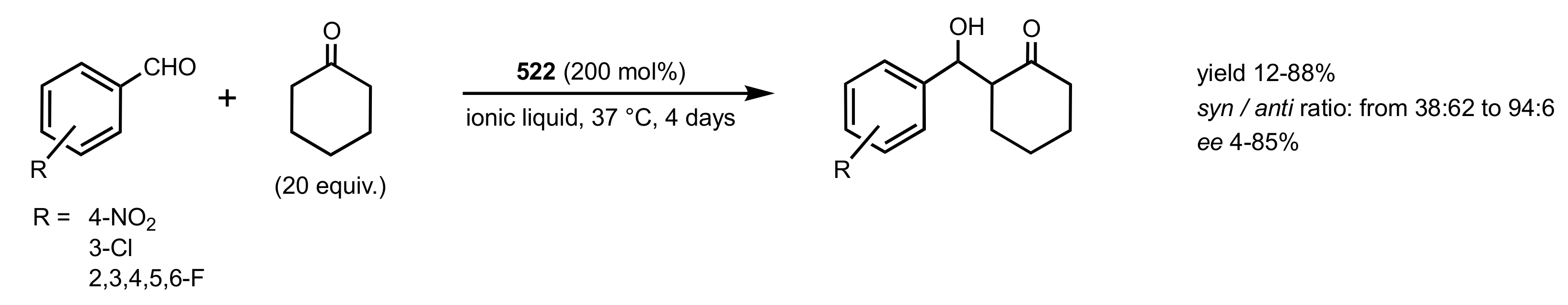
Scheme 141.
Aldol reactions catalysed by chitosan 522 in ionic liquids.
Scheme 141.
Aldol reactions catalysed by chitosan 522 in ionic liquids.
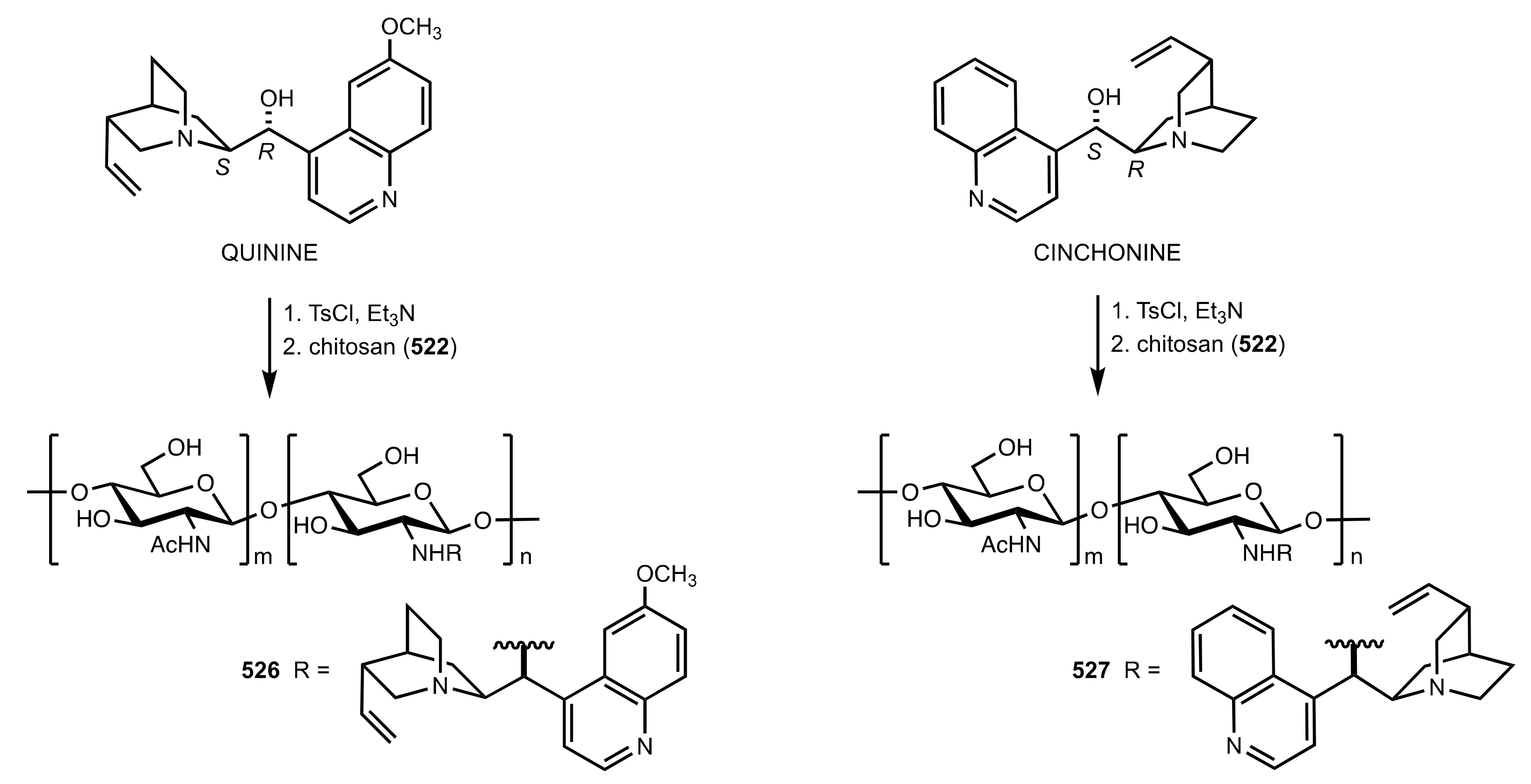
Scheme 142.
The synthesis of chitosan-quinine 529 and chitosan-cinchonine 530 organocatalysts.
Scheme 142.
The synthesis of chitosan-quinine 529 and chitosan-cinchonine 530 organocatalysts.
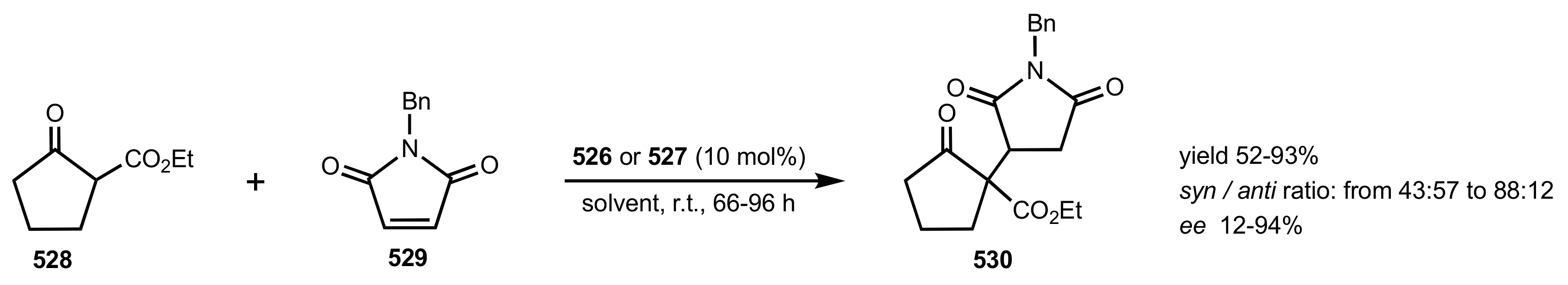
Scheme 143.
The optimization of the Michael addition to N-benzyl-maleimide.
Scheme 143.
The optimization of the Michael addition to N-benzyl-maleimide.
Figure 41.
1,3-Dicarbonyl compounds employed in the Michael addition to N-benzyl-maleimide.
Figure 41.
1,3-Dicarbonyl compounds employed in the Michael addition to N-benzyl-maleimide.
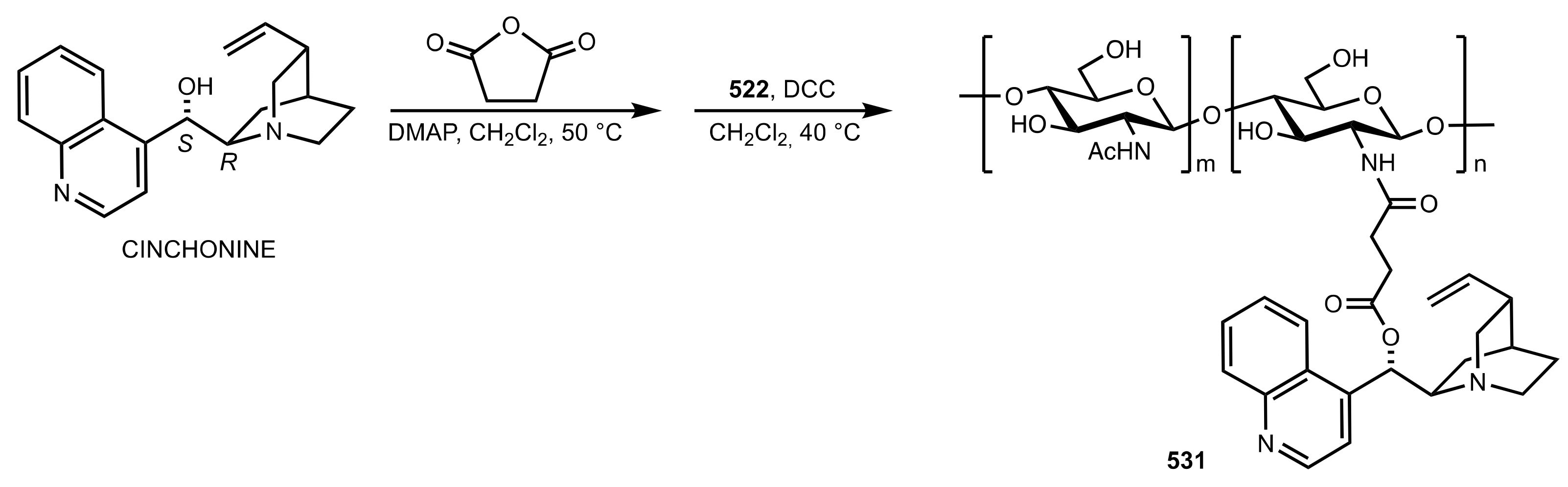
Scheme 144.
The synthesis of the chitosan-cinchonine organocatalyst 531.
Scheme 144.
The synthesis of the chitosan-cinchonine organocatalyst 531.
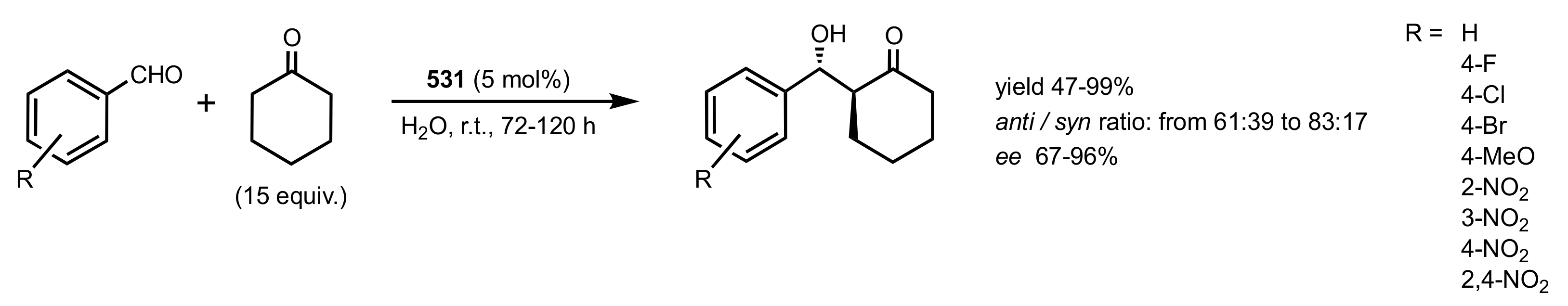
Scheme 145.
The aldol reactions catalysed by chitosan-cinchonine 531.
Scheme 145.
The aldol reactions catalysed by chitosan-cinchonine 531.
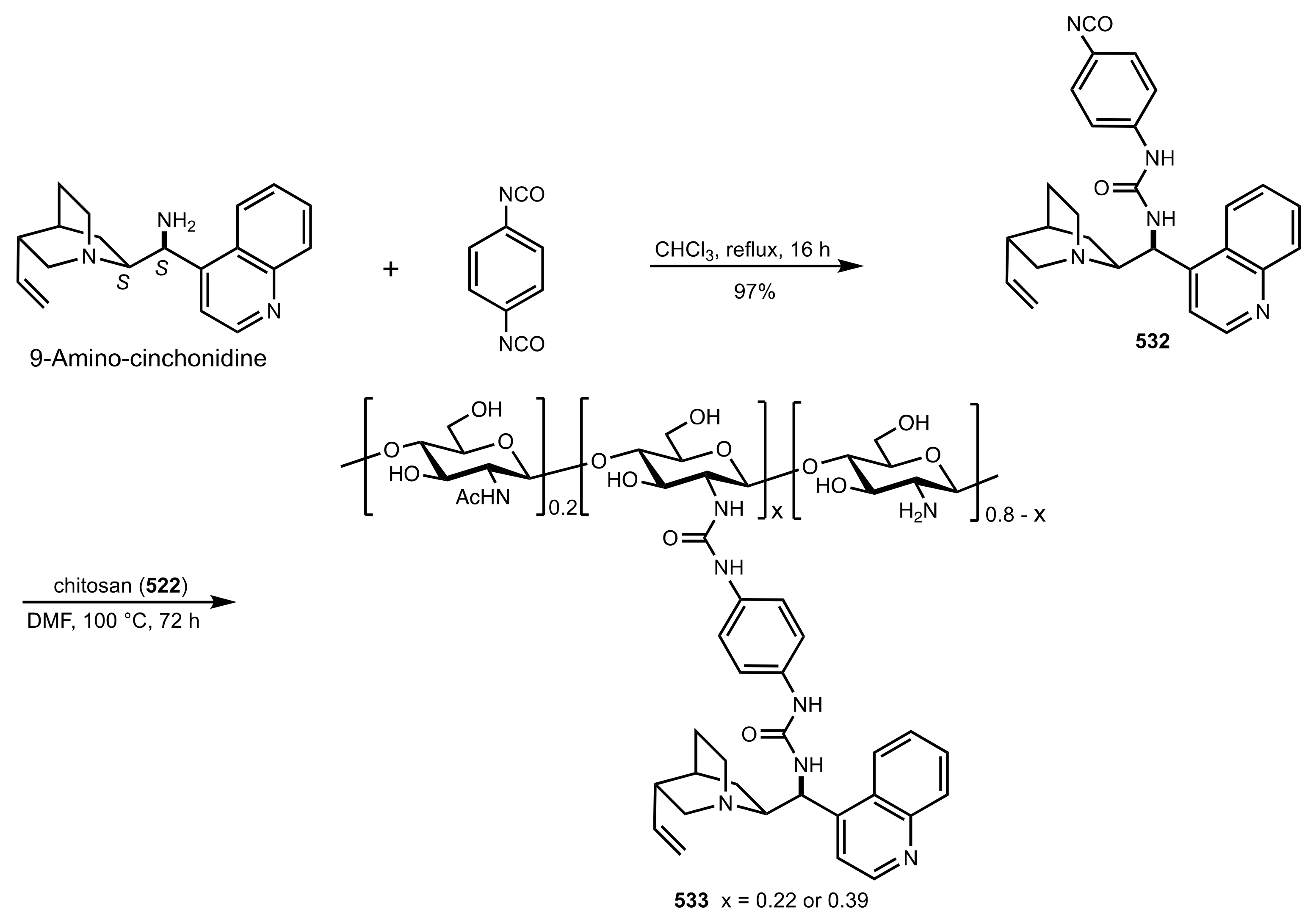
Scheme 146.
The synthesis of the chitosan-aminocinchonidine 533 organocatalyst.
Scheme 146.
The synthesis of the chitosan-aminocinchonidine 533 organocatalyst.
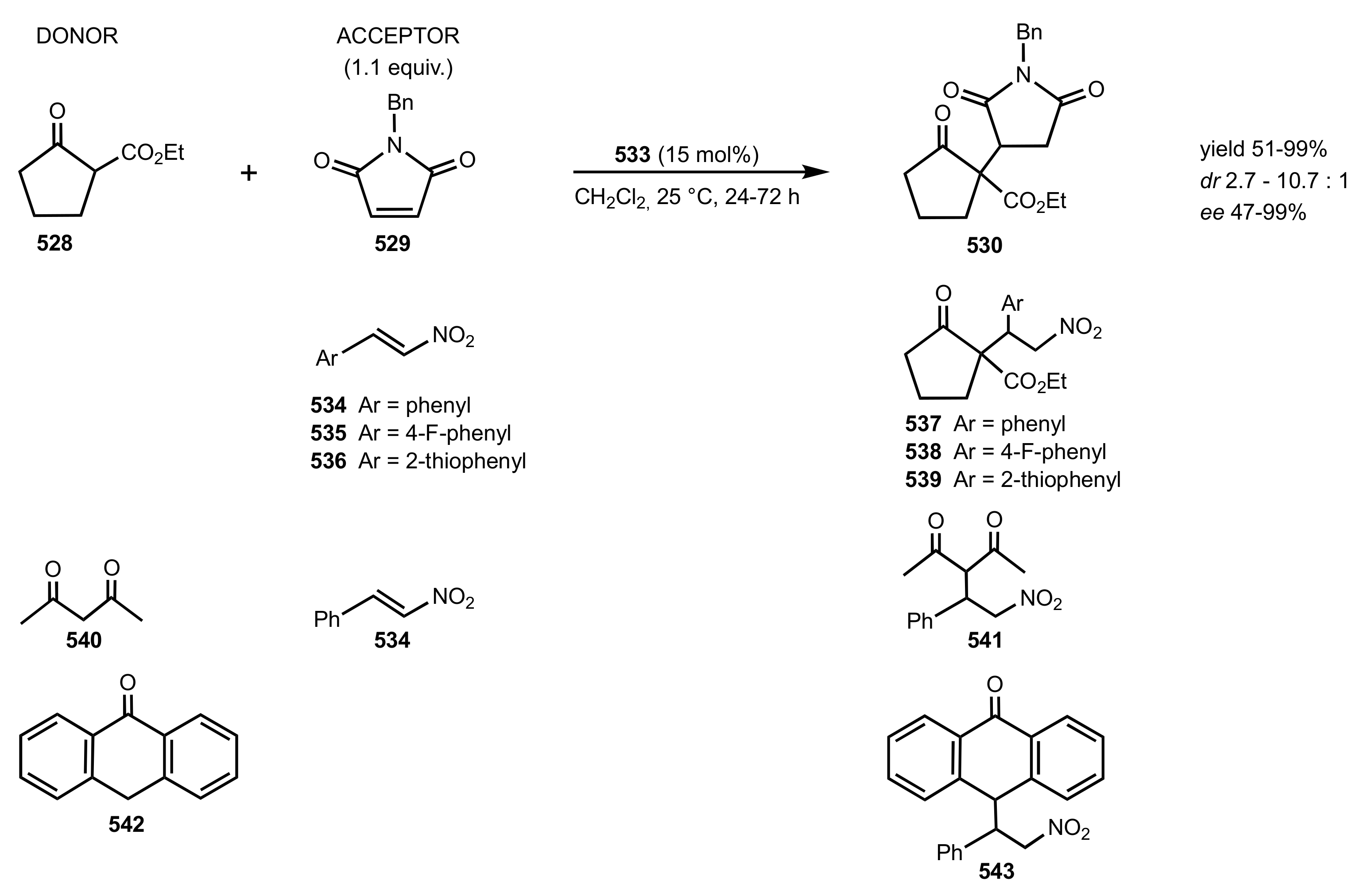
Scheme 147.
The Michael addition catalysed by the chitosan-aminocinchonidine 533 organocatalyst.
Scheme 147.
The Michael addition catalysed by the chitosan-aminocinchonidine 533 organocatalyst.
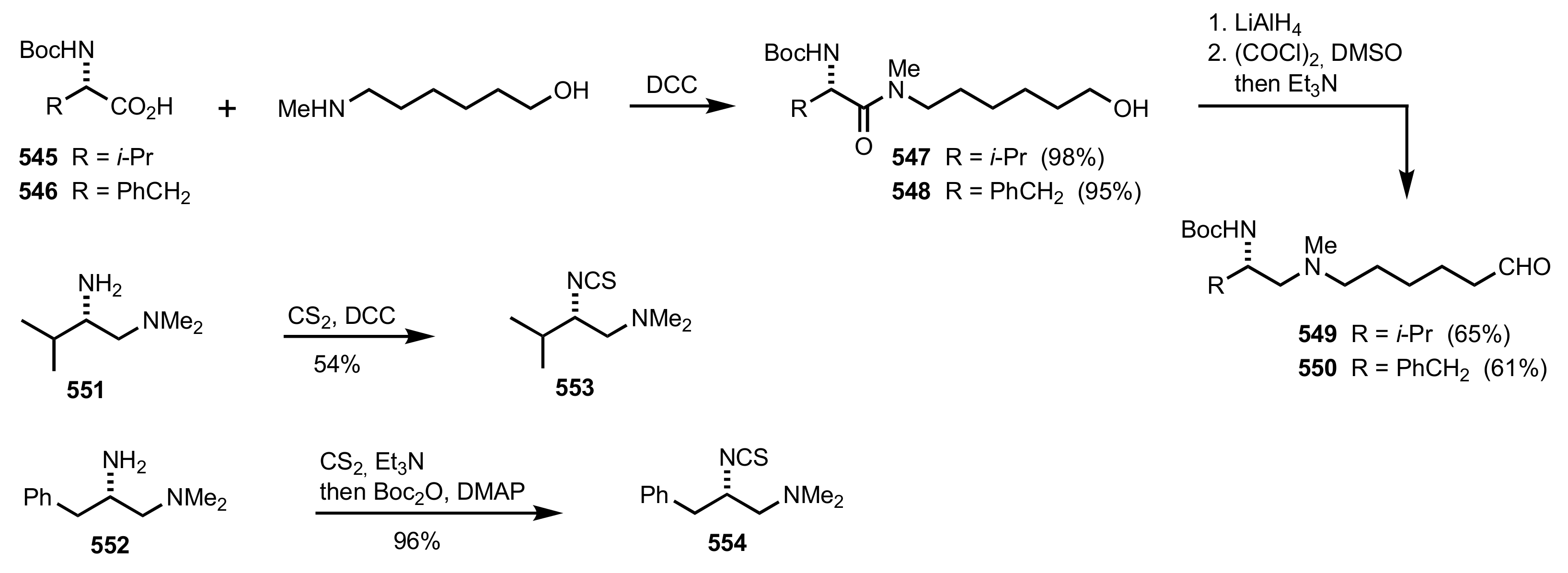
Scheme 148.
The synthesis of the chitosan prolinamide 544 organocatalyst.
Scheme 148.
The synthesis of the chitosan prolinamide 544 organocatalyst.
Scheme 149.
Aldol reactions catalysed by chitosan prolinamide 547.
Scheme 149.
Aldol reactions catalysed by chitosan prolinamide 547.
Scheme 150.
The synthesis of the chiral isocyanates.
Scheme 150.
The synthesis of the chiral isocyanates.
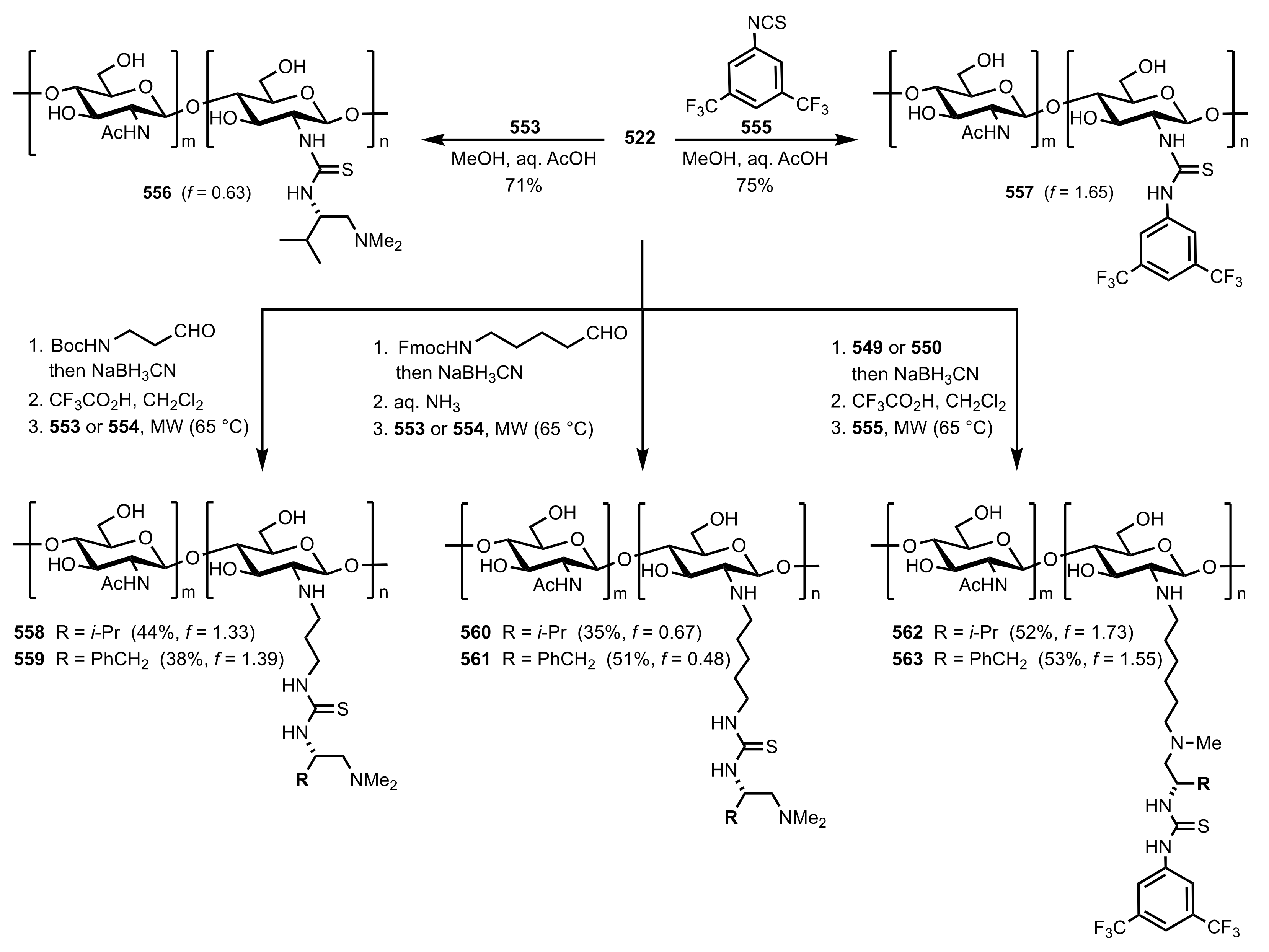
Scheme 151.
The synthesis of the chitosan-thiourea organocatalysts.
Scheme 151.
The synthesis of the chitosan-thiourea organocatalysts.
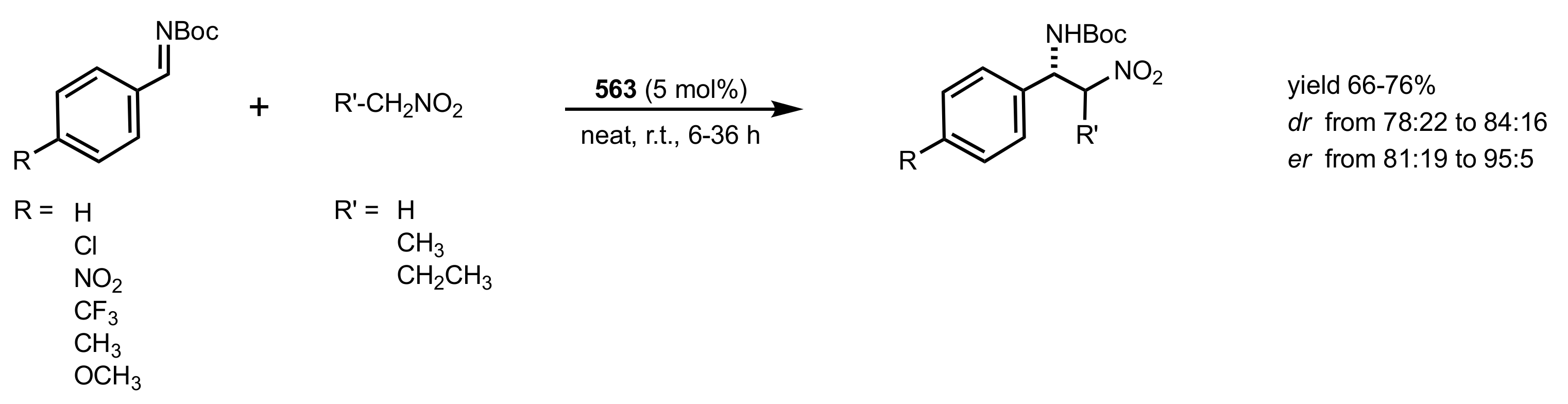
Scheme 152.
The Aza–Henry reaction catalysed by the chitosan-thiourea organocatalyst 566.
Scheme 152.
The Aza–Henry reaction catalysed by the chitosan-thiourea organocatalyst 566.
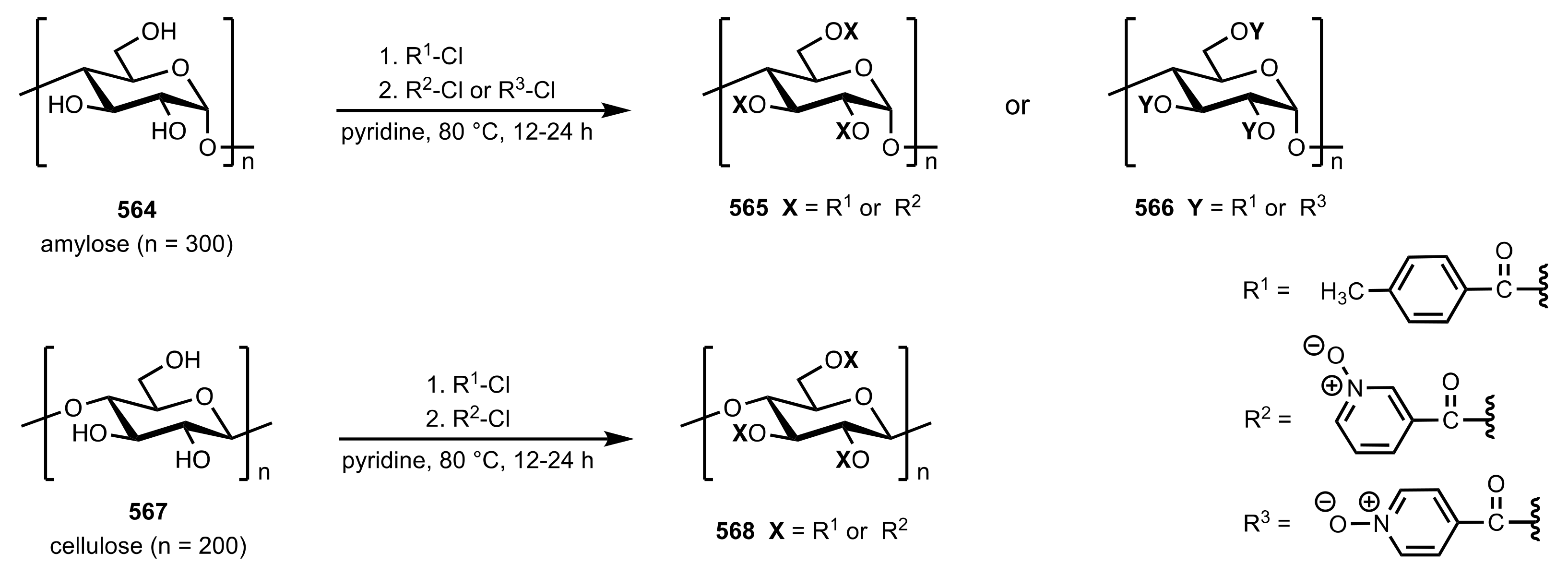
Scheme 153.
The synthesis of the amylose and cellulose pyridine oxide organocatalysts.
Scheme 153.
The synthesis of the amylose and cellulose pyridine oxide organocatalysts.
Scheme 154.
The allylation of benzaldehyde catalysed by the amylose and cellulose pyridine oxide organocatalysts.
Scheme 154.
The allylation of benzaldehyde catalysed by the amylose and cellulose pyridine oxide organocatalysts.
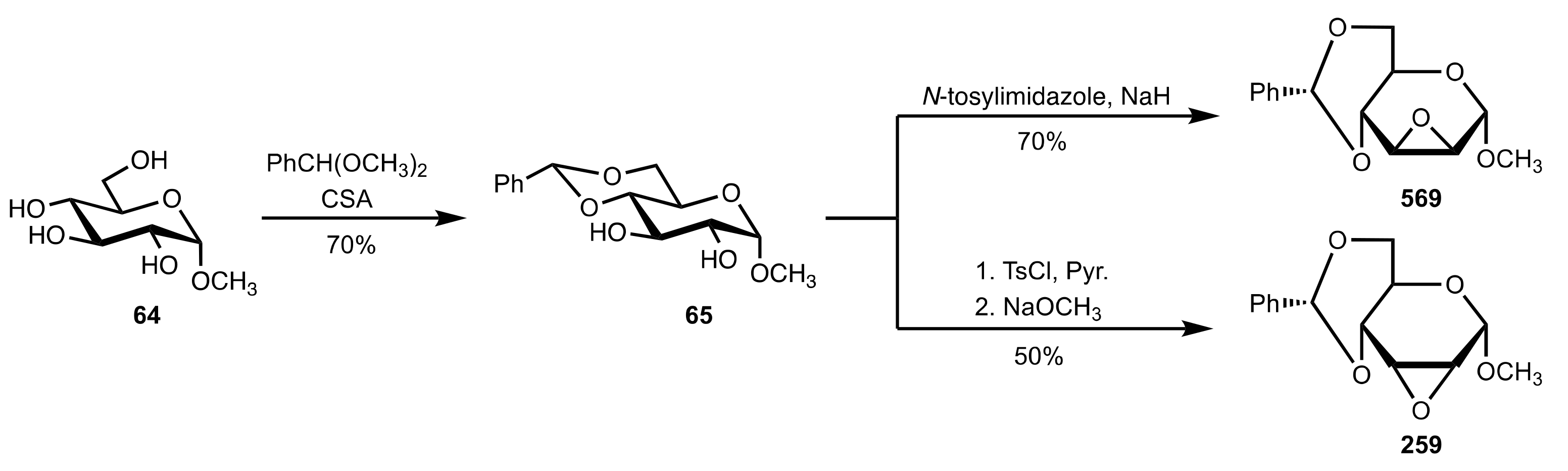
Scheme 155.
The synthesis of protected sugar epoxides.
Scheme 155.
The synthesis of protected sugar epoxides.
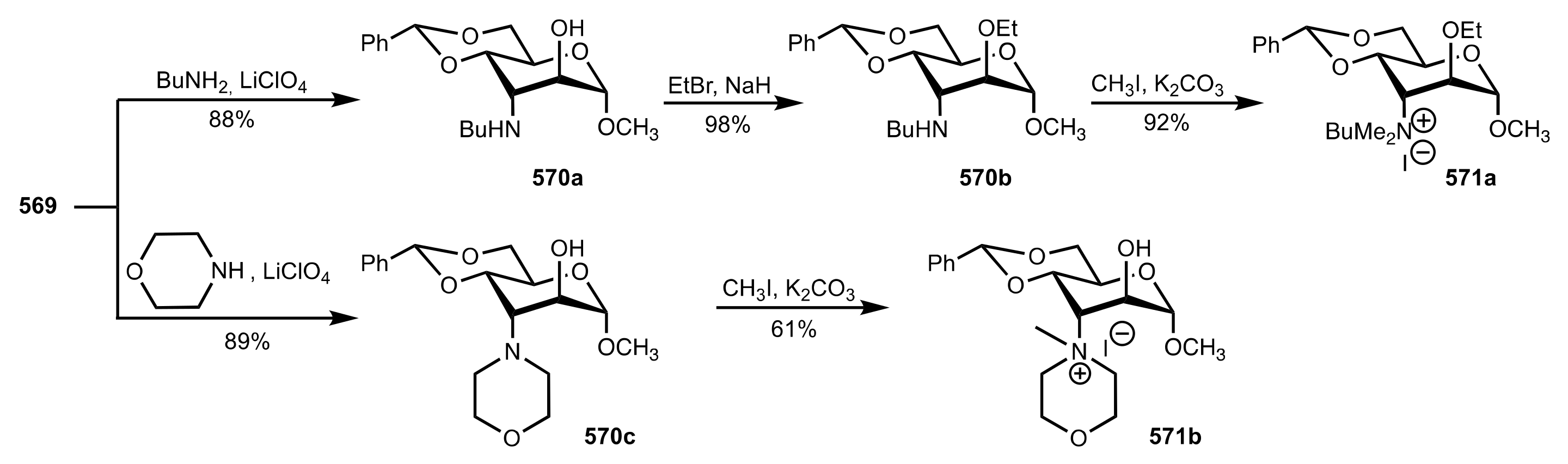
Scheme 156.
The synthesis of altrose-based quaternary ammonium salts.
Scheme 156.
The synthesis of altrose-based quaternary ammonium salts.
Figure 42.
Sugar-based quaternary ammonium salts.
Figure 42.
Sugar-based quaternary ammonium salts.
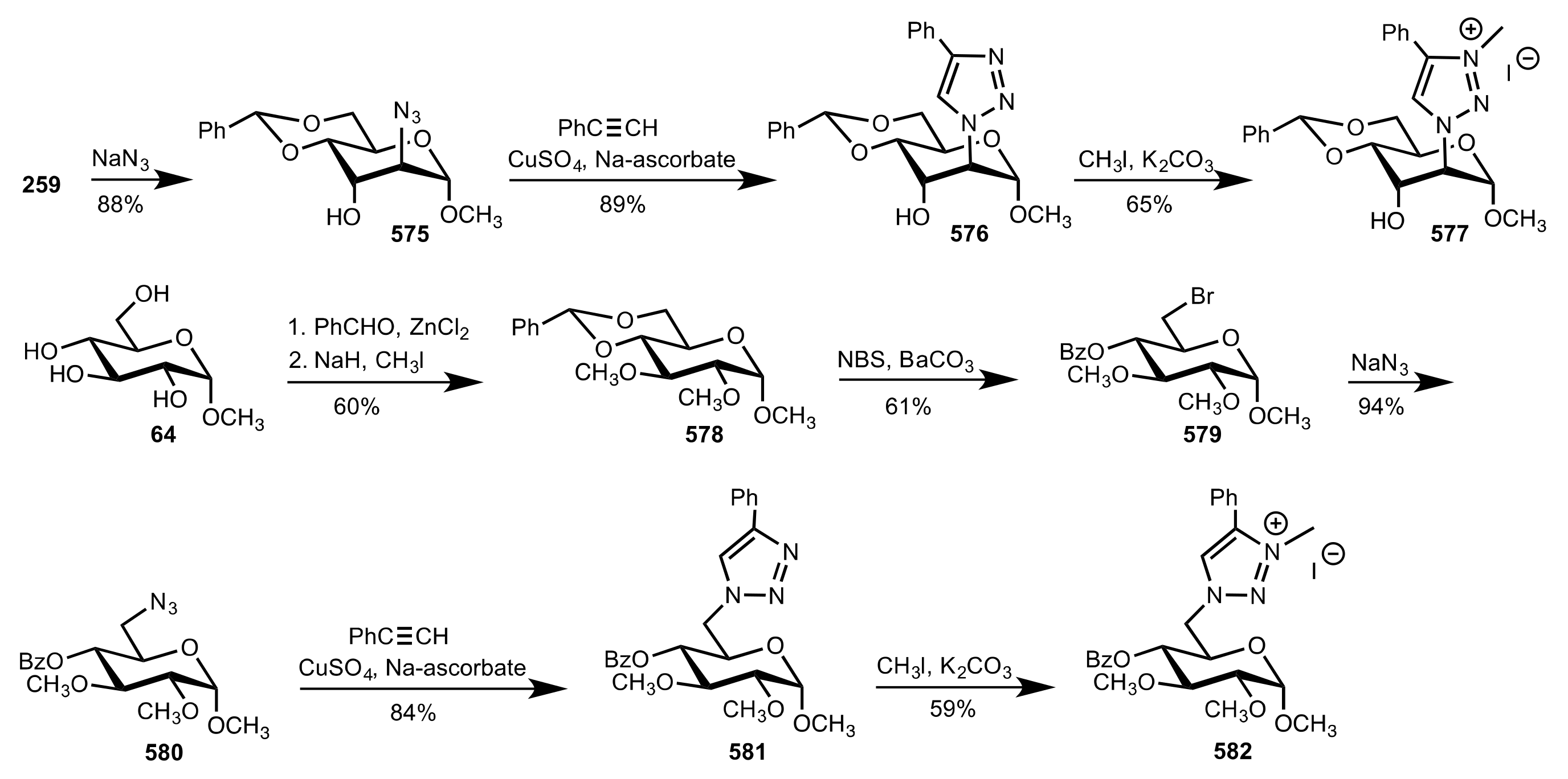
Scheme 157.
The synthesis of sugar triazolium salts.
Scheme 157.
The synthesis of sugar triazolium salts.
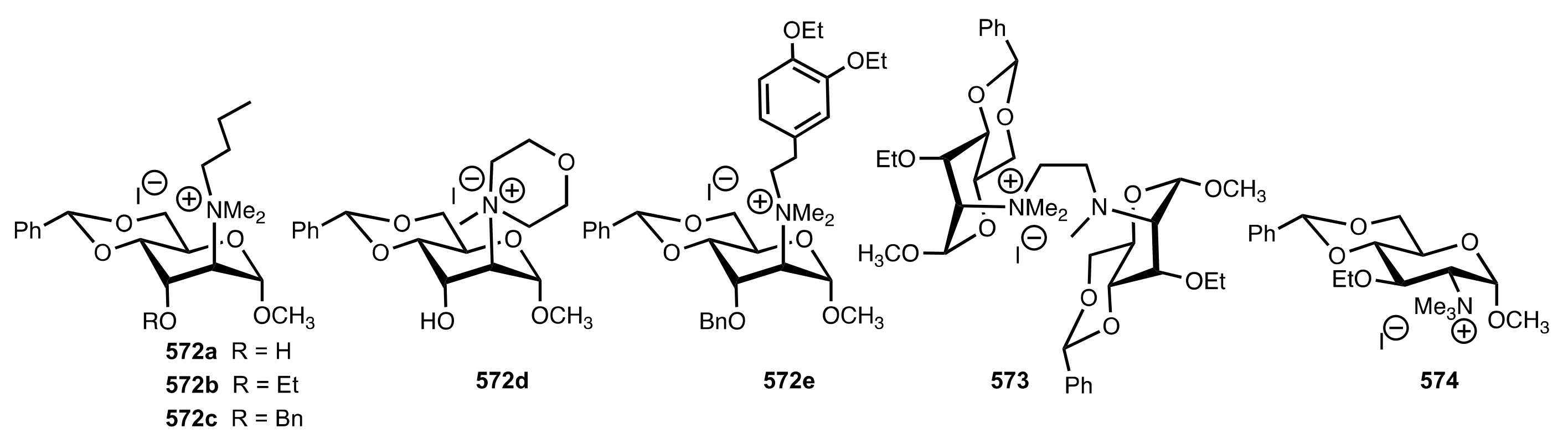
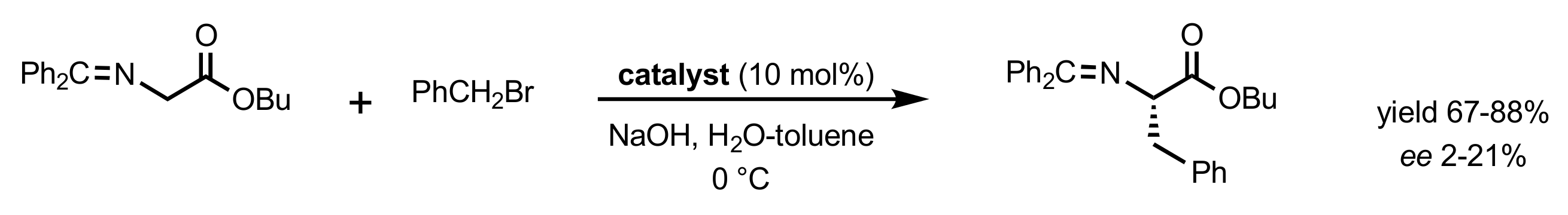
Scheme 158.
Alkylation of glycine ester catalysed by quaternary ammonium (571a,b, 572a-e, 573, 574) or triazolium (577 and 582) salts.
Scheme 158.
Alkylation of glycine ester catalysed by quaternary ammonium (571a,b, 572a-e, 573, 574) or triazolium (577 and 582) salts.
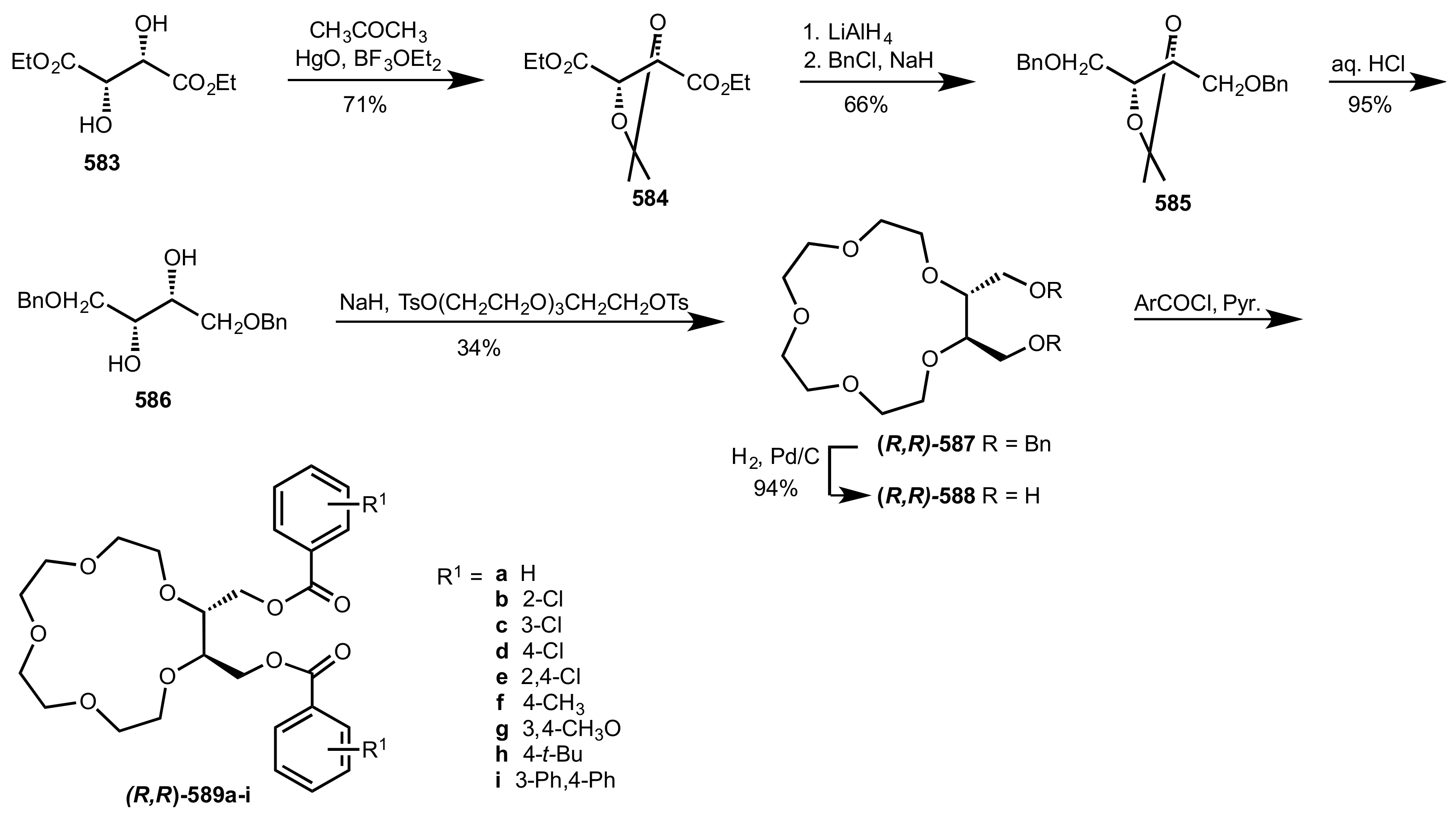
Scheme 159.
The synthesis of chiral crown ethers from l-tartaric ethyl ester.
Scheme 159.
The synthesis of chiral crown ethers from l-tartaric ethyl ester.
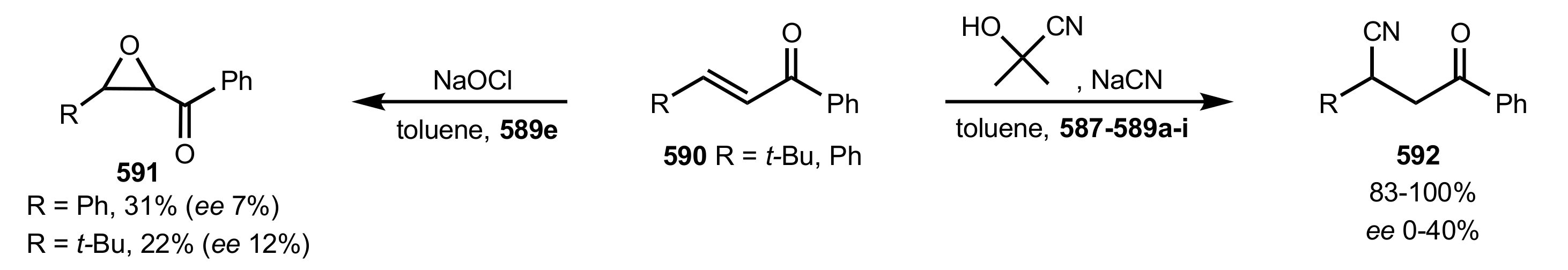
Scheme 160.
The epoxidation and cyanide addition to enones catalysed by 587, 588 and 589a-i.
Scheme 160.
The epoxidation and cyanide addition to enones catalysed by 587, 588 and 589a-i.
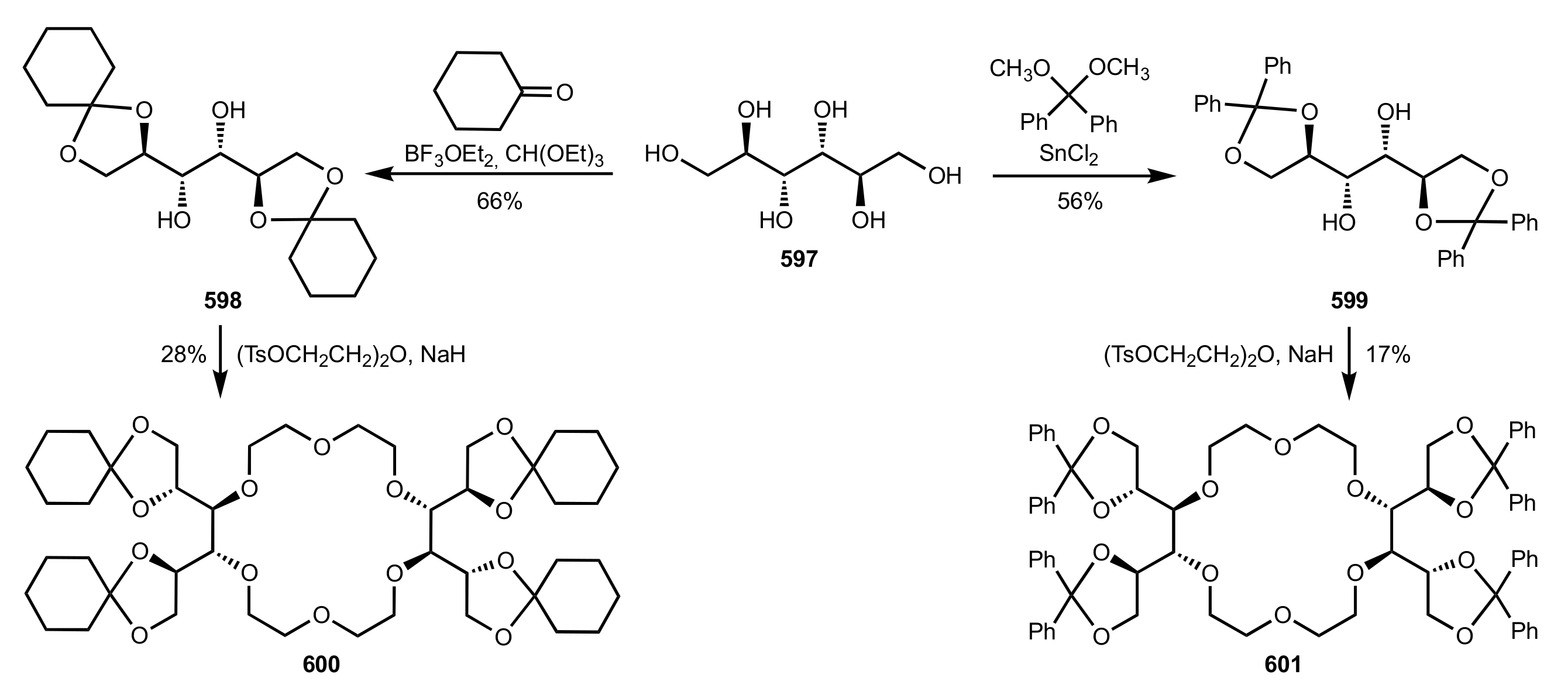
Scheme 161.
The synthesis of bis-diisopropylidene 18-crown-6 599.
Scheme 161.
The synthesis of bis-diisopropylidene 18-crown-6 599.
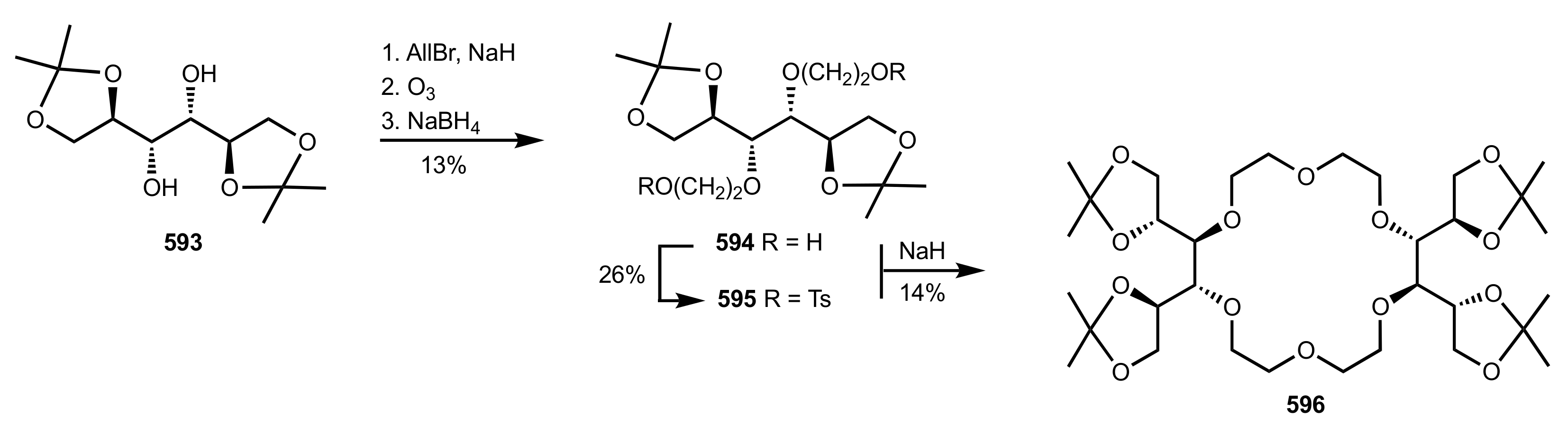
Scheme 162.
Crown ethers 600 and 601 synthesised by Bakó and co-workers.
Scheme 162.
Crown ethers 600 and 601 synthesised by Bakó and co-workers.
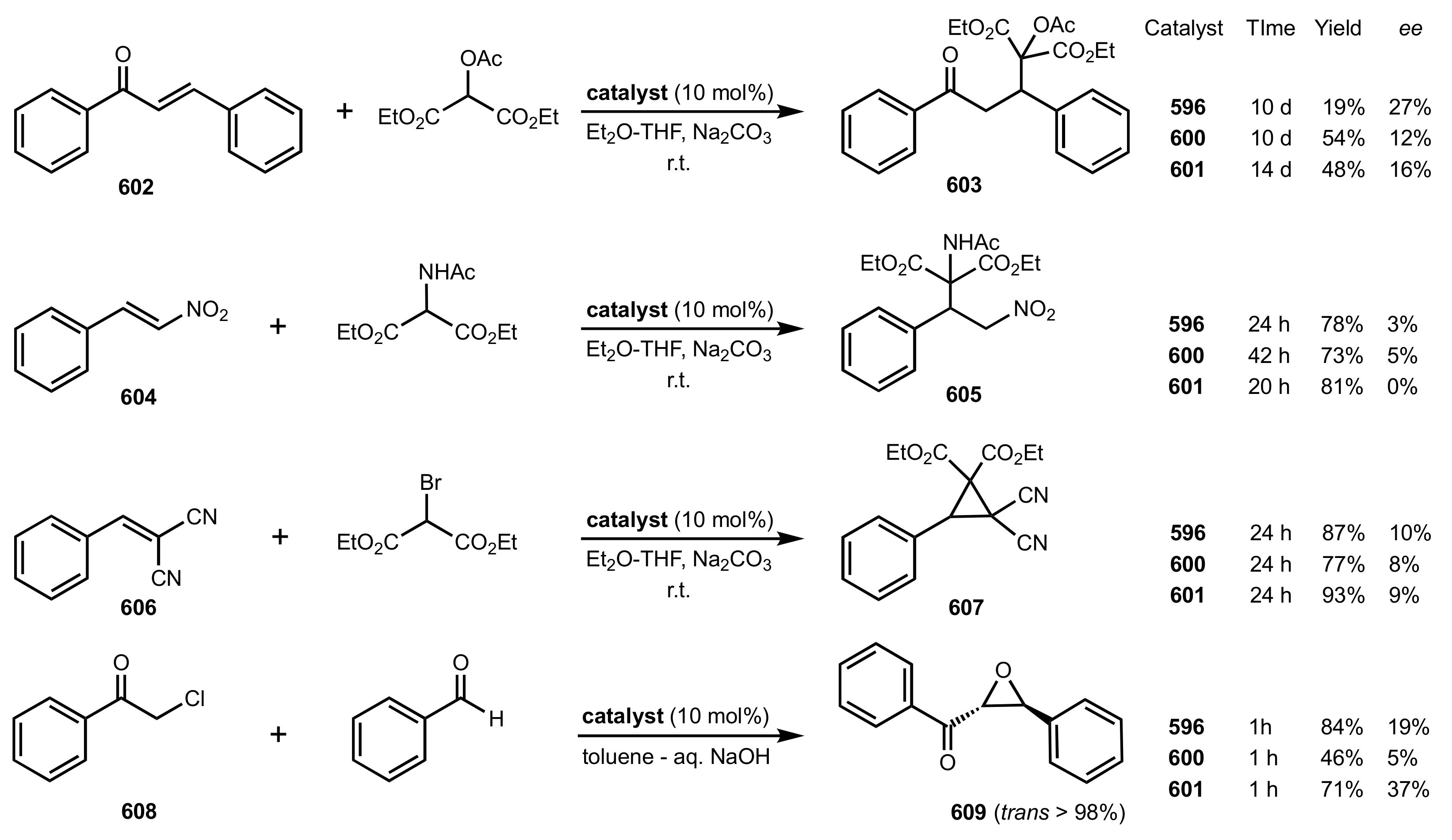
Scheme 163.
The Michael additions and Darzens condensation catalysed by 596, 600 and 601.
Scheme 163.
The Michael additions and Darzens condensation catalysed by 596, 600 and 601.
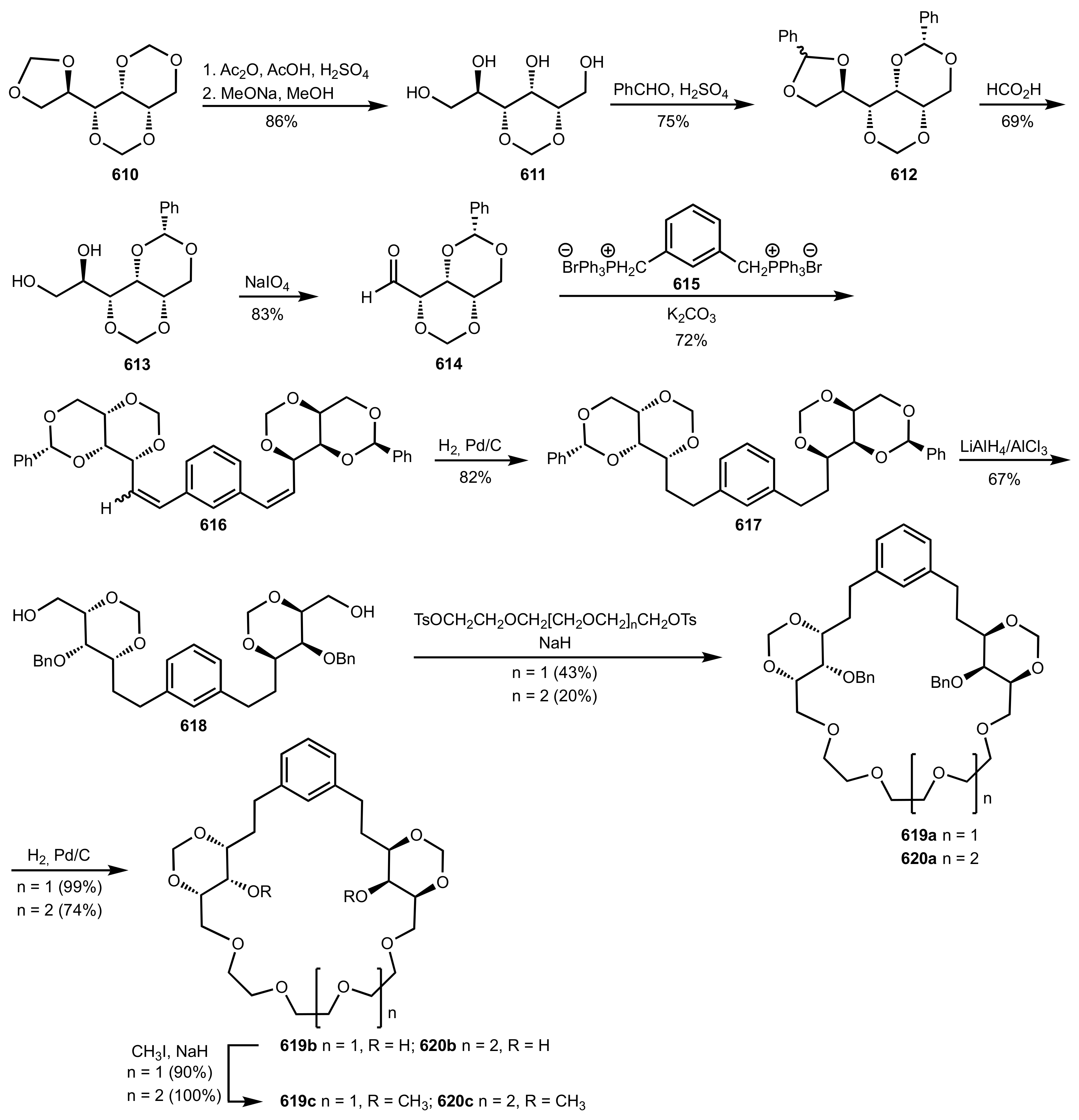
Scheme 164.
The synthesis of the alditol-based crown ethers 619c and 620c.
Scheme 164.
The synthesis of the alditol-based crown ethers 619c and 620c.
Scheme 165.
The synthesis of the permethylated crown ether 624.
Scheme 165.
The synthesis of the permethylated crown ether 624.
Scheme 166.
The model Michael addition catalysed by crown ethers 619a-c, 620a,b and 624.
Scheme 166.
The model Michael addition catalysed by crown ethers 619a-c, 620a,b and 624.
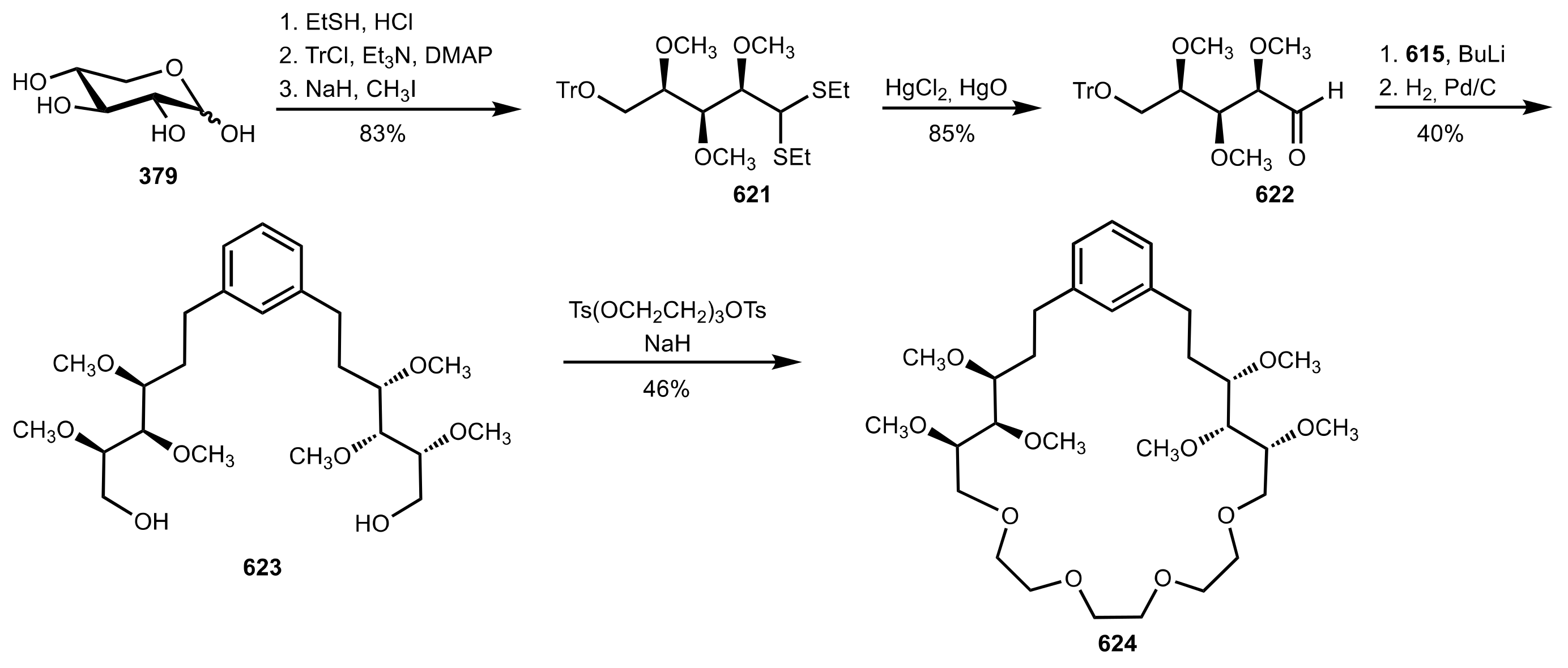
Scheme 167.
The synthesis of the glucose-based crown ether 633.
Scheme 167.
The synthesis of the glucose-based crown ether 633.
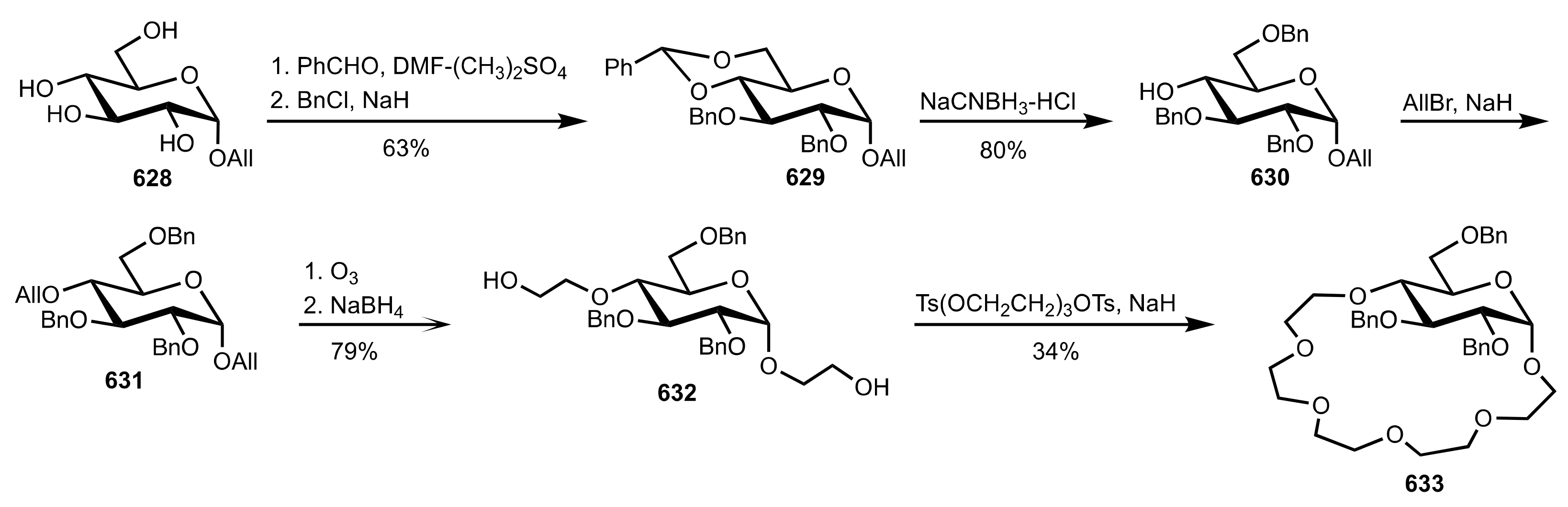
Scheme 168.
The synthesis of crown ethers bearing two and four d-glucose units.
Scheme 168.
The synthesis of crown ethers bearing two and four d-glucose units.
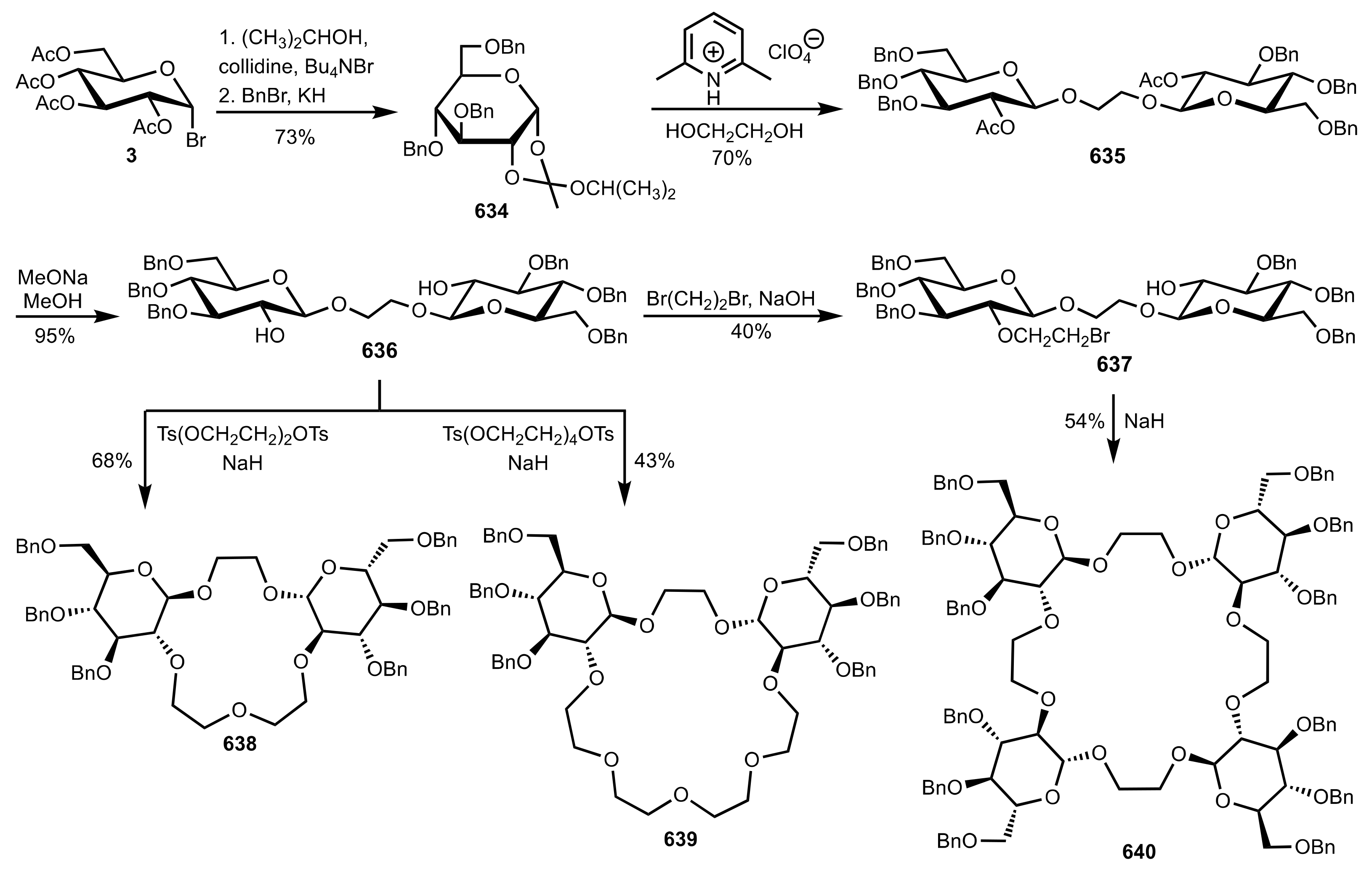
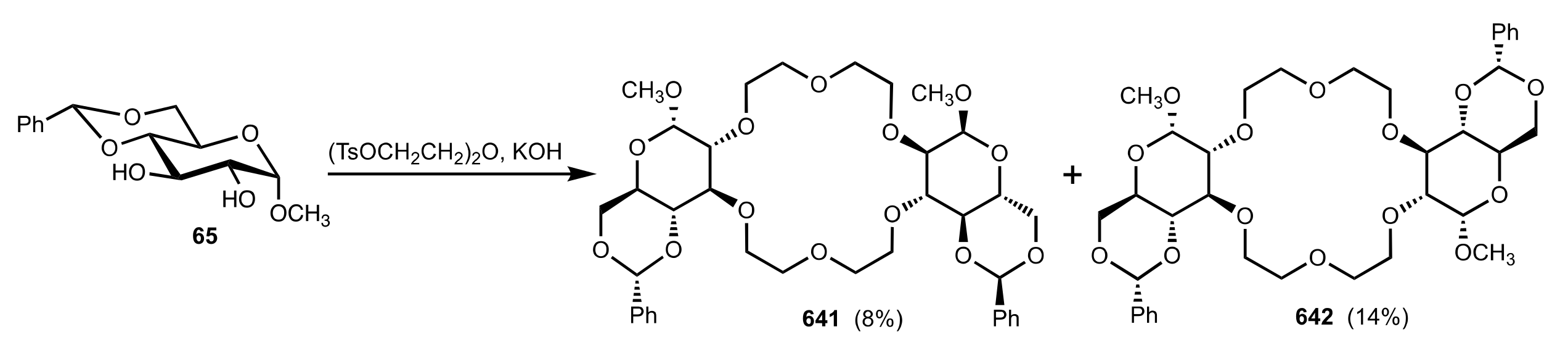
Scheme 169.
The synthesis of d-glucose-based C2-symmetric crown ethers.
Scheme 169.
The synthesis of d-glucose-based C2-symmetric crown ethers.
Figure 43.
The synthesis of a series of C2-symmetric crown ethers.
Figure 43.
The synthesis of a series of C2-symmetric crown ethers.
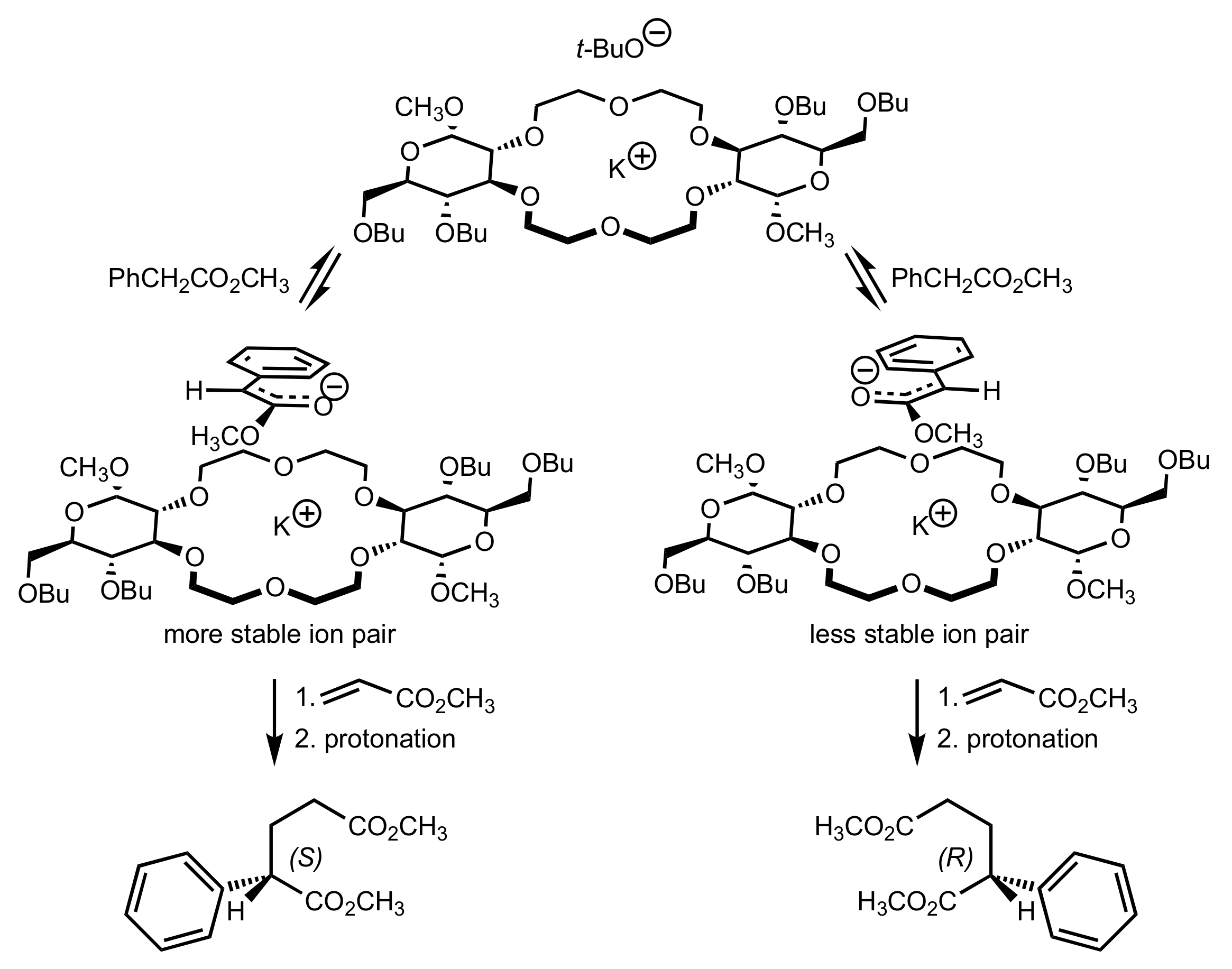
Scheme 170.
The proposed mechanism for the (S)-enantioselectivity of the Michael addition.
Scheme 170.
The proposed mechanism for the (S)-enantioselectivity of the Michael addition.
Scheme 171.
The synthesis of d-lactoside-based crown ethers.
Scheme 171.
The synthesis of d-lactoside-based crown ethers.
Scheme 172.
The synthesis of crown ethers containing two d-lactoside units.
Scheme 172.
The synthesis of crown ethers containing two d-lactoside units.
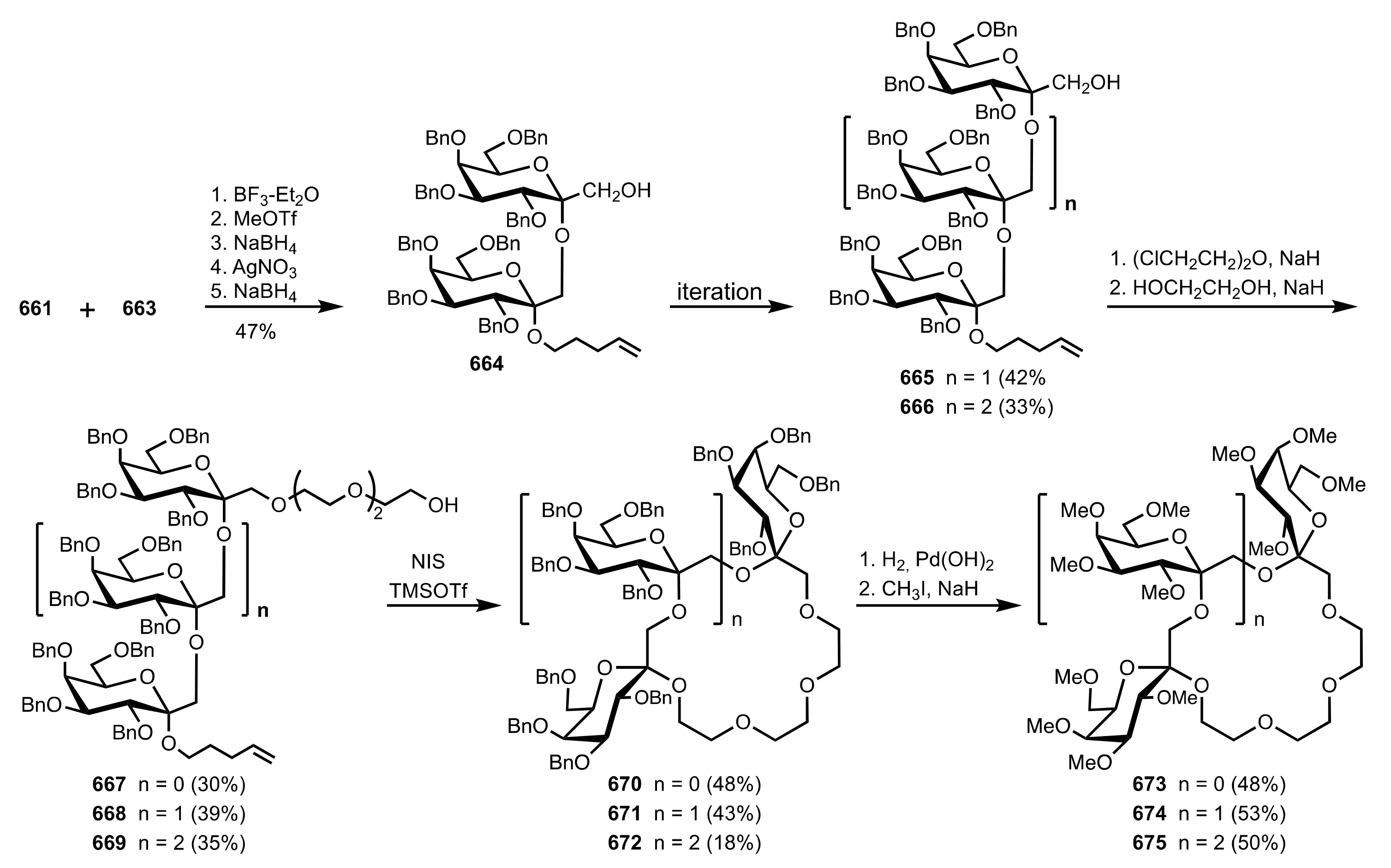
Scheme 173.
The synthesis of the glycosyl donor and glycosyl acceptor required for the preparation of the di-, tri- and tetrasaccharidic ketosides.
Scheme 173.
The synthesis of the glycosyl donor and glycosyl acceptor required for the preparation of the di-, tri- and tetrasaccharidic ketosides.
Scheme 174.
The synthesis of the oligoketoside-based crown ethers.
Scheme 174.
The synthesis of the oligoketoside-based crown ethers.
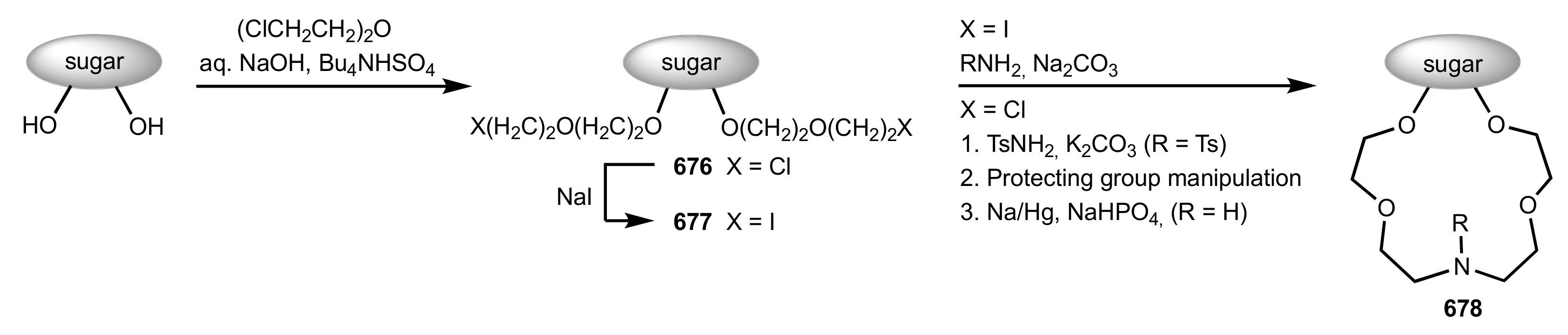
Scheme 175.
The general synthetic approach to sugar-based aza-crown ethers.
Scheme 175.
The general synthetic approach to sugar-based aza-crown ethers.
Figure 44.
Series of prepared mannitol-based aza-crown ethers.
Figure 44.
Series of prepared mannitol-based aza-crown ethers.
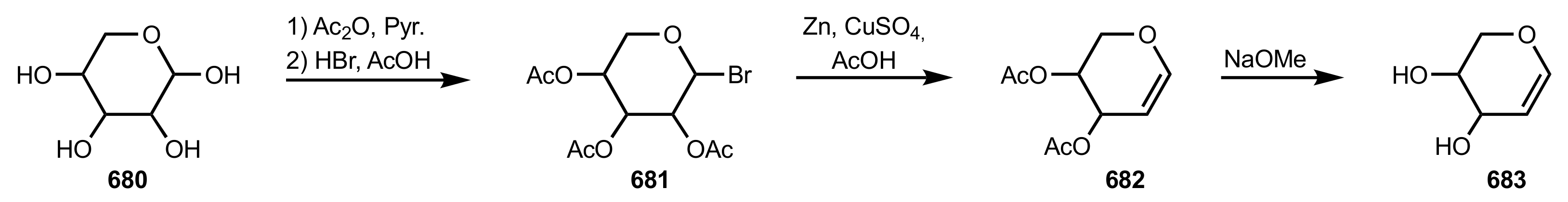
Scheme 176.
The general synthetic approach to deprotected glycals.
Scheme 176.
The general synthetic approach to deprotected glycals.
Figure 45.
Glycal-based aza-crown ethers prepared by Bakó and co-workers.
Figure 45.
Glycal-based aza-crown ethers prepared by Bakó and co-workers.
Scheme 177.
Epoxidations, Michael additions and Darzens reactions catalysed by 684–687.
Scheme 177.
Epoxidations, Michael additions and Darzens reactions catalysed by 684–687.
Scheme 178.
The general synthesis of glucose-based aza-crown ethers (for R, see
Table 2).
Scheme 178.
The general synthesis of glucose-based aza-crown ethers (for R, see
Table 2).
Scheme 179.
The synthesis of the supported glucose-based aza-15-crown-5 ether.
Scheme 179.
The synthesis of the supported glucose-based aza-15-crown-5 ether.
Figure 46.
Newly synthesised aza-crown ethers bearing a methyl α-d-glucoside moiety.
Figure 46.
Newly synthesised aza-crown ethers bearing a methyl α-d-glucoside moiety.
Scheme 180.
The general synthesis of aza-crown ethers bearing a β-
d-glucoside moiety (for X, see
Table 3).
Scheme 180.
The general synthesis of aza-crown ethers bearing a β-
d-glucoside moiety (for X, see
Table 3).
Scheme 181.
The general synthesis of aza-crown ethers bearing a
d-galactoside moiety (for X, see
Table 4).
Scheme 181.
The general synthesis of aza-crown ethers bearing a
d-galactoside moiety (for X, see
Table 4).
Figure 47.
Aza-crown ether prepared by Bakó and co-workers from various glycosides.
Figure 47.
Aza-crown ether prepared by Bakó and co-workers from various glycosides.
Scheme 182.
The synthesis of the altrose-based crown-amines.
Scheme 182.
The synthesis of the altrose-based crown-amines.
Figure 48.
Aza-crown ether containing a pyridine ring prepared by Bakó and co-workers.
Figure 48.
Aza-crown ether containing a pyridine ring prepared by Bakó and co-workers.
Scheme 183.
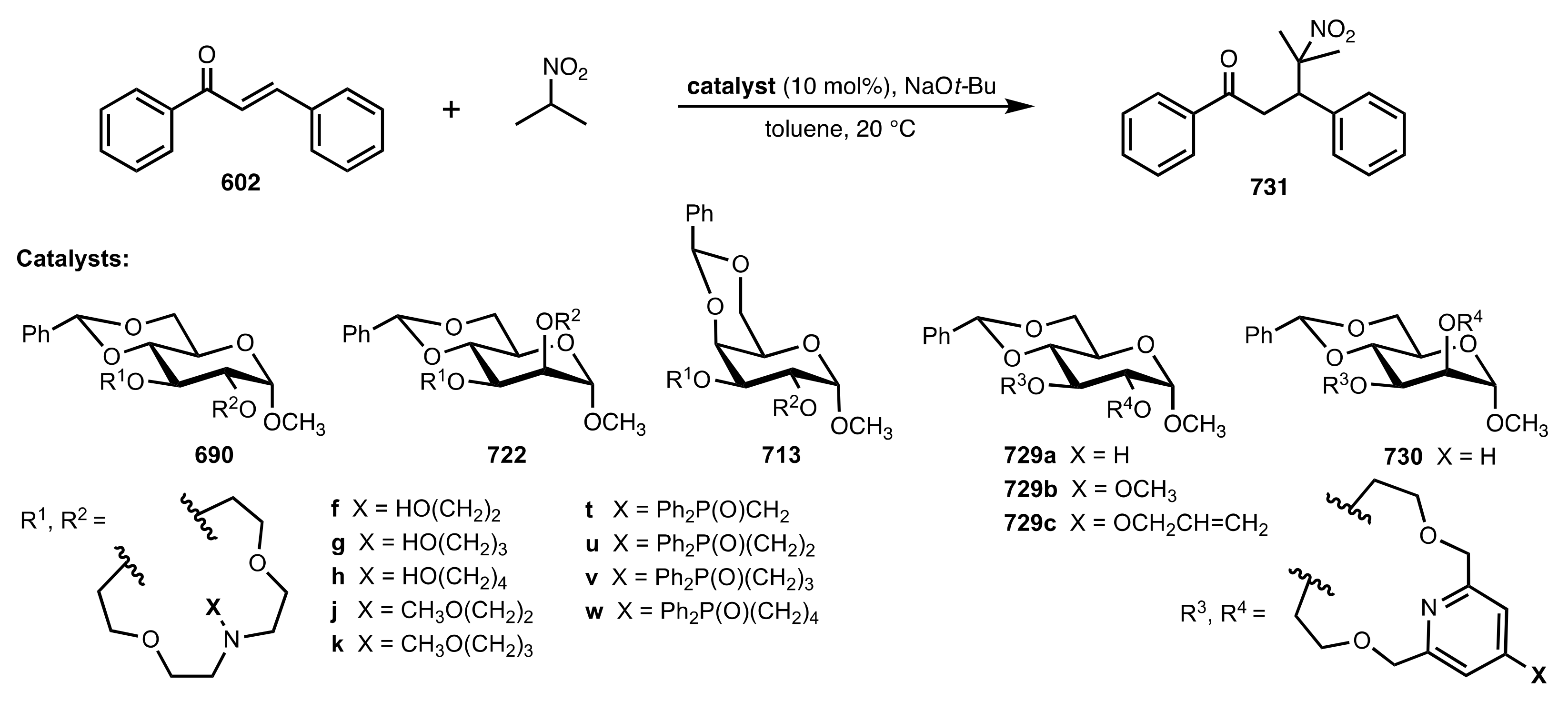
The Michael addition catalysed by glucoside-, mannoside- and galactoside-based aza-crown ethers.
Scheme 183.
The Michael addition catalysed by glucoside-, mannoside- and galactoside-based aza-crown ethers.
Scheme 184.
Other Michael additions catalysed by sugar-based aza-crown ethers.
Scheme 184.
Other Michael additions catalysed by sugar-based aza-crown ethers.
Scheme 185.
The Michael-initiated ring closure reaction catalysed by sugar-based aza-crown ethers.
Scheme 185.
The Michael-initiated ring closure reaction catalysed by sugar-based aza-crown ethers.
Scheme 186.
Cyclopropanation catalysed by sugar-based aza-crown ethers (nd: not determined).
Scheme 186.
Cyclopropanation catalysed by sugar-based aza-crown ethers (nd: not determined).
Scheme 187.
Cyclopropanation catalysed by sugar-based aza-crown ethers.
Scheme 187.
Cyclopropanation catalysed by sugar-based aza-crown ethers.
Scheme 188.
Cyclopropanation of 2-arylidene-1,3-indandiones catalysed by aza-crown ethers.
Scheme 188.
Cyclopropanation of 2-arylidene-1,3-indandiones catalysed by aza-crown ethers.
Scheme 189.
The Michael-initiated ring closure reaction catalysed by sugar-based aza-crown ethers.
Scheme 189.
The Michael-initiated ring closure reaction catalysed by sugar-based aza-crown ethers.
Scheme 190.
The epoxidation of chalcone catalysed by sugar-based aza-crown ethers functionalised with hydroxypropyl side arm.
Scheme 190.
The epoxidation of chalcone catalysed by sugar-based aza-crown ethers functionalised with hydroxypropyl side arm.
Scheme 191.
Darzens condensation catalysed by aldose-based aza-crown ethers.
Scheme 191.
Darzens condensation catalysed by aldose-based aza-crown ethers.
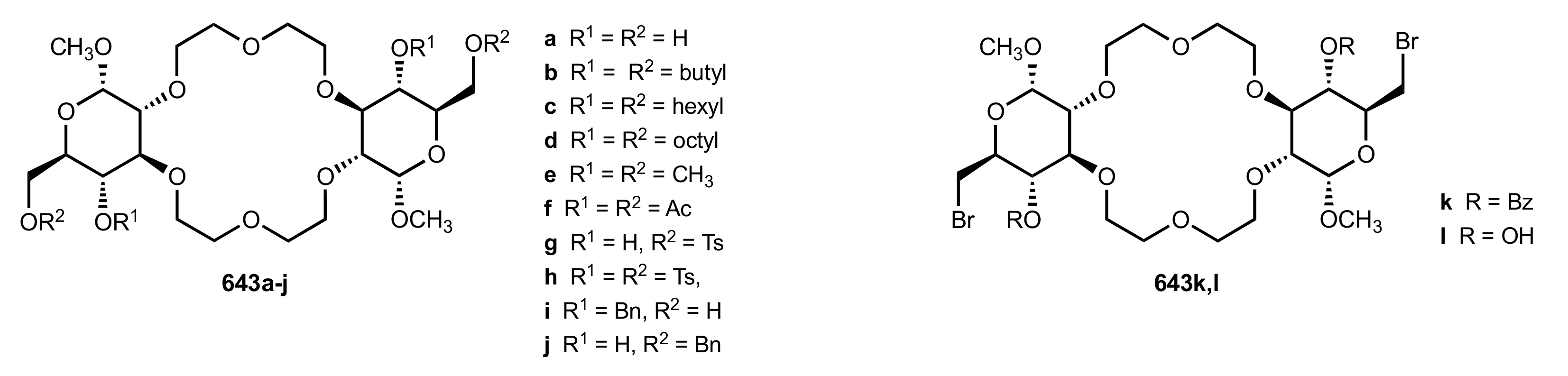
Table 1.
The Michael addition of methyl phenylacetate (625, 1.3 equiv.) to methyl acrylate (626) in the presence of the crown ethers bearing two glucose units (6 mol%) and potassium tert-butoxide (0.34 equiv.). The reactions were conducted at −78 °C in toluene for 8 min if not specified otherwise.
Table 1.
The Michael addition of methyl phenylacetate (625, 1.3 equiv.) to methyl acrylate (626) in the presence of the crown ethers bearing two glucose units (6 mol%) and potassium tert-butoxide (0.34 equiv.). The reactions were conducted at −78 °C in toluene for 8 min if not specified otherwise.
| Entry | Catalyst | Yield (%) | ee % (S) |
|---|
| 1 | 642 | 59 | 17 |
| 2 | 643a | 44 | 7 |
| 3 | 643ba | 82 | 84 |
| 4 | 643b | 100 | 80 |
| 5 | 643bb | 100 | 76 |
| 6 | 643c | 69 | 46 |
| 7 | 643d | 69 | 29 |
| 8 | 643e | 100 | 76 |
| 9 | 643f | 75 | 17 |
| 10 | 643g | 67 | 0.4 |
| 11 | 643h | 31 | 1.7 |
| 12 | 643i | 66 | 0.2 |
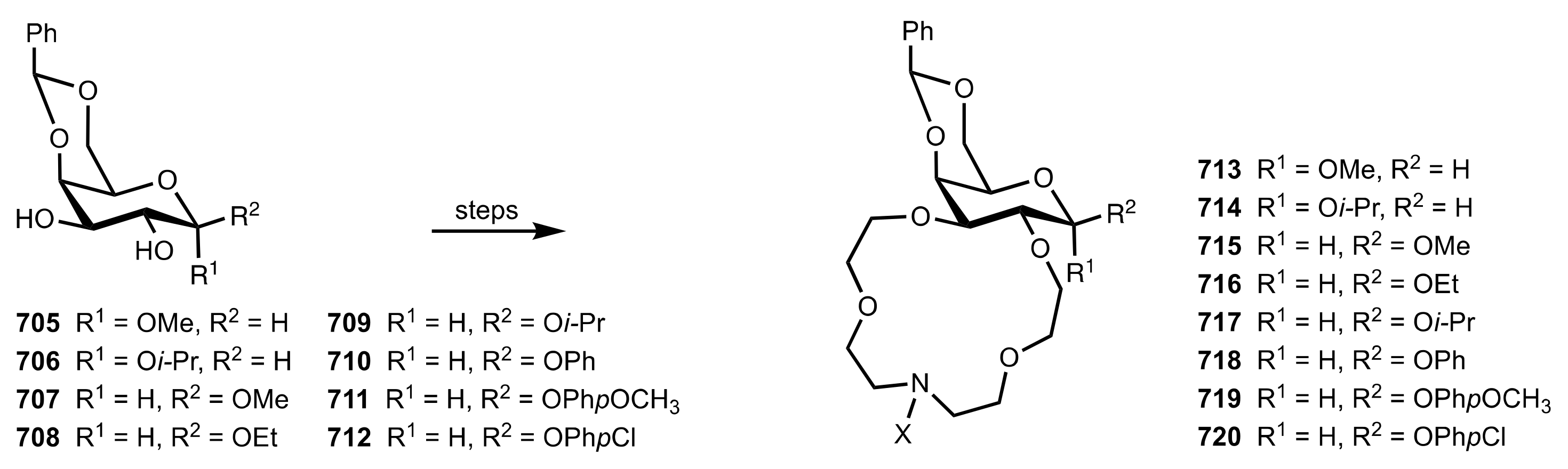
Table 2.
A series of glucose-based aza-crown ethers synthesised by Bakó and co-workers.
Table 3.
A series of β-d-glucoside-based aza-crown ethers synthesised by Bakó and co-workers.
Table 3.
A series of β-d-glucoside-based aza-crown ethers synthesised by Bakó and co-workers.
Table 4.
A series of d-galactoside-based aza-crown ethers synthesised by Bakó and co-workers.
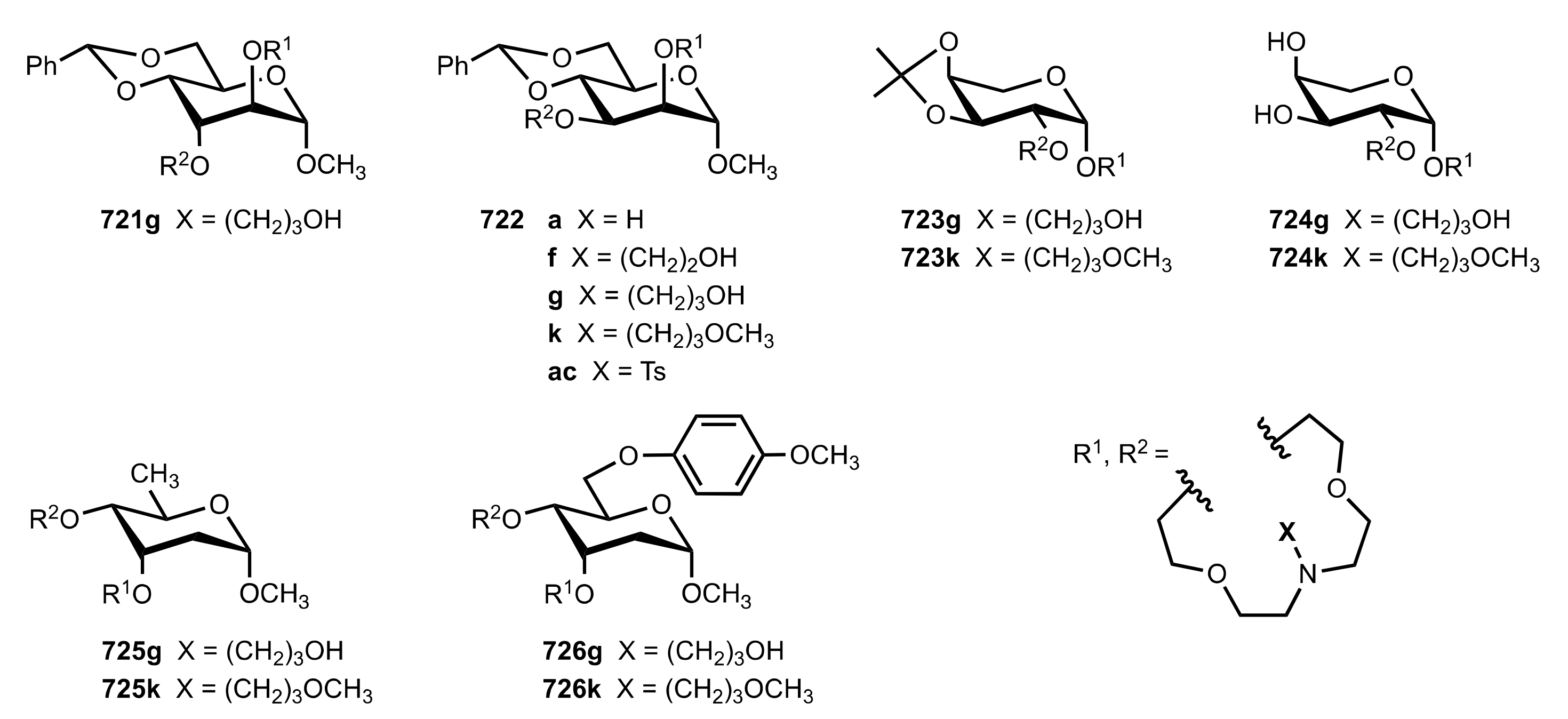
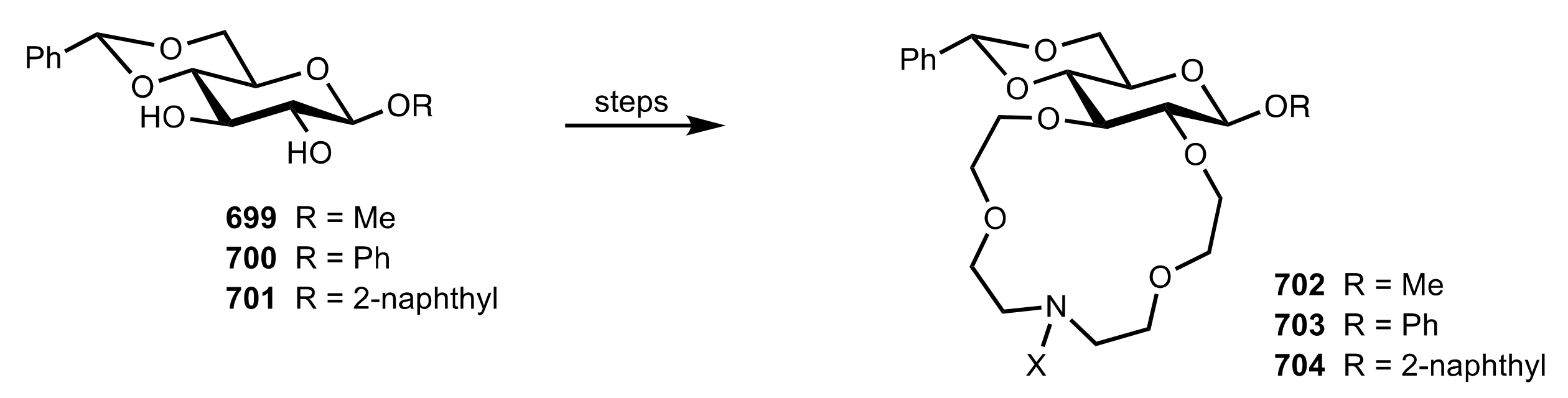
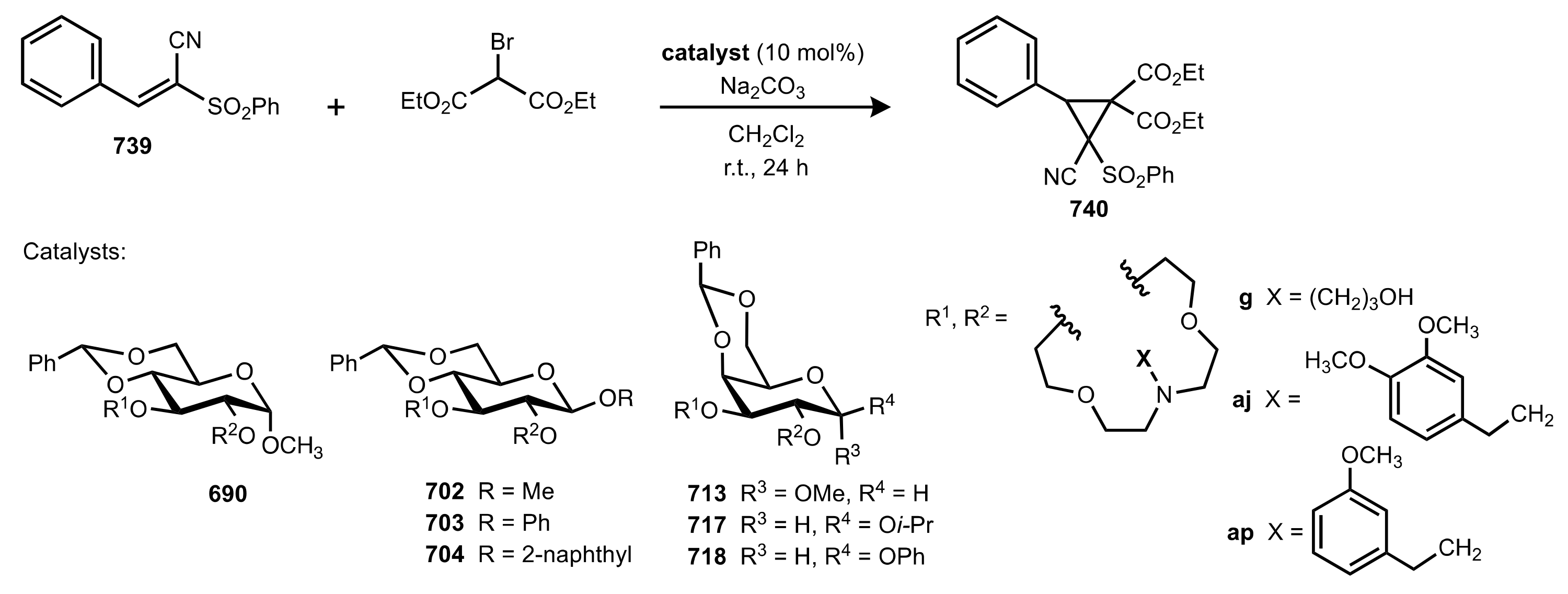
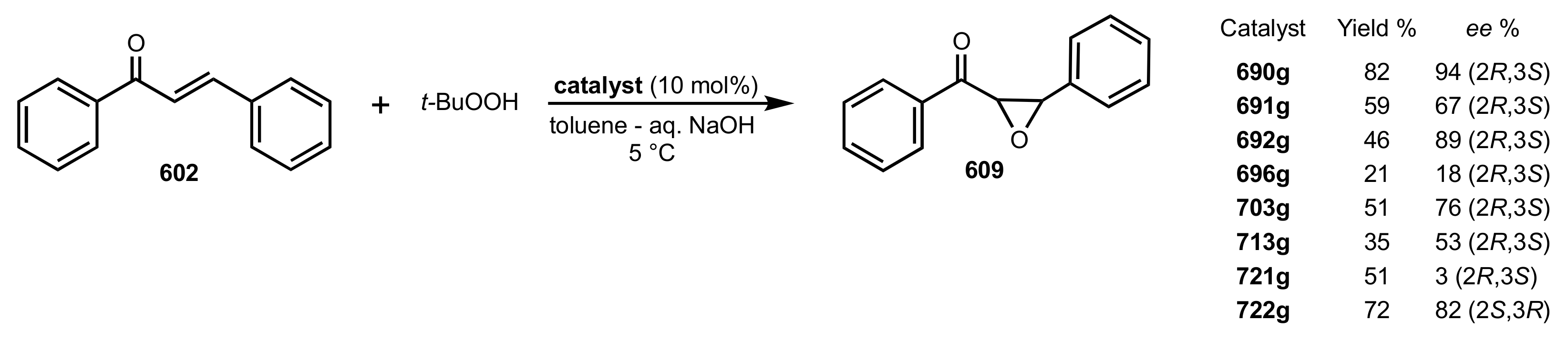
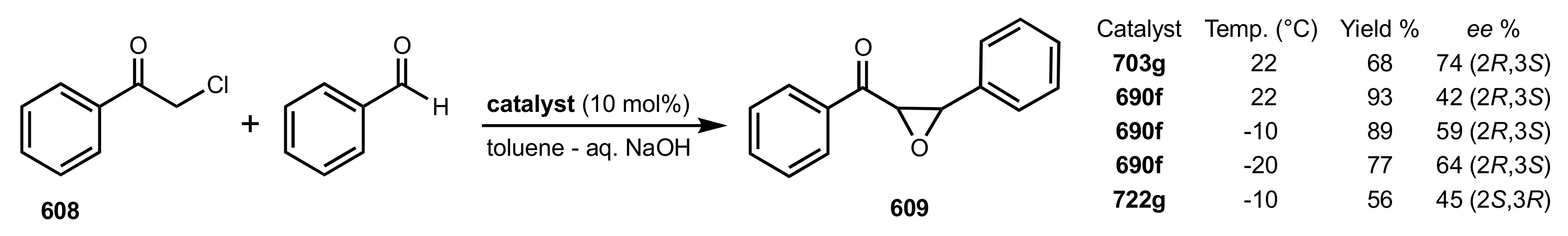
Table 5.
The enantioselective Michael addition shown in
Scheme 183.
Table 5.
The enantioselective Michael addition shown in
Scheme 183.
| Entry | Catalyst | Time (h) | Yield (%) | ee (%) | Ref. |
|---|
| 1 | 690f | 20 | 51 | 62 (R) a | [286,313] |
| 2 | 690g | 28 | 53 | 85 (R) a | [286,293,309] |
| 3 | 690j | 40 | 45 | 87 (R) | [286,310] |
| 4 | 690t | 48 | 39 | 60 (R) | [288] |
| 5 | 690u | 48 | 41 | 74 (R) | [288] |
| 6 | 690v | 48 | 32 | 77 (R) | [288] |
| 7 | 690w | 32 | 43 | 94 (R) | [285,288,310] |
| 8 | 722f | 44 | 32 | 70 (S) | [305] |
| 9 | 722g | 52 | 37 | 92 (S) | [305,309] |
| 10 | 722h | 58 | 40 | 63(S) | [305] |
| 11 | 722k | 50 | 37 | 77 (S) | [305] |
| 12 | 713j | 38 | 34 | 52 (R) | [286] |
| 13 | 729a | 24 | 48 | 72 (S) | [308] |
| 14 | 729b | 30 | 47 | 76 (S) | [308] |
| 15 | 729c | 30 | 51 | 67 (S) | [308] |
| 16 | 730 | 25 | 50 | 80 (R) | [308] |
Table 6.
The Michael-initiated ring closure reaction shown in
Scheme 189.
Table 6.
The Michael-initiated ring closure reaction shown in
Scheme 189.
| Entry | Catalyst | Yield (%) | ee trans (%) |
|---|
| 1 | 690g | 89 | 50 |
| 2 | 690aj | 94 | 73 |
| 3 | 702aj | 91 | 58 |
| 4 | 704g | 88 | 18 |
| 5 | 704aj | 90 | 35 |
| 6 | 713g | 85 | 62 |
| 7 | 713aj | 93 | 80 |
| 8 | 713ap | 95 | 76 |
| 9 | 717g | 93 | 61 |
| 10 | 718g | 91 | 43 |
| 11 | 718aj | 90 | 72 |


 ,
, 















































































































































































































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
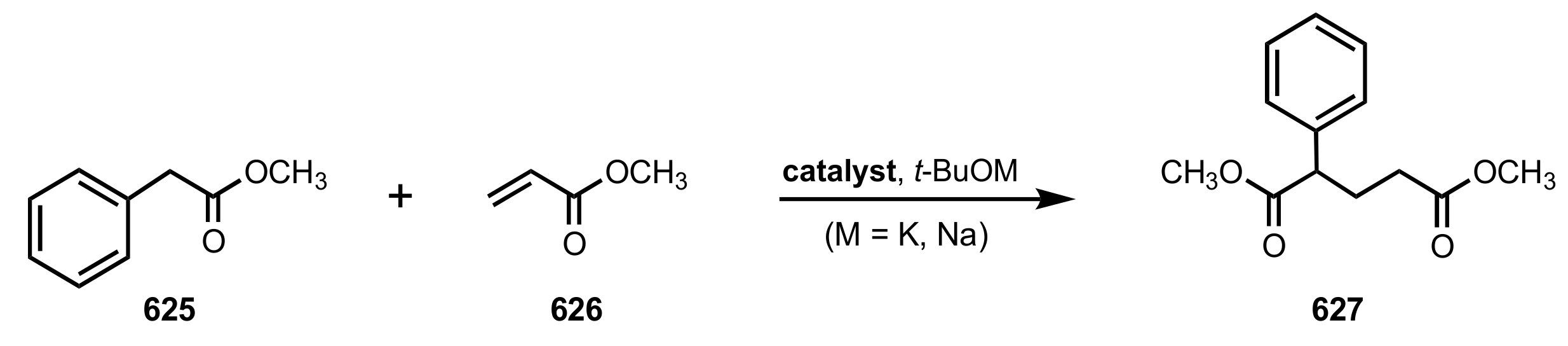{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











