
Virtual Screening for Potential Phytobioactives as Therapeutic Leads to Inhibit NQO1 for Selective Anticancer Therapy

 , , , , , ,
, , , , , ,  ,
,  , and
, and 
Abstract
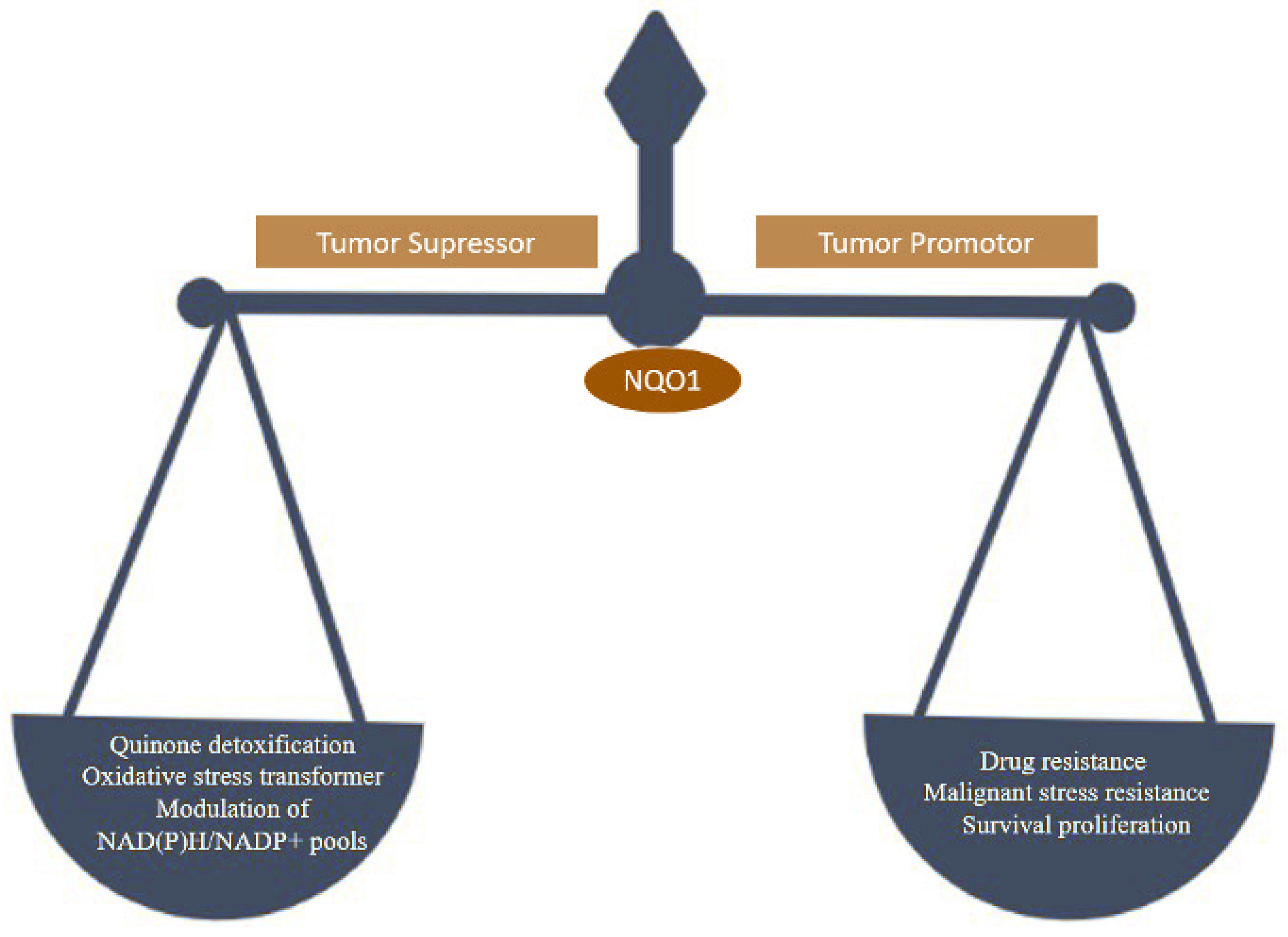
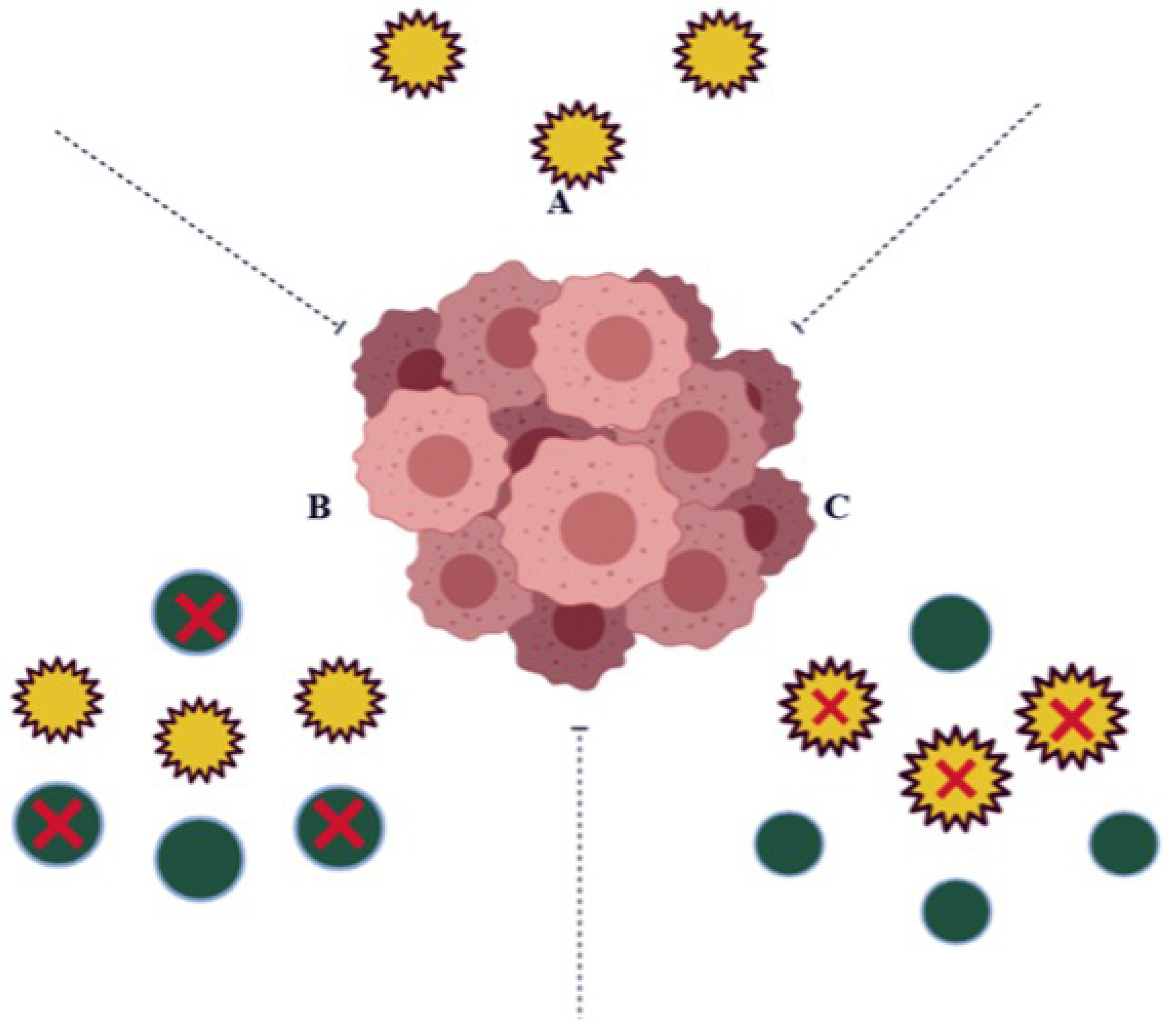
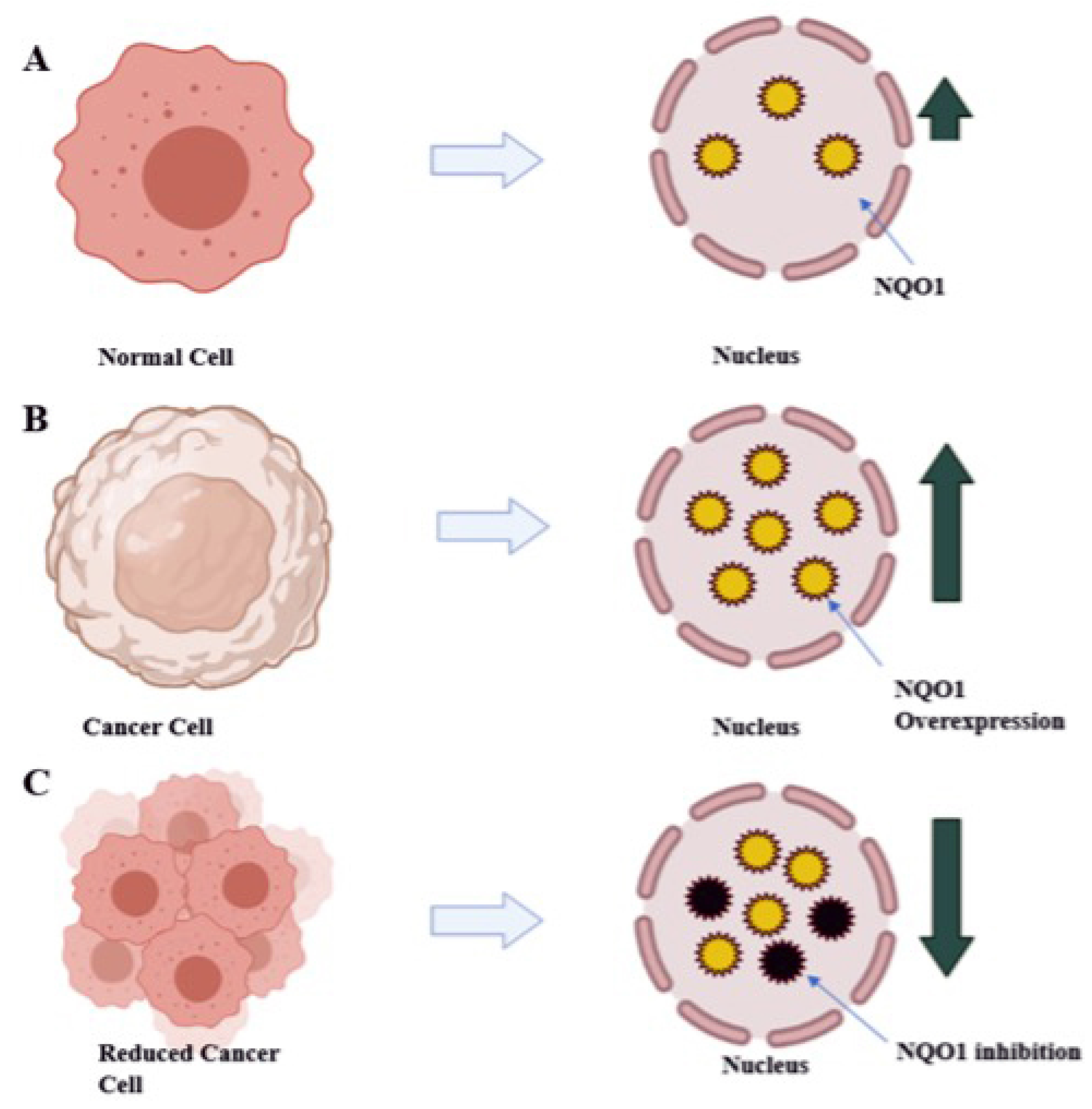
:1. Introduction
2. Materials and Methods
2.1. Collection of Phytobioactives
2.2. Preparation of Ligand
2.3. Protein Preparation
2.4. Prediction of the Binding Pockets
2.5. Protein Refinement and Structure Validation
2.6. Molecular Docking Studies
2.7. Molecular Docking Visualization
2.8. Molecular Dynamics
2.9. ADMET
2.10. Conceptual DFT Studies
3. Results and Discussion
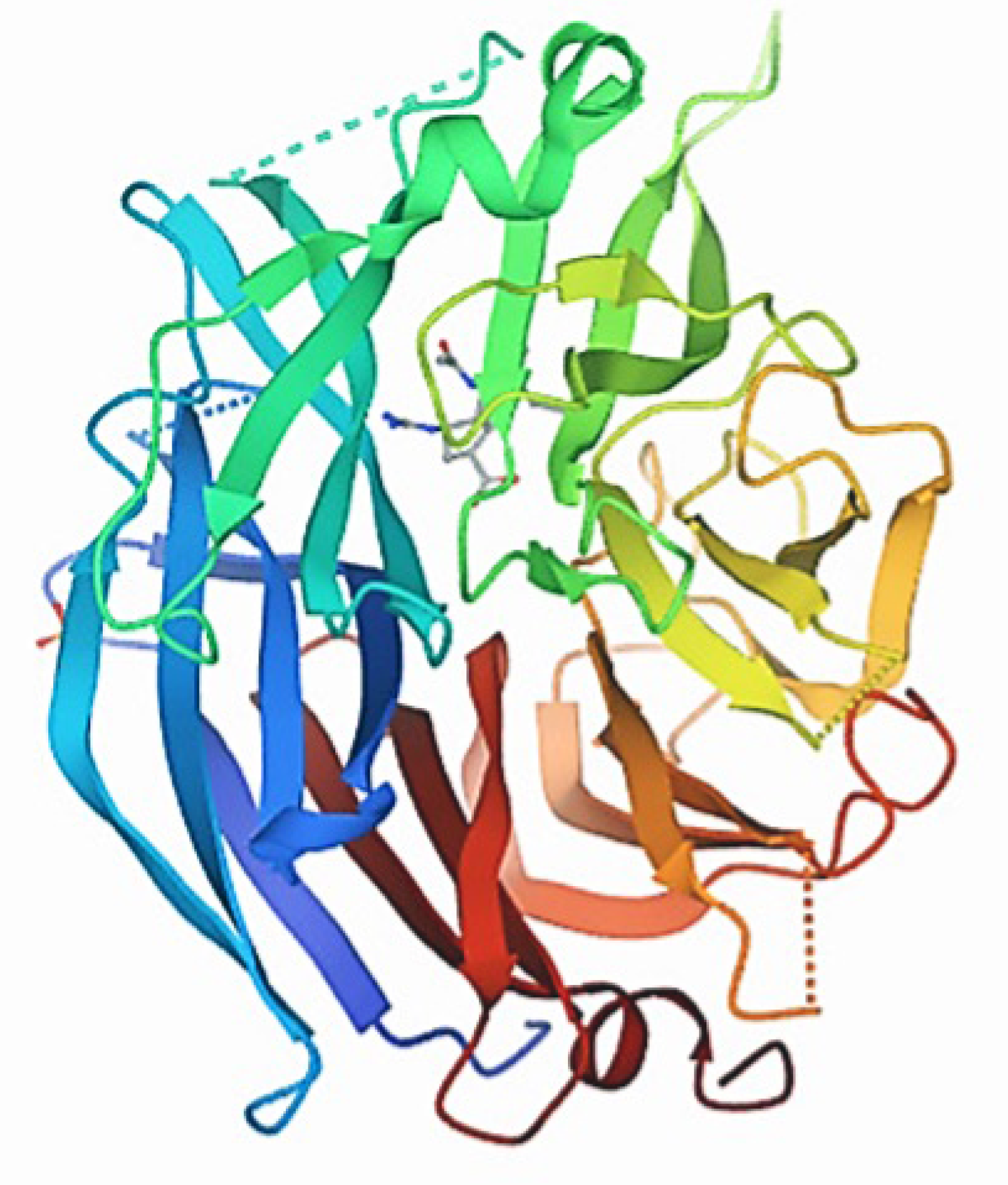
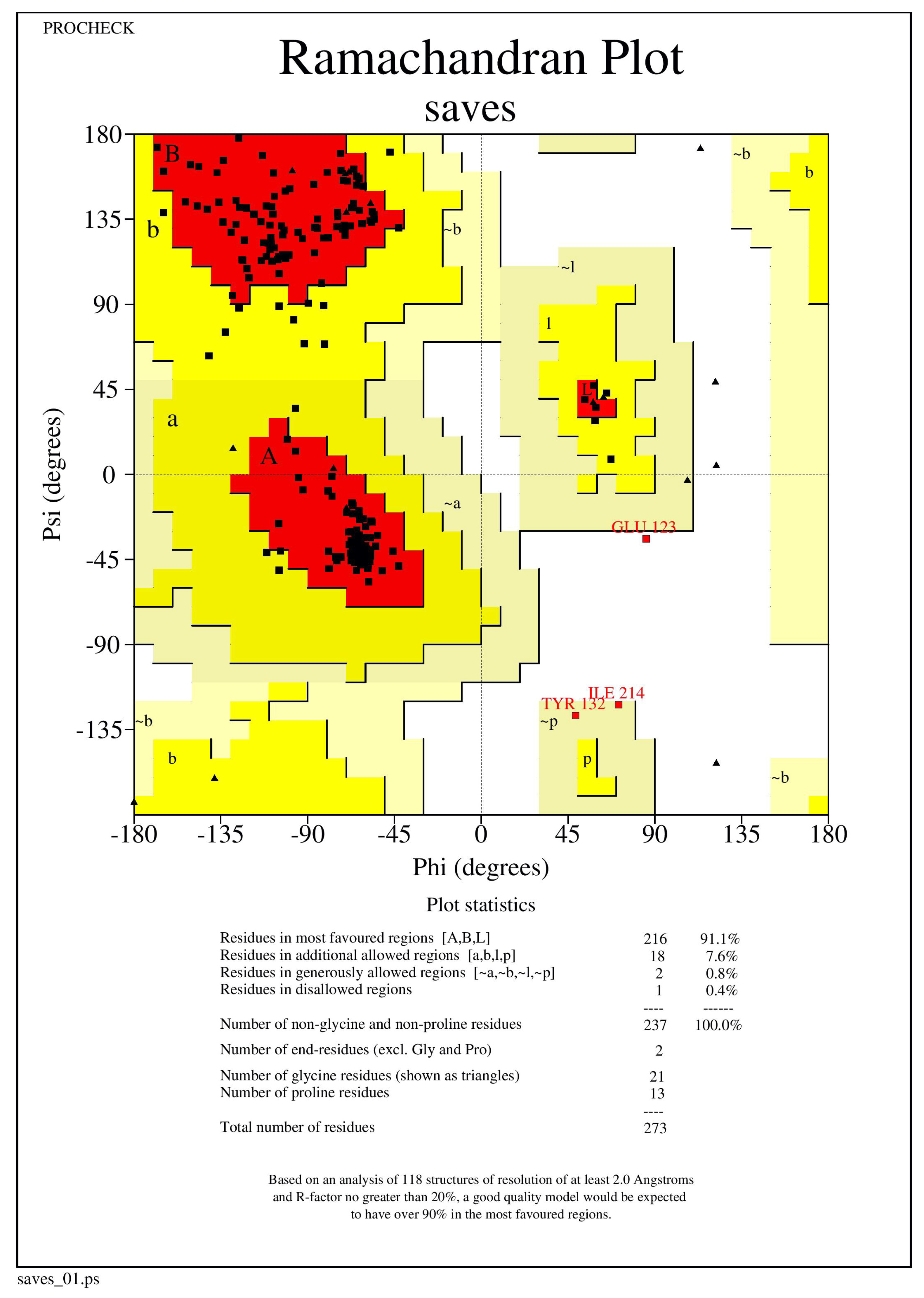
3.1. Protein Structure Validation
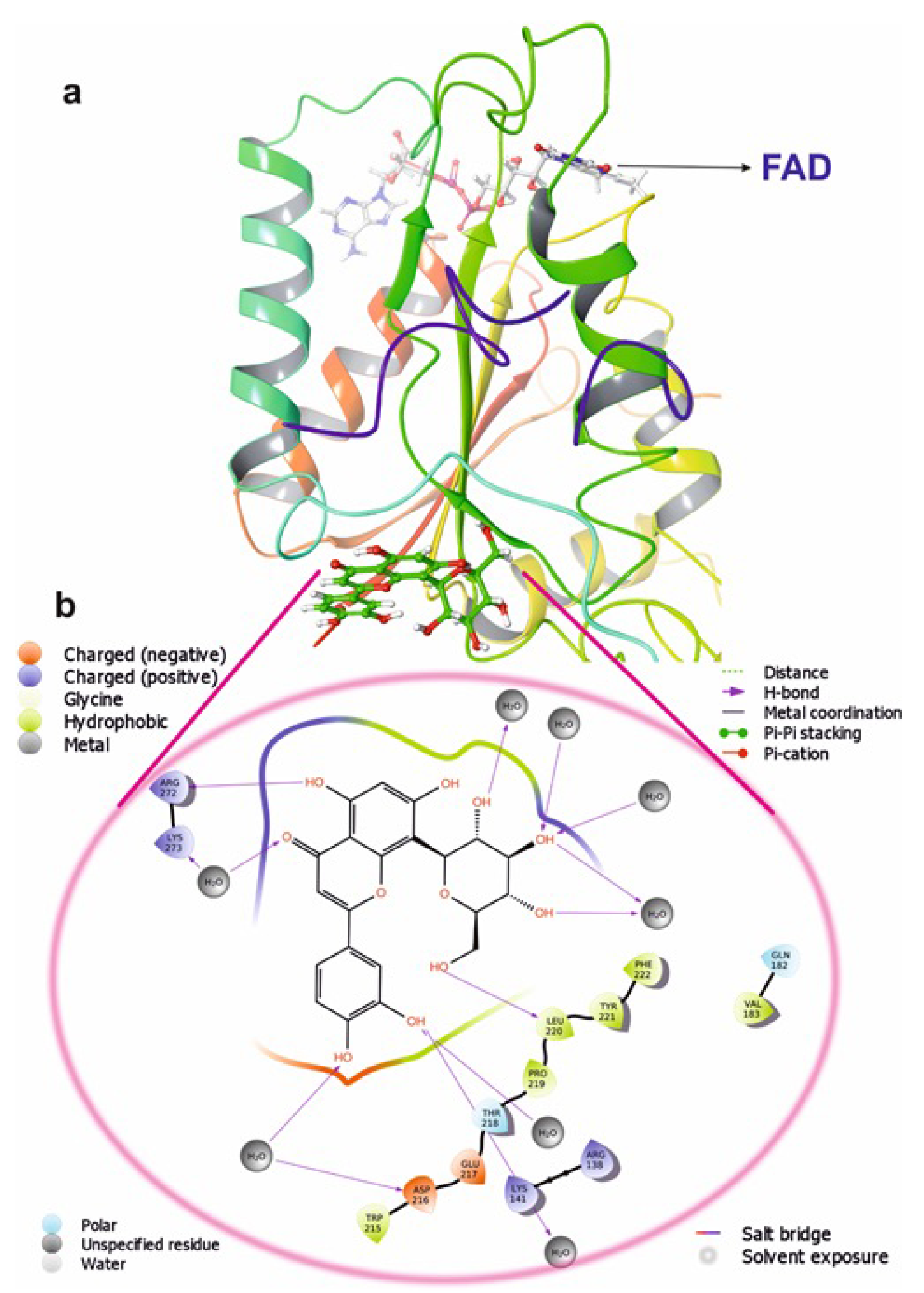
3.2. Protein-Ligand Interaction
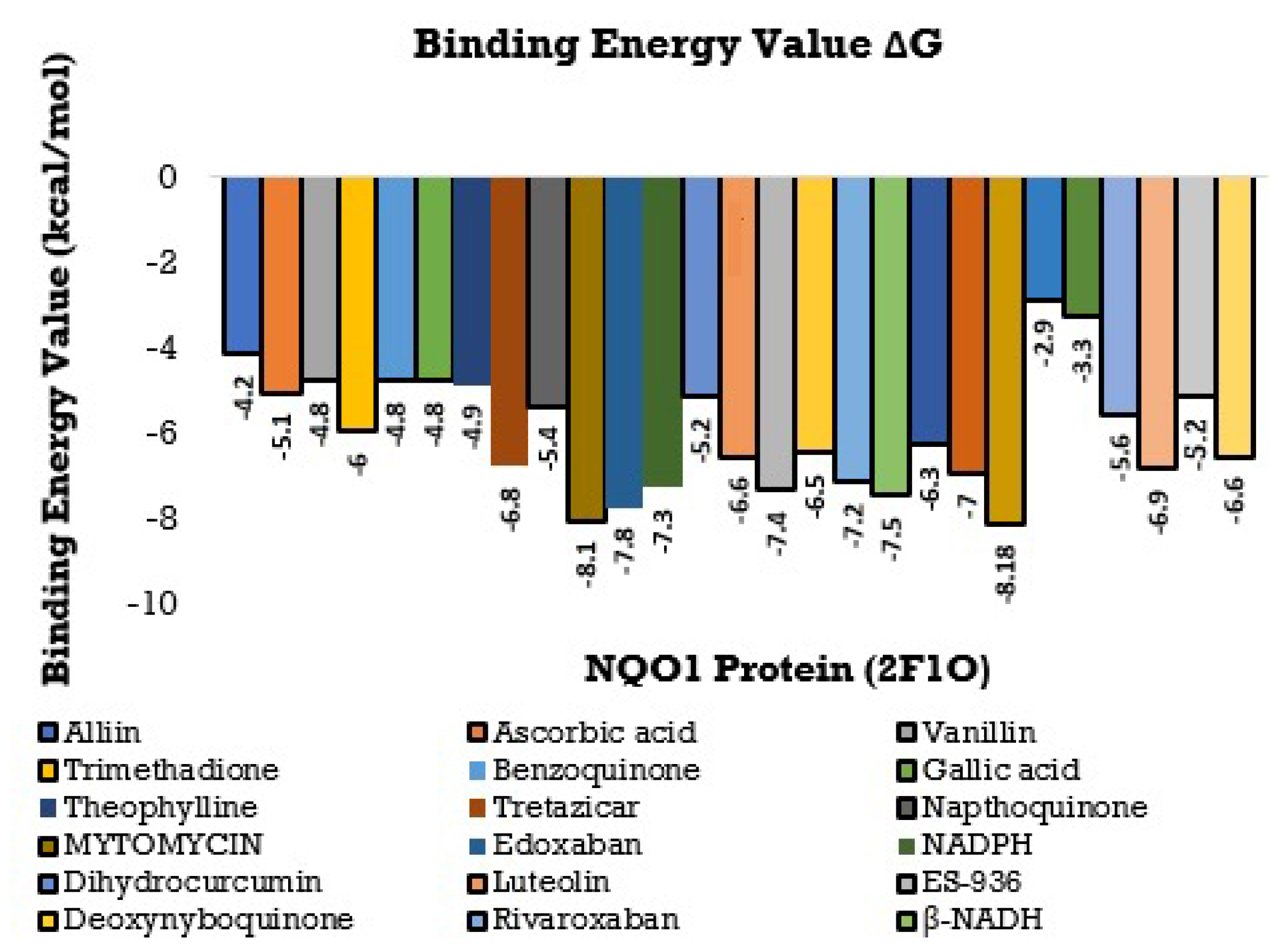
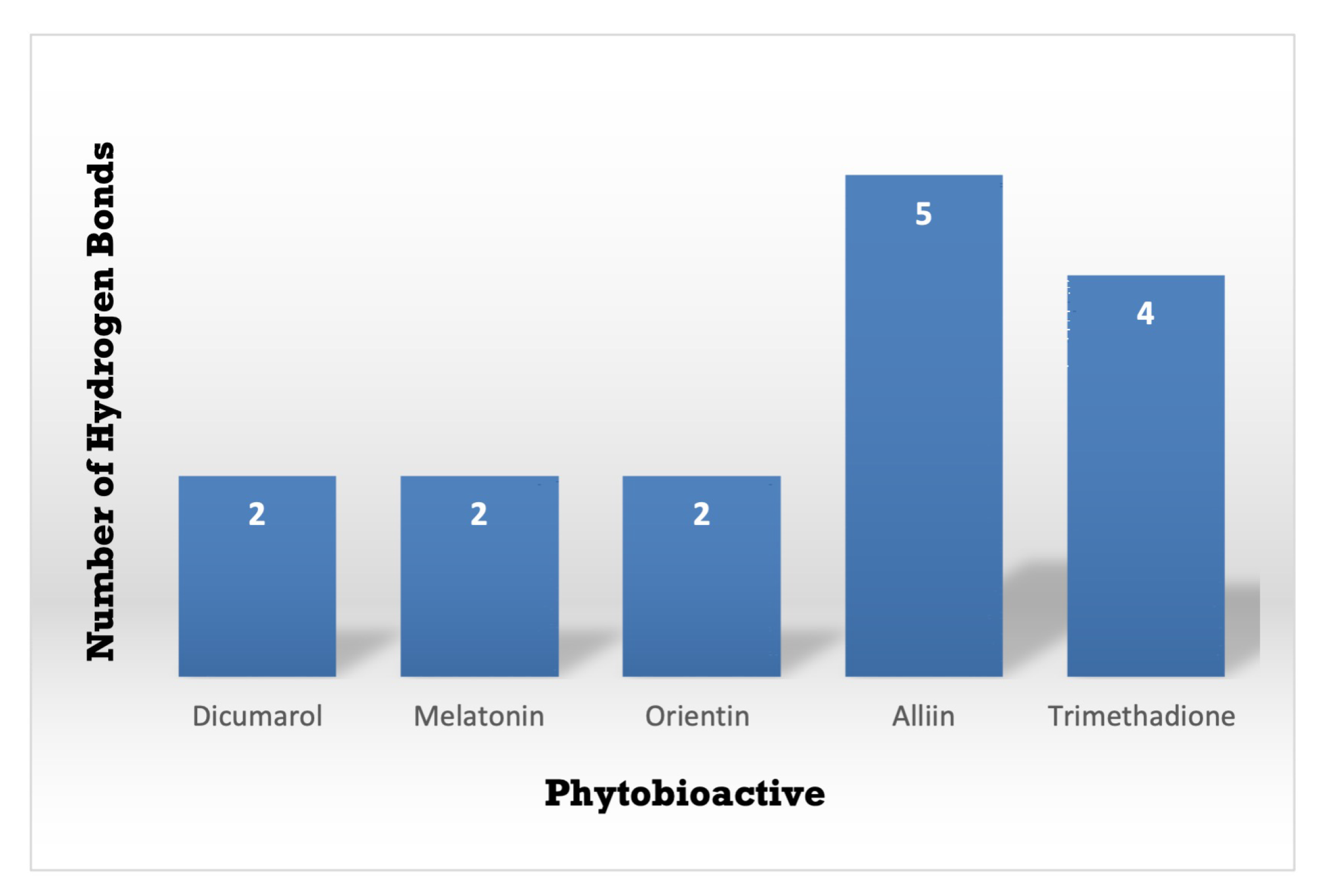
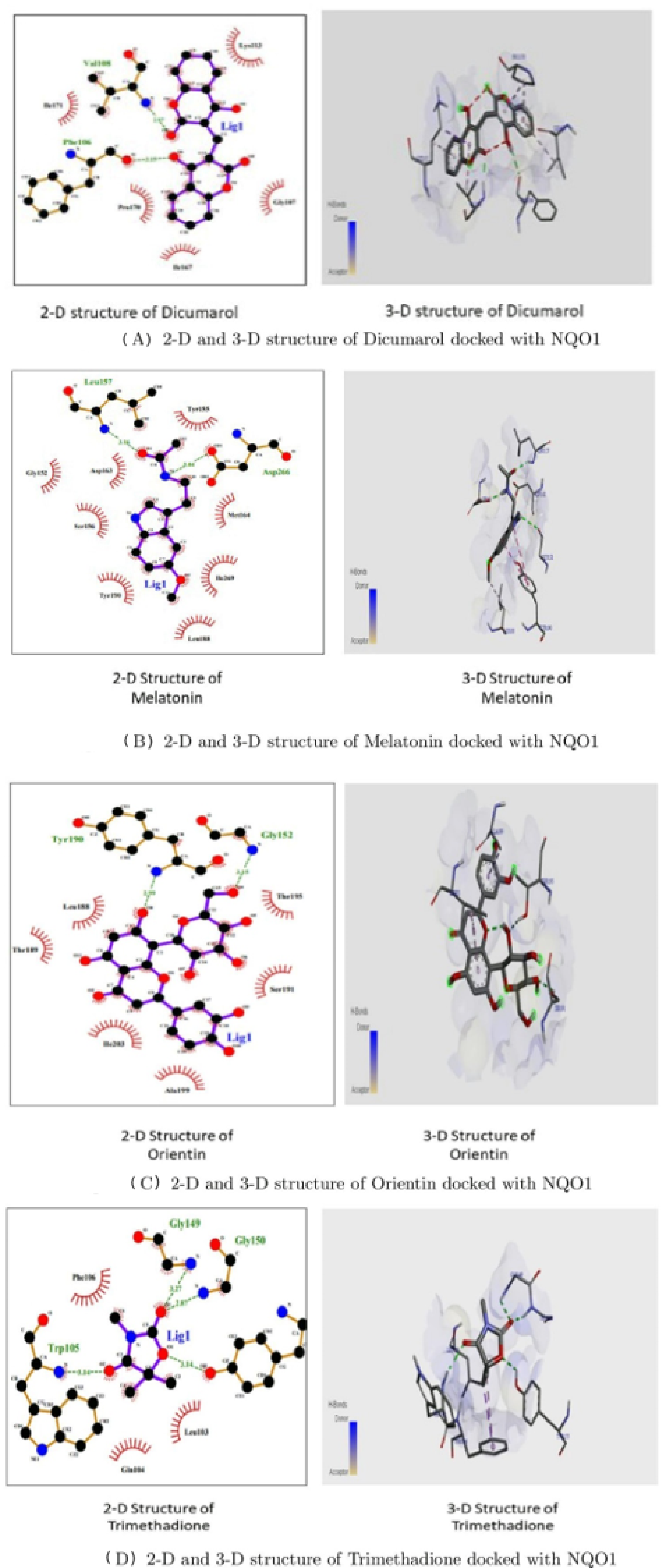
3.3. Molecular Docking Interactions of 2F1O Protein with Selected Phytobioactives
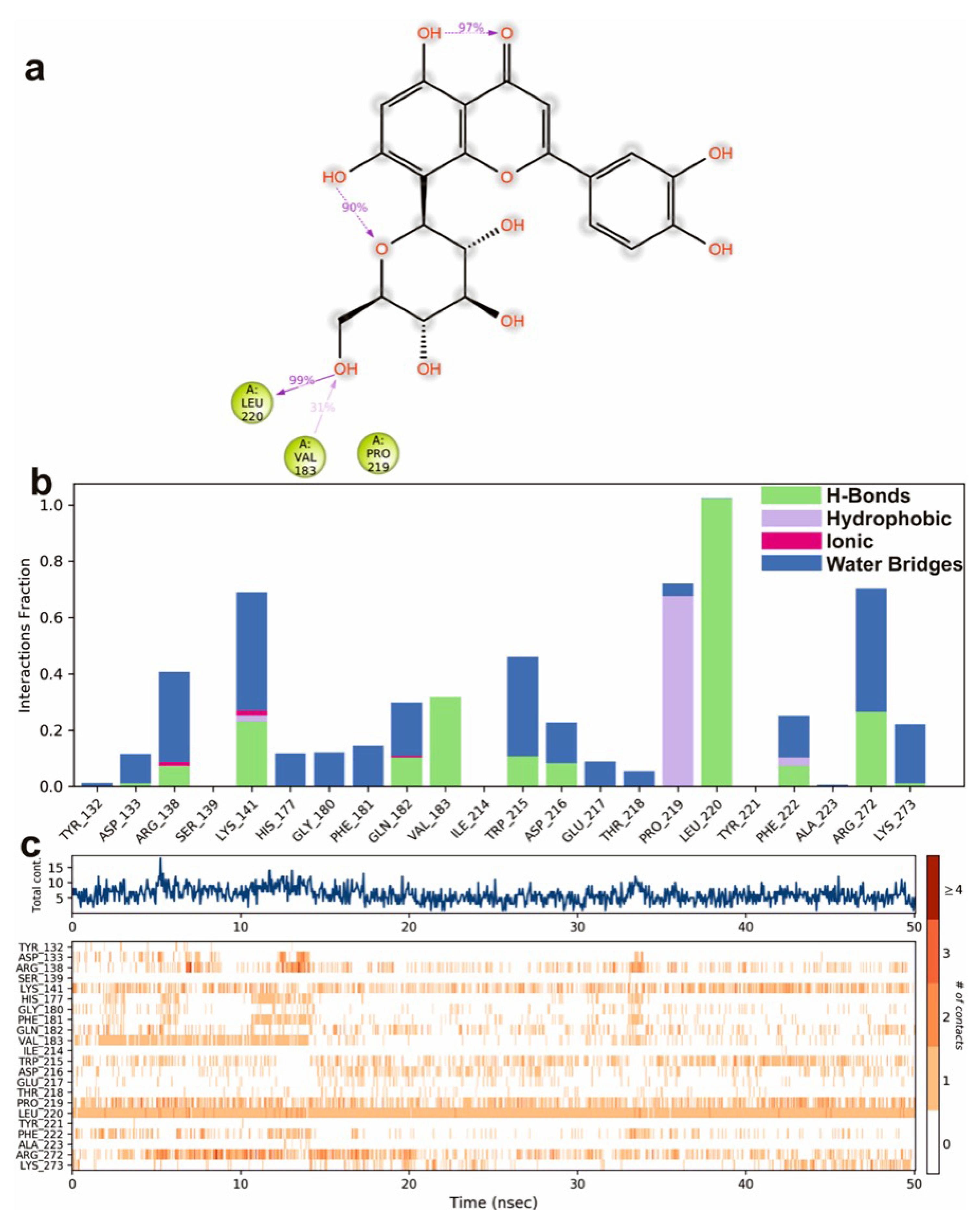
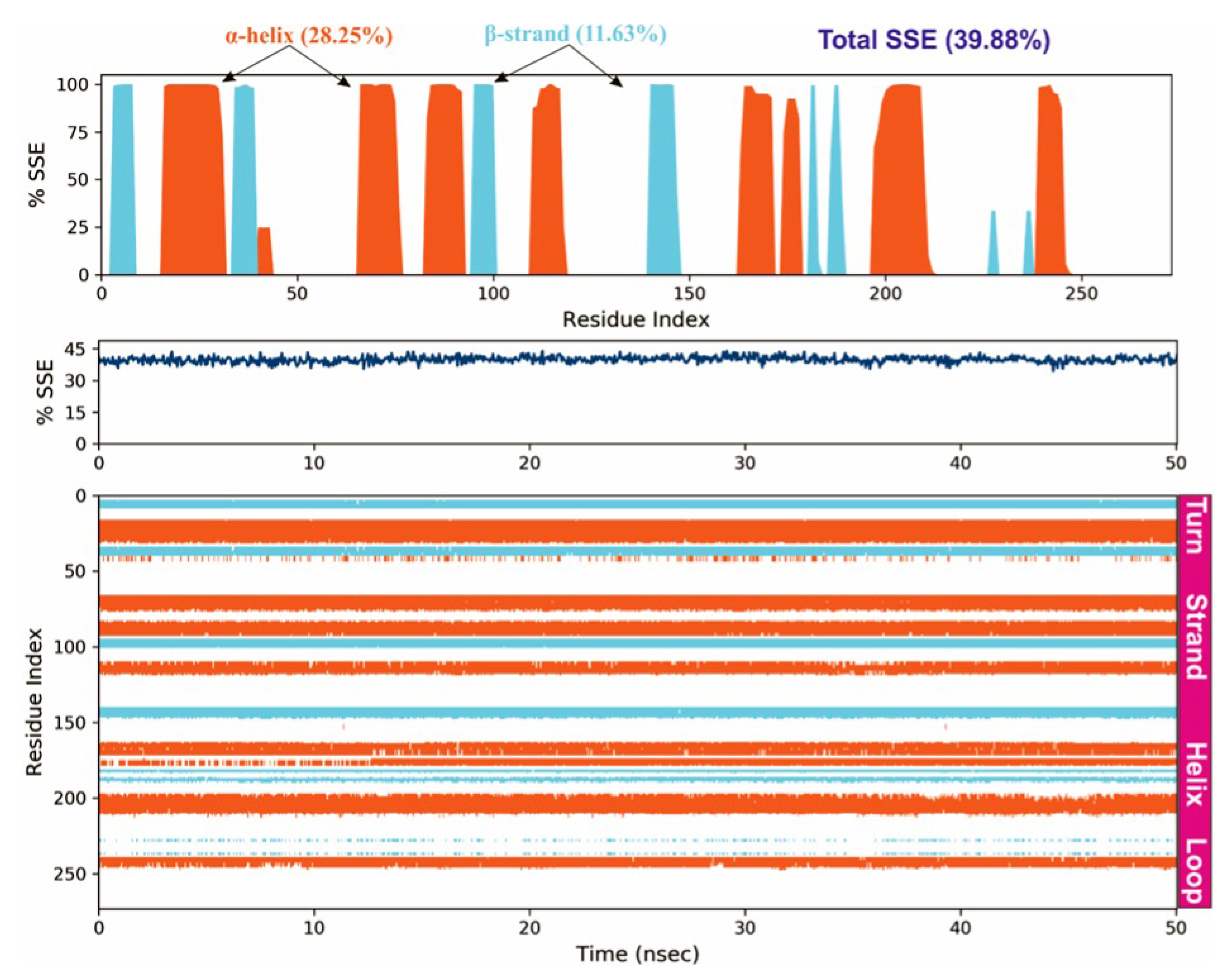
3.4. Molecular Dynamics Simulations
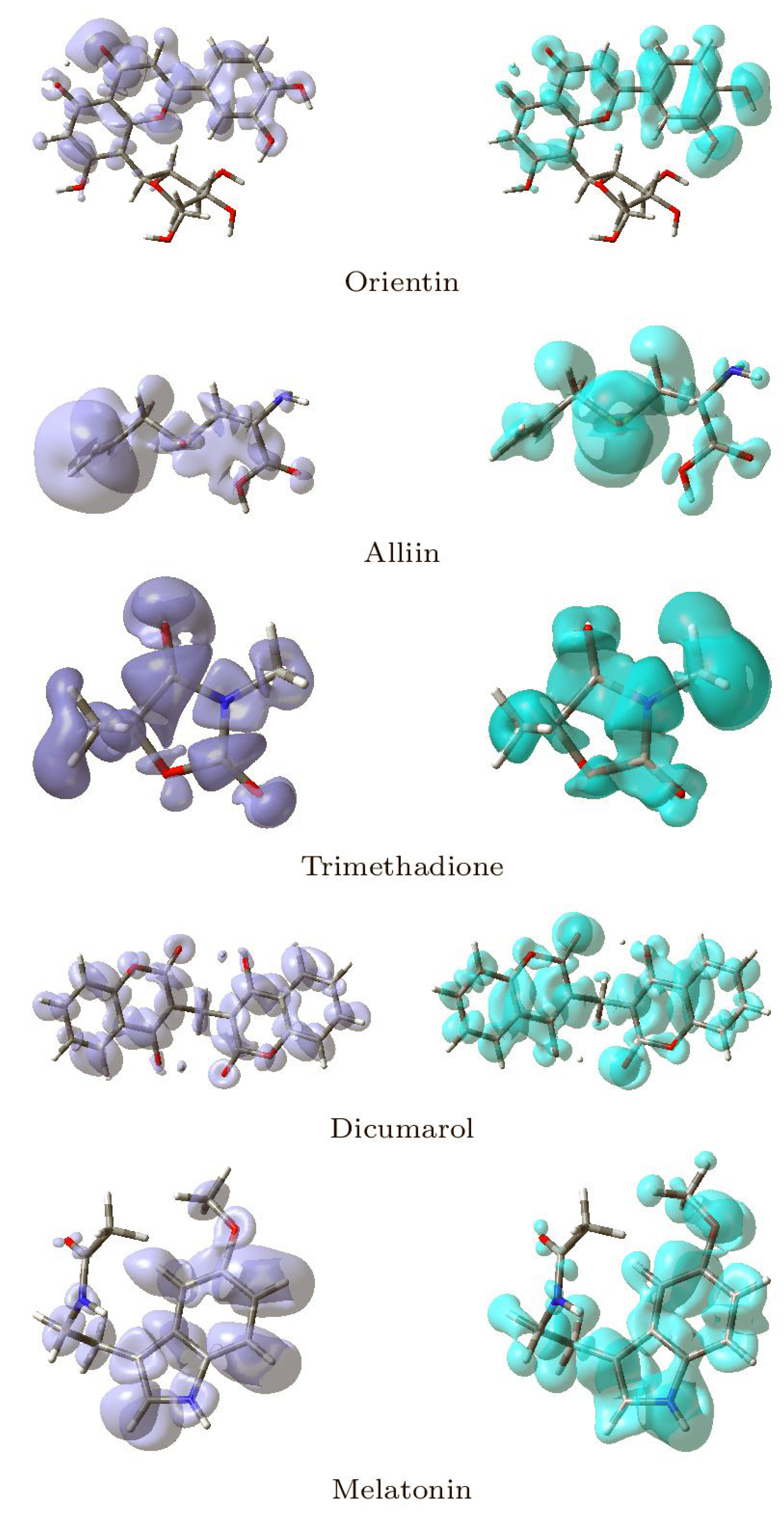
3.5. Conceptual DFT Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dinkova-Kostova, A.T.; Talalay, P. NAD(P)H:quinone Acceptor Oxidoreductase 1 (NQO1), a Multifunctional Antioxidant Enzyme and Exceptionally Versatile Cytoprotector. Arch. Biochem. Biophys. 2010, 501, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Ross, D.; Siegel, D. Functions of NQO1 in Cellular Protection and CoQ10 Metabolism and its Potential Role as a Redox Sensitive Molecular Switch. Front. Physiol. 2017, 8, 595. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.T.; Park, H.J. Implications of NQO1 in Cancer Therapy. BMB Rep. 2015, 48, 609–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asher, G.; Dym, O.; Tsvetkov, P.; Adler, J.; Shaul, Y. The Crystal Structure of NAD(P)H Quinone Oxidoreductase 1 in Complex with Its Potent Inhibitor Dicoumarol. Biochemistry 2006, 45, 6372–6378. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.; Ross, D. Immunodetection of NAD(P)H:quinone Oxidoreductase 1 (NQO1) in Human Tissues. Free Radic. Biol. Med. 2000, 29, 246–253. [Google Scholar] [CrossRef]
- Siegel, D.; Franklin, W.A.; Ross, D. Immunohistochemical Detection of NAD(P)H:quinone Oxidoreductase in Human Lung and Lung Tumors. Clin. Cancer Res. 1998, 4, 2065–2070. [Google Scholar] [PubMed]
- Kaspar, J.W.; Jaiswal, A.K. Antioxidant-induced Phosphorylation of Tyrosine 486 Leads to Rapid Nuclear Export of Bach1 That Allows Nrf2 to Bind to the Antioxidant Response Element and Activate Defensive Gene Expression. J. Biol. Chem. 2010, 285, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Li, Y. NAD(P)H:Quinone Oxidoreductase 1 and its Potential Protective Role in Cardiovascular Diseases and Related Conditions. Cardiovasc. Toxicol. 2011, 12, 39–45. [Google Scholar] [CrossRef]
- di Pietro, A.; Koster, R.; van Eck, W.B.; Dam, W.A.; Mulder, N.H.; Gietema, J.A.; de Vries, E.G.; de Jong, S. Pro- and Anti-Apoptotic Effects of p53 in Cisplatin-treated Human Testicular Cancer are Cell Context-Dependent. Cell Cycle 2012, 11, 4552–4562. [Google Scholar] [CrossRef] [Green Version]
- Patrick, B.A.; Gong, X.; Jaiswal, A.K. Retraction Note: Disruption of NAD(P)H:quinone Oxidoreductase 1 Gene in Mice Leads to 20S Proteosomal Degradation of p63 Resulting in Thinning of Epithelium and Chemical-induced Skin Cancer. Oncogene 2017, 36, 2919. [Google Scholar] [CrossRef] [Green Version]
- Srijiwangsa, P.; Bangchang, K.N. Roles of NAD (P) H-Quinone Oxidoreductase 1 (NQO1) On Cancer Progression and Chemoresistance. J. Clin. Exp. Oncol. 2017, 6, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Siegel, D.; Yan, C.; Ross, D. NAD(P)H:quinone oxidoreductase 1 (NQO1) in the Sensitivity and Resistance to Antitumor Quinones. Biochem. Pharmacol. 2012, 83, 1033–1040. [Google Scholar] [CrossRef] [Green Version]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sánchez-Rivera, F.J.; et al. Keap1 Loss Promotes Kras-driven Lung Cancer and Results in Dependence on Glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Chen, D.; Ma, K.; Wu, X.; Hao, H.; Jiang, S. NAD(P)H:Quinone Oxidoreductase 1 (NQO1) as a Therapeutic and Diagnostic Target in Cancer. J. Med. Chem. 2018, 61, 6983–7003. [Google Scholar] [CrossRef]
- Hamed, A.R.; Hegazy, M.E.F.; Higgins, M.; Mohamed, T.A.; Abdel-Azim, N.S.; Pare, P.W.; Dinkova-Kostova, A.T. Potency of Extracts from Selected Egyptian Plants as Inducers of the Nrf2-dependent Chemopreventive Enzyme NQO1. J. Nat. Med. 2016, 70, 683–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatem, N.; Lara, H.W.; Safaa, B.; Nabil, N.; Nelly, A.A. Antimicrobial Activity Of Essential Oil From Achillea Fragrantissima (Forssk.) Sch.Bip.(Asteraceae) Growing Wild In North Bekaa, Lebanon. Int. J. Eng. Sci. Res. Technol. 2018, 7, 115–124. [Google Scholar] [CrossRef]
- Panossian, A.; Wikman, G.; Wagner, H. Plant Adaptogens III. Earlier and More Recent Aspects and Concepts on their Mode of Action. Phytomedicine 1999, 6, 287–300. [Google Scholar] [CrossRef]
- Brekhman, I.I.; Dardymov, I.V. New Substances of Plant Origin which Increase Nonspecific Resistance. Annu. Rev. Pharmacol. 1969, 9, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, A.A.; Gottlieb, M.S.; Scholes, V.E. Reconstitution of Immune Function in AIDS/ARC. Concepts Immunopathol. 1987, 4, 261–274. [Google Scholar]
- Colucci, M.A.; Reigan, P.; Siegel, D.; Chilloux, A.; Ross, D.; Moody, C.J. Synthesis and Evaluation of 3-Aryloxymethyl-1, 2-dimethylindole-4, 7-diones as Mechanism-Based Inhibitors of NAD(P)H:Quinone Oxidoreductase 1 (NQO1) Activity. J. Med. Chem. 2007, 50, 5780–5789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolan, K.A.; Zhao, H.; Faulder, P.F.; Frenkel, A.D.; Timson, D.J.; Siegel, D.; Ross, D.; Burke, T.R., Jr.; Stratford, I.J.; Bryce, R.A. Coumarin-Based Inhibitors of Human NAD(P)H:Quinone Oxidoreductase-1. Identification, Structure-Activity, Off-Target Effects and In Vitro Human Pancreatic Cancer Toxicity. J. Med. Chem. 2007, 50, 6316–6325. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Asher, G.; Reiss, V.; Shaul, Y.; Sachs, L.; Lotem, J. Inhibition of NAD(P)H:quinone Oxidoreductase 1 Activity and Induction of p53 Degradation by the Natural Phenolic Compound Curcumin. Proc. Natl. Acad. Sci. USA 2005, 102, 5535–5540. [Google Scholar] [CrossRef] [Green Version]
- Diwanay, S.; Chitre, D.; Patwardhan, B. Immunoprotection by Botanical Drugs in Cancer Chemotherapy. J. Ethnopharmacol. 2004, 90, 49–55. [Google Scholar] [CrossRef]
- Watanabe, J.; Nishiyama, H.; Matsui, Y.; Ito, M.; Kawanishi, H.; Kamoto, T.; Ogawa, O. Dicoumarol Potentiates Cisplatin-induced Apoptosis Mediated by c-Jun N-terminal Kinase in p53 Wild-type Urogenital Cancer Cell Lines. Oncogene 2006, 25, 2500–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehn, D.L.; Siegel, D.; Swann, E.; Moody, C.J.; Ross, D. Biochemical, Cytotoxic, and Genotoxic Effects of ES936, a Mechanism-Based Inhibitor of NAD(P)H:quinone Oxidoreductase 1, in Cellular Systems. Mol. Pharmacol. 2003, 64, 714–720. [Google Scholar] [CrossRef]
- López-Lira, C.; Alzate-Morales, J.H.; Paulino, M.; Mella-Raipán, J.; Salas, C.O.; Tapia, R.A.; Soto-Delgado, J. Combined Molecular Modelling and 3D-QSAR Study for Understanding the Inhibition of NQO1 by Heterocyclic Quinone Derivatives. Chem. Biol. Drug Des. 2017, 91, 29–38. [Google Scholar] [CrossRef]
- Megarity, C.F.; Bettley, H.A.A.; Caraher, M.C.; Scott, K.A.; Whitehead, R.C.; Jowitt, T.A.; Gutierrez, A.; Bryce, R.A.; Nolan, K.A.; Stratford, I.J.; et al. Negative Cooperativity in NAD(P)H Quinone Oxidoreductase 1 (NQO1). ChemBioChem 2019, 20, 2841–2849. [Google Scholar] [CrossRef] [PubMed]
- Pey, A.L.; Megarity, C.F.; Timson, D.J. NAD(P)H Quinone Oxidoreductase (NQO1): An Enzyme Which Needs Just Enough Mobility, in Just the Right Places. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.Y.; Chew, E.H.; Go, M.L. Functionalized Aurones as Inducers of NAD(P)H:Quinone Oxidoreductase 1 that Activate AhR/XRE and Nrf2/ARE Signaling Pathways: Synthesis, Evaluation and SAR. Eur. J. Med. Chem. 2010, 45, 2957–2971. [Google Scholar] [CrossRef]
- Kumar, D.T.; Susmita, B.; Judith, E.; Christy, J.P.; Doss, C.G.P.; Zayed, H. Elucidating the Role of Interacting Residues of the MSH2-MSH6 Complex in DNA Repair Mechanism: A Computational Approach. In Advances in Protein Chemistry and Structural Biology—DNA Repair; Donev, R., Ed.; Academic Press: Cambridge, MA, USA, 2019; Chapter 10; pp. 325–350. [Google Scholar] [CrossRef]
- Torktaz, I.; Najafi, A.; Golmohamadi, R.; Hassani, S. Molecular Dynamics Simulation (MDS) Analysis of Vibrio Cholerae ToxT Virulence Factor Complexed Docked Potential Inhib. Bioinformation 2018, 14, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Aamir, M.; Singh, V.K.; Dubey, M.K.; Meena, M.; Kashyap, S.P.; Katari, S.K.; Upadhyay, R.S.; Umamaheswari, A.; Singh, S. In silico Prediction, Characterization, Molecular Docking, and Dynamic Studies on Fungal SDRs as Novel Targets for Searching Potential Fungicides Against Fusarium Wilt in Tomato. Front. Pharmacol. 2018, 9, 1038. [Google Scholar] [CrossRef] [PubMed]
- Lewars, E. Computational Chemistry—Introduction to the Theory and Applications of Molecular and Quantum Mechanics; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2003. [Google Scholar]
- Young, D. Computational Chemistry—A Practical Guide for Applying Techniques to Real-World Problems; John Wiley & Sons: New York, NY, USA, 2001. [Google Scholar]
- Jensen, F. Introduction to Computational Chemistry, 2nd ed.; John Wiley & Sons: Chichester, UK, 2007. [Google Scholar]
- Cramer, C. Essentials of Computational Chemistry—Theories and Models, 2nd ed.; John Wiley & Sons: Chichester, UK, 2004. [Google Scholar]
- Parr, R.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Chermette, H. Chemical Reactivity Indexes in Density Functional Theory. J. Comput. Chem. 1999, 20, 129–154. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1873. [Google Scholar] [CrossRef]
- Geerlings, P.; Chamorro, E.; Chattaraj, P.K.; Proft, F.D.; Gázquez, J.L.; Liu, S.; Morell, C.; Toro-Labbé, A.; Vela, A.; Ayers, P. Conceptual Density Functional Theory: Status, Prospects, Issues. Theor. Chem. Acc. 2020, 139, 36. [Google Scholar] [CrossRef]
- Toro-Labbé, A. (Ed.) Theoretical Aspects of Chemical Reactivity; Elsevier Science: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Chattaraj, P.K. (Ed.) Chemical Reactivity Theory—A Density Functional View; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2009. [Google Scholar]
- Chakraborty, D.; Chattaraj, P.K. Conceptual Density Functional Theory Based Electronic Structure Principles. Chem. Sci. 2021, 12, 6264–6279. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck Molecular Force Field. I. Basis, Form, Scope, Parameterization, and Performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck Molecular Force Field. II. MMFF94 van der Waals and Electrostatic Parameters for Intermolecular Interactions. J. Comput. Chem. 1996, 17, 520–552. [Google Scholar] [CrossRef]
- Halgren, T.A. MMFF VI. MMFF94s Option for Energy Minimization Studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Halgren, T.A.; Nachbar, R.B. Merck Molecular Force Field. IV. Conformational Energies and Geometries for MMFF94. J. Comput. Chem. 1996, 17, 587–615. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck Molecular Force field. V. Extension of MMFF94 Using Experimental Data, Additional Computational Data, and Empirical Rules. J. Comput. Chem. 1996, 17, 616–641. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Peverati, R.; Truhlar, D.G. Screened-Exchange Density Functionals with Broad Accuracy for Chemistry and Solid-State Physics. Phys. Chem. Chem. Phys. 2012, 14, 16187–16191. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting Basis Sets for H to R. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Computational Peptidology Assisted by Conceptual Density Functional Theory for the Study of Five New Antifungal Tripeptides. ACS Omega 2019, 4, 12555–12560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Conceptual DFT as a Helpful Chemoinformatics Tool for the Study of the Clavanin Family of Antimicrobial Marine Peptides. In Density Functional Theory Calculations; IntechOpen: Rijetia, Croatia, 2019; pp. 1–11. [Google Scholar]
- Frau, J.; Glossman-Mitnik, D. Molecular Reactivity and Absorption Properties of Melanoidin Blue-G1 through Conceptual DFT. Molecules 2018, 23, 559. [Google Scholar] [CrossRef] [Green Version]
- Frau, J.; Glossman-Mitnik, D. Conceptual DFT Study of the Local Chemical Reactivity of the Dilysyldipyrrolones A and B Intermediate Melanoidins. Theor. Chem. Acc. 2018, 137, 1210. [Google Scholar] [CrossRef]
- Frau, J.; Glossman-Mitnik, D. Conceptual DFT Study of the Local Chemical Reactivity of the Colored BISARG Melanoidin and Its Protonated Derivative. Front. Chem. 2018, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Frau, J.; Glossman-Mitnik, D. Computational Study of the Chemical Reactivity of the Blue-M1 Intermediate Melanoidin. Comput. Theor. Chem. 2018, 1134, 22–29. [Google Scholar] [CrossRef]
- Frau, J.; Glossman-Mitnik, D. Chemical Reactivity Theory Applied to the Calculation of the Local Reactivity Descriptors of a Colored Maillard Reaction Product. Chem. Sci. Int. J. 2018, 22, 1–14. [Google Scholar] [CrossRef]
- Frau, J.; Glossman-Mitnik, D. Blue M2: An Intermediate Melanoidin Studied via Conceptual DFT. J. Mol. Model. 2018, 24, 1–13. [Google Scholar] [CrossRef]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Chemical-Reactivity Properties, Drug Likeness, and Bioactivity Scores of Seragamides A–F Anticancer Marine Peptides: Conceptual Density Functional Theory Viewpoint. Computation 2019, 7, 52. [Google Scholar] [CrossRef] [Green Version]
- Frau, J.; Flores-Holguín, N.; Glossman-Mitnik, D. Chemical Reactivity Theory and Empirical Bioactivity Scores as Computational Peptidology Alternative Tools for the Study of Two Anticancer Peptides of Marine Origin. Molecules 2019, 24, 1115. [Google Scholar] [CrossRef] [Green Version]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Computational Prediction of Bioactivity Scores and Chemical Reactivity Properties of the Parasin I Therapeutic Peptide of Marine Origin Through the Calculation of Global and Local Conceptual DFT Descriptors. Theor. Chem. Acc. 2019, 138, 78. [Google Scholar] [CrossRef]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. A Fast and Simple Evaluation of the Chemical Reactivity Properties of the Pristinamycin Family of Antimicrobial Peptides. Chem. Phys. Lett. 2020, 739, 137021. [Google Scholar] [CrossRef]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Conceptual DFT-Based Computational Peptidology of Marine Natural Compounds: Discodermins A–H. Molecules 2020, 25, 4158. [Google Scholar] [CrossRef]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Virtual Screening of Marine Natural Compounds by Means of Chemoinformatics and CDFT-Based Computational Peptidology. Mar. Drugs 2020, 18, 478. [Google Scholar] [CrossRef]
- Flores-Holguín, N.; Frau, J.; Glossman-Mitnik, D. Conceptual DFT as a Helpful Chemoinformatics Tool for the Study of the Clavanin Family of Antimicrobial Marine Peptides. In Density Functional Theory; De Lazaro, S.R., Da Silveira Lacerda, L.H., Pontes Ribeiro, R.A., Eds.; IntechOpen: London, UK, 2021; Chapter 3; pp. 57–67. [Google Scholar]
- Marenich, A.; Cramer, C.; Truhlar, D. Universal Solvation Model Based on Solute Electron Density and a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Dharmashekara, C.; Pradeep, S.; Prasad, S.K.; Jain, A.S.; Syed, A.; Prasad, K.S.; Patil, S.S.; Beelagi, M.S.; Srinivasa, C.; Shivamallu, C. Virtual Screening of Potential Phyto-Candidates as Therapeutic Leads Against SARS-CoV-2 Infection. Environ. Chall. 2021, 4, 100136. [Google Scholar] [CrossRef]
- Lei, K.; Gu, X.; Alvarado, A.G.; Du, Y.; Luo, S.; Ahn, E.H.; Kang, S.S.; Ji, B.; Liu, X.; Mao, H.; et al. Discovery of a Dual Inhibitor of NQO1 and GSTP1 for Treating Glioblastoma. J. Hematol. Oncol. 2020, 13. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [Green Version]
- Domingo, L.R.; Aurell, M.; Pérez, P.; Contreras, R. Quantitative Characterization of the Global Electrophilicity Power of Common diene/Dienophile Pairs in Diels-Alder Reactions. Tetrahedron 2002, 58, 4417–4423. [Google Scholar] [CrossRef]
- Domingo, L.R.; Sáez, J.A. Understanding the Mechanism of Polar Diels-Alder Reactions. Org. Biomol. Chem. 2009, 7, 3576–3583. [Google Scholar] [CrossRef]
- Pérez, P.; Domingo, L.R.; Aurell, M.J.; Contreras, R. Quantitative Characterization of the Global Electrophilicity Pattern of Some Reagents Involved in 1,3-Dipolar Cycloaddition Reactions. Tetrahedron 2003, 59, 3117–3125. [Google Scholar] [CrossRef]
- Morell, C.; Grand, A.; Toro-Labbé, A. New Dual Descriptor for Chemical Reactivity. J. Phys. Chem. A 2005, 109, 205–212. [Google Scholar] [CrossRef]
- Morell, C.; Grand, A.; Toro-Labbé, A. Theoretical Support for Using the Δf(r) Descriptor. Chem. Phys. Lett. 2006, 425, 342–346. [Google Scholar] [CrossRef]
- Martínez-Araya, J.I. Revisiting Caffeate’s Capabilities as a Complexation Agent to Silver Cation in Mining Processes by means of the Dual Descriptor – A Conceptual DFT Approach. J. Mol. Model. 2012, 18, 4299–4307. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Araya, J.I. Explaining Reaction Mechanisms Using the Dual Descriptor: A Complementary Tool to the Molecular Electrostatic Potential. J. Mol. Model. 2012, 19, 2715–2722. [Google Scholar] [CrossRef]
- Martínez-Araya, J.I. Why is the Dual Descriptor a More Accurate Local Reactivity Descriptor than Fukui Functions? J. Math. Chem. 2015, 53, 451–465. [Google Scholar] [CrossRef]
- Lam, K.Y.; Ling, A.P.K.; Koh, R.Y.; Wong, Y.P.; Say, Y.H. A Review on Medicinal Properties of Orientin. Adv. Pharmacol. Sci. 2016, 2016, 1–9. [Google Scholar] [CrossRef] [Green Version]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SI | CID | Compound Name | SI | CID | Compound Name | SI | CID | Compound Name |
|---|---|---|---|---|---|---|---|---|
| 1 | 637563 | Anethol | 21 | 460 | Guaiacol | 41 | 442882 | Justicidin B |
| 2 | 160512 | Ar-Turmerone | 22 | 6549 | Linalool | 42 | 637520 | Methyl Cinnamate |
| 3 | 9793905 | Sallylcysteine | 23 | 8815 | Estragol | 43 | 5280343 | Quercetin |
| 4 | 11128 | Linamarin | 24 | 10281 | Thymoquinone | 44 | 5280346 | Ubiquinone |
| 5 | 20657680 | S-allylmercaptocystein | 25 | 11617 | Dyallyl Sulfide | 45 | 5280445 | Luteolin |
| 6 | 10494 | Oleanolic acid | 26 | 29746 | Geosmin | 46 | 5280489 | -Carotene |
| 7 | 64945 | Ursolic acid | 27 | 6654 | -Pinene | 47 | 5281672 | Myricetin |
| 8 | 5315615 | Rosemarinic acid | 28 | 7794 | Citronellol | 48 | 5281675 | Orientin |
| 9 | 370 | Gallic acid | 29 | 65036 | Allicin | 49 | 5315472 | Bisdemethoxycurcumin |
| 10 | 10364 | Carvacrol | 30 | 70308 | 2,2-Dimethylpropiophenone | 50 | 5386591 | Ajoene |
| 11 | 5281515 | -Caryophyllene | 31 | 87014 | Anthraquinone | 51 | 7794 | Citronellol |
| 12 | 2537 | Camphor | 32 | 87310 | Alliin | 52 | 1042933 | Dihydrocurcumin |
| 13 | 2519 | Caffeine | 33 | 92139 | -Curcumin | 53 | 1125088 | NADH |
| 14 | 2758 | Eucalyptol | 34 | 323 | Coumarin | 54 | 12302243 | -Calacorene |
| 15 | 3314 | Eugenol | 35 | 442793 | Gingerols | 55 | 17753965 | Benzopyrone |
| 16 | 3885 | -Lapachone | 36 | 160512 | Ar-Turmerone | 56 | 54670067 | L-Ascorbic acid |
| 17 | 5281166 | Jasmonic acid | 37 | 167812 | Curcumenol | 57 | 643779 | Neral |
| 18 | 5429 | Theobromine | 38 | 196216 | -Turmerone | 58 | 54736423 | Dimethyl-3-naphtalene |
| 19 | 5746 | Mitomycin | 39 | 442402 | Thujopsene | 59 | 31211 | Zingerone |
| 20 | 6616 | Camphene | 40 | 86895 | Cuparene | 60 | 117587706 | Dihydronaphtalene |
| SI No | Name of | Taxonomical | Chemical | Name of the Phytobioactive | Molecular Weight | Chemical |
|---|---|---|---|---|---|---|
| the Plant | Name | Formula | Components | (g/mol) | Class | |
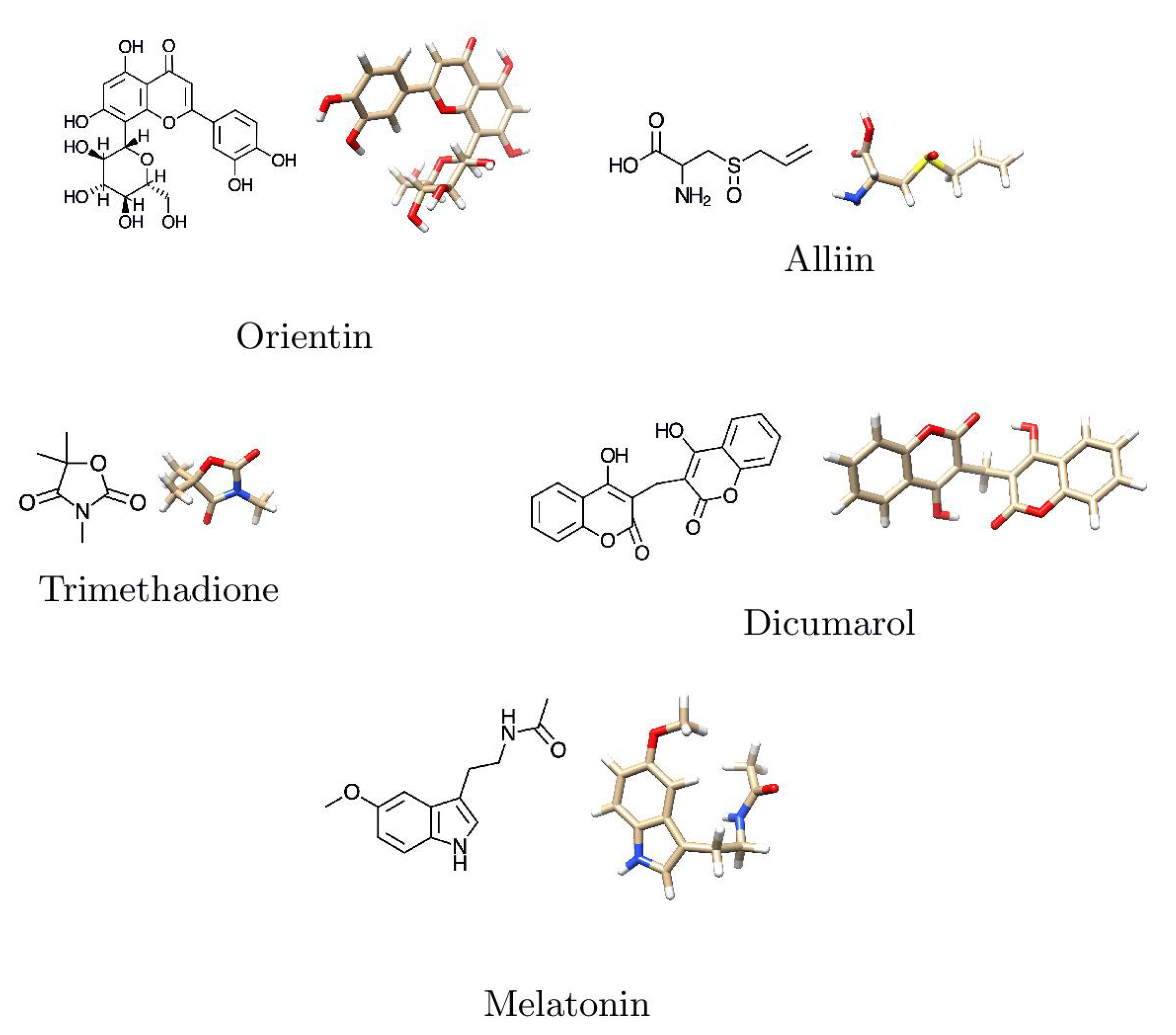
| A | Holy Tulsi | Ocinum tenuiflorum | C21H20O11 | Orientin | 448.40 | Flavone |
| B | Garlic | Allium sativum | C6H10OS2 | Alliin | 177.22 | Tioester |
| C | Punarnava | Boerhavia diffusa | C6H9NO3 | Trimethadione | 143.14 | Oxazolidinedione |
| SI No | Protein Structure | Number of Residues | Number of Residues | Number of Residues in |
|---|---|---|---|---|
| of NQO1 | in Favored Regions (%) | in Allowed Regions (%) | Disallowed Regions (%) | |
| 1 | 2F1O | 92.4 | 6.8 | 0.0 |
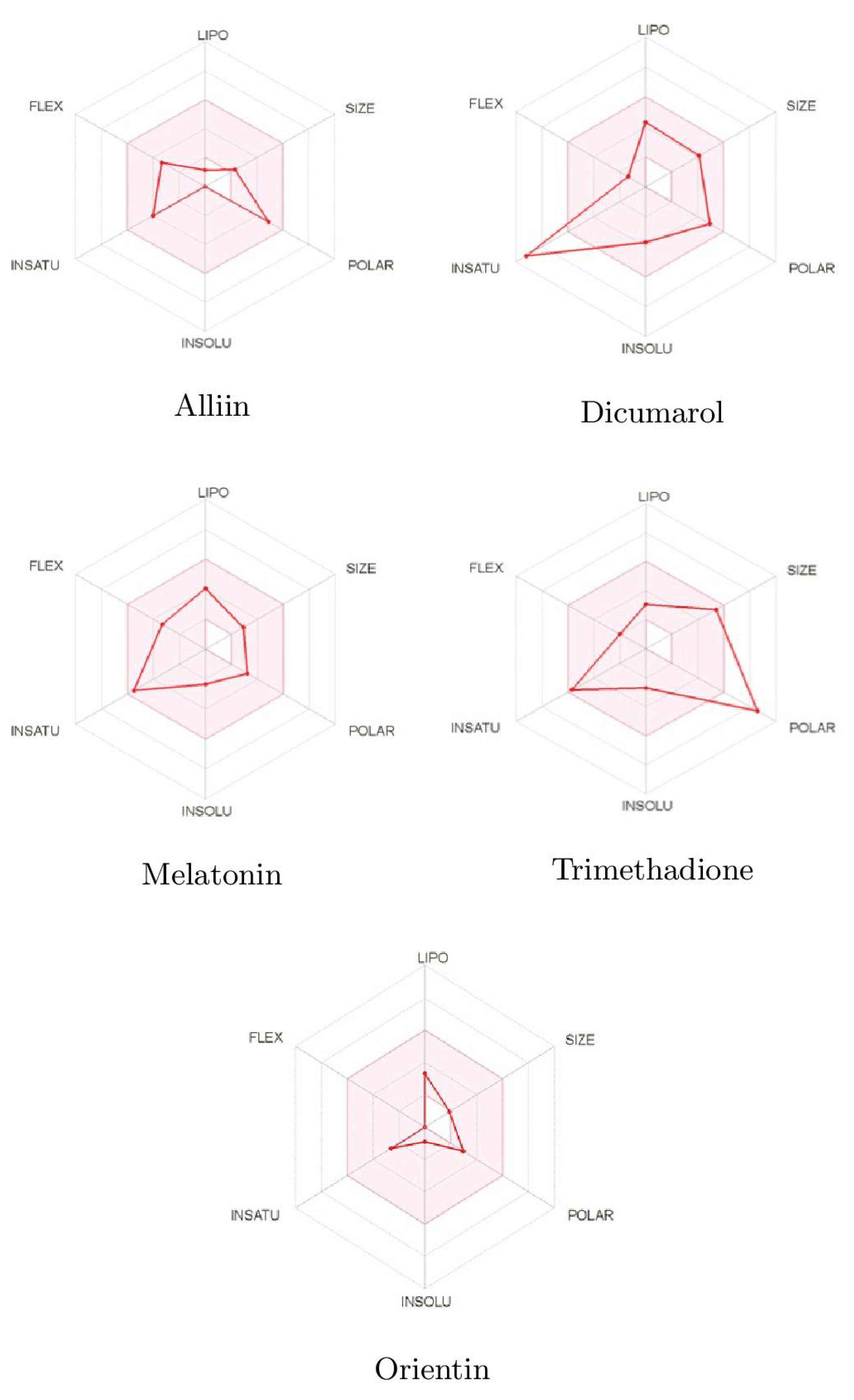
| ADMET Entry | Alliin | Dicumarol | Melatonin | Trimethadione | Orientin |
|---|---|---|---|---|---|
| Drug Likeness | |||||
| Lipinski | Yes | Yes | Yes | Yes | No |
| Bioavailability Score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 |
| Solubility | |||||
| Water Solubility | Yes | Yes | Yes | Yes | No |
| Absorption | |||||
| Intestinal Absorption | High | High | High | High | Low |
| Skin Permeability | −9.14 | 6.88 | 6.59 | −6.96 | −9.14 |
| P-glycoprotein Substrate | No | No | No | No | No |
| Distribution | |||||
| BBB Permeability | No | No | Yes | No | No |
| CYP1SA2 Inhibitor | No | Yes | Yes | No | No |
| CYP12C19 Inhibitor | No | No | No | No | No |
| CYP2C9 Inhibitor | No | No | No | No | No |
| CYP2D6 Inhibitor | No | No | No | No | No |
| CYP3A4 Inhibitor | No | No | No | No | No |
| Toxicity | |||||
| AMES Toxicity | No | No | No | No | No |
| Molecule | S | N | ||||||
|---|---|---|---|---|---|---|---|---|
| Orientin | 4.15 | 3.73 | 2.31 | 0.27 | 2.78 | 6.93 | 2.78 | 9.71 |
| Alliin | 3.44 | 5.96 | 0.99 | 0.17 | 2.37 | 4.09 | 0.64 | 4.73 |
| Trimethadione | 4.43 | 7.16 | 1.37 | 0.14 | 0.78 | 5.41 | 0.98 | 6.39 |
| Dicumarol | 4.32 | 4.26 | 2.19 | 0.23 | 2.34 | 6.81 | 2.49 | 9.29 |
| Melatonin | 3.23 | 4.71 | 1.11 | 0.21 | 3.21 | 4.12 | 0.89 | 5.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shreevatsa, B.; Dharmashekara, C.; Swamy, V.H.; Gowda, M.V.; Achar, R.R.; Kameshwar, V.H.; Thimmulappa, R.K.; Syed, A.; Elgorban, A.M.; Al-Rejaie, S.S.; et al. Virtual Screening for Potential Phytobioactives as Therapeutic Leads to Inhibit NQO1 for Selective Anticancer Therapy. Molecules 2021, 26, 6863. https://doi.org/10.3390/molecules26226863
Shreevatsa B, Dharmashekara C, Swamy VH, Gowda MV, Achar RR, Kameshwar VH, Thimmulappa RK, Syed A, Elgorban AM, Al-Rejaie SS, et al. Virtual Screening for Potential Phytobioactives as Therapeutic Leads to Inhibit NQO1 for Selective Anticancer Therapy. Molecules. 2021; 26(22):6863. https://doi.org/10.3390/molecules26226863
Chicago/Turabian StyleShreevatsa, Bhargav, Chandan Dharmashekara, Vikas Halasumane Swamy, Meghana V. Gowda, Raghu Ram Achar, Vivek Hamse Kameshwar, Rajesh Kumar Thimmulappa, Asad Syed, Abdallah M. Elgorban, Salim S. Al-Rejaie, and et al. 2021. "Virtual Screening for Potential Phytobioactives as Therapeutic Leads to Inhibit NQO1 for Selective Anticancer Therapy" Molecules 26, no. 22: 6863. https://doi.org/10.3390/molecules26226863