4.14. Preparation and Characterisation of Final Compounds and Intermediates
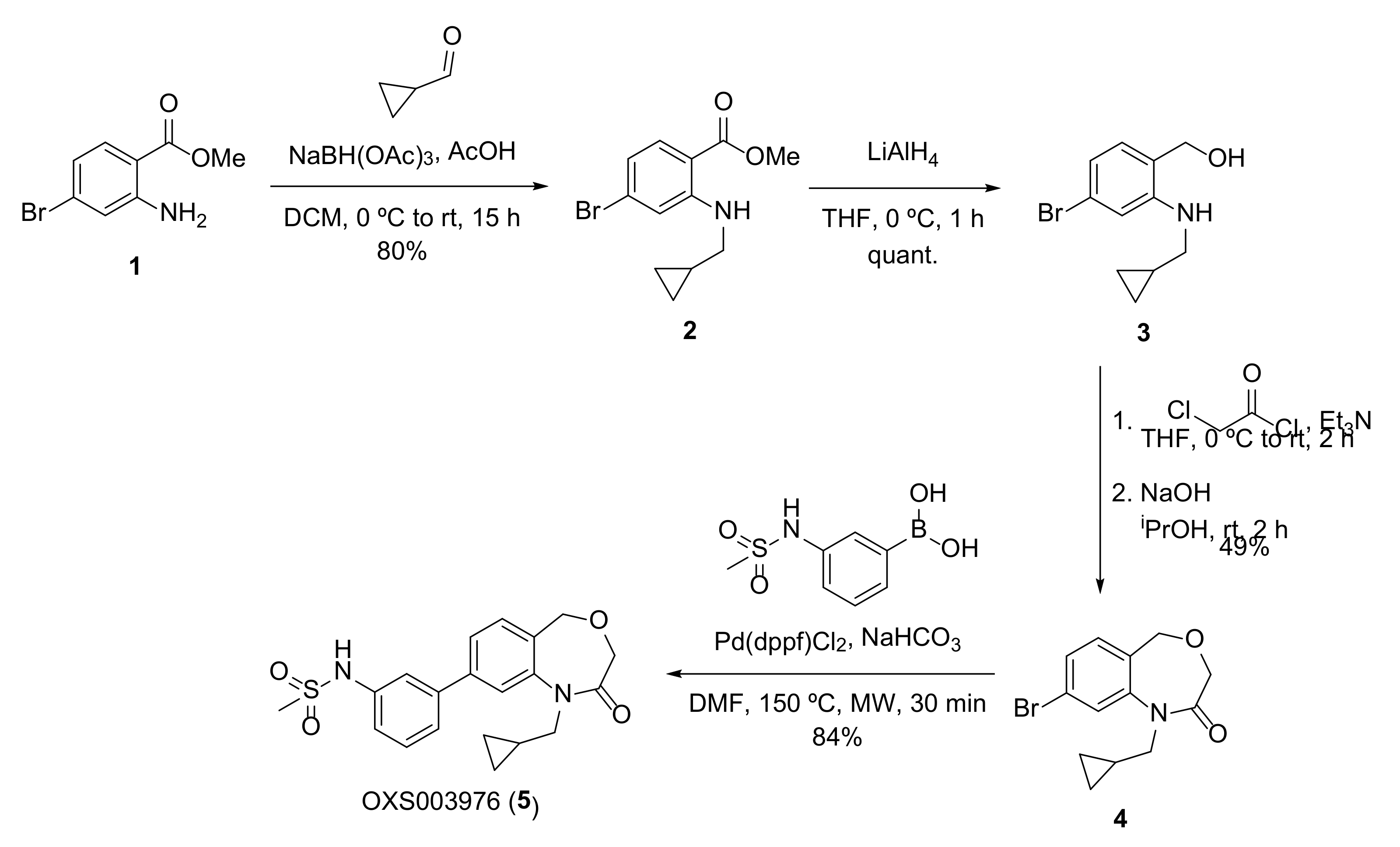
4.14.1. Methyl 4-bromo-2-((cyclopropylmethyl)amino)benzoate (2)
General procedure A; yield 80% (989 mg, 3.48 mmol); brown oil. 1H NMR (400 MHz, CDCl3) δ 7.86 (s, 1H), 7.73 (d, J = 8.5 Hz, 1H), 6.79 (d, J = 1.9 Hz, 1H), 6.68 (dd, J = 8.6, 1.8 Hz, 1H), 3.85 (s, 3H), 3.01 (d, J = 6.9 Hz, 2H), 1.19–1.09 (m, 1H), 0.65–0.56 (m, 2H), 0.28 (dt, J = 5.9, 4.6 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 169.1, 152.2, 133.3, 130.2, 118.0, 114.4, 109.1, 52.0, 48.3, 10.9, 4.1; m/z (ESI+) 284.0 ([M+H]+, 100%); HRMS (ESI+) C12H15NO2Br+ ([M+H]+) requires 284.0281; found 284.0282.
4.14.2. (4-Bromo-2-((cyclopropylmethyl)amino)phenyl)methanol (3)
General procedure B from 2 (950 mg, 3.43 mmol); yield quant. (863 mg, 3.37 mmol); light brown solid. 1H NMR (400 MHz, CDCl3) δ 6.88 (dd, J = 7.6, 1.9 Hz, 1H), 6.79–6.70 (m, 2H), 4.60 (s, 2H), 2.94 (dd, J = 6.9, 1.9 Hz, 2H), 1.20–1.04 (m, 1H), 0.62–0.53 (m, 2H), 0.32–0.20 (m, 2H), NH and OH not visible; 13C NMR (100 MHz, CDCl3) δ 149.0, 130.3, 123.7, 123.0, 119.0, 113.6, 64.3, 48.7, 10.8, 3.7; m/z (ESI+) 256.0 ([M+H]+, 100%); HRMS (ESI+) C11H15NOBr+ ([M+H]+) requires 256.03315; found 256.03331.
4.14.3. 8-Bromo-1-(cyclopropylmethyl)-1,5-dihydrobenzo[e][1,4]oxazepin-2(3H)-one (4)
General procedure C.1 from 3 (840 mg, 3.28 mmol); yield 49% (472 mg, 1.59 mmol); light yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.46 (d, J = 1.9 Hz, 1H), 7.41 (dd, J = 8.0, 1.9 Hz, 1H), 7.24 (d, J = 8.1 Hz, 1H), 4.65 (s, 2H), 3.97 (s, 2H), 3.80 (d, J = 7.2 Hz, 2H), 1.11–0.98 (m, 1H), 0.48–0.41 (m, 2H), 0.31–0.22 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 168.0, 144.1, 131.9, 129.8, 128.9, 125.2, 123.6, 67.5, 67.4, 51.6, 10.3, 4.1; m/z (ESI+) 296.0 ([M+H]+, 70%); HRMS (ESI+) C13H15NO2Br+ ([M+H]+) requires 296.02807; found 296.02819.
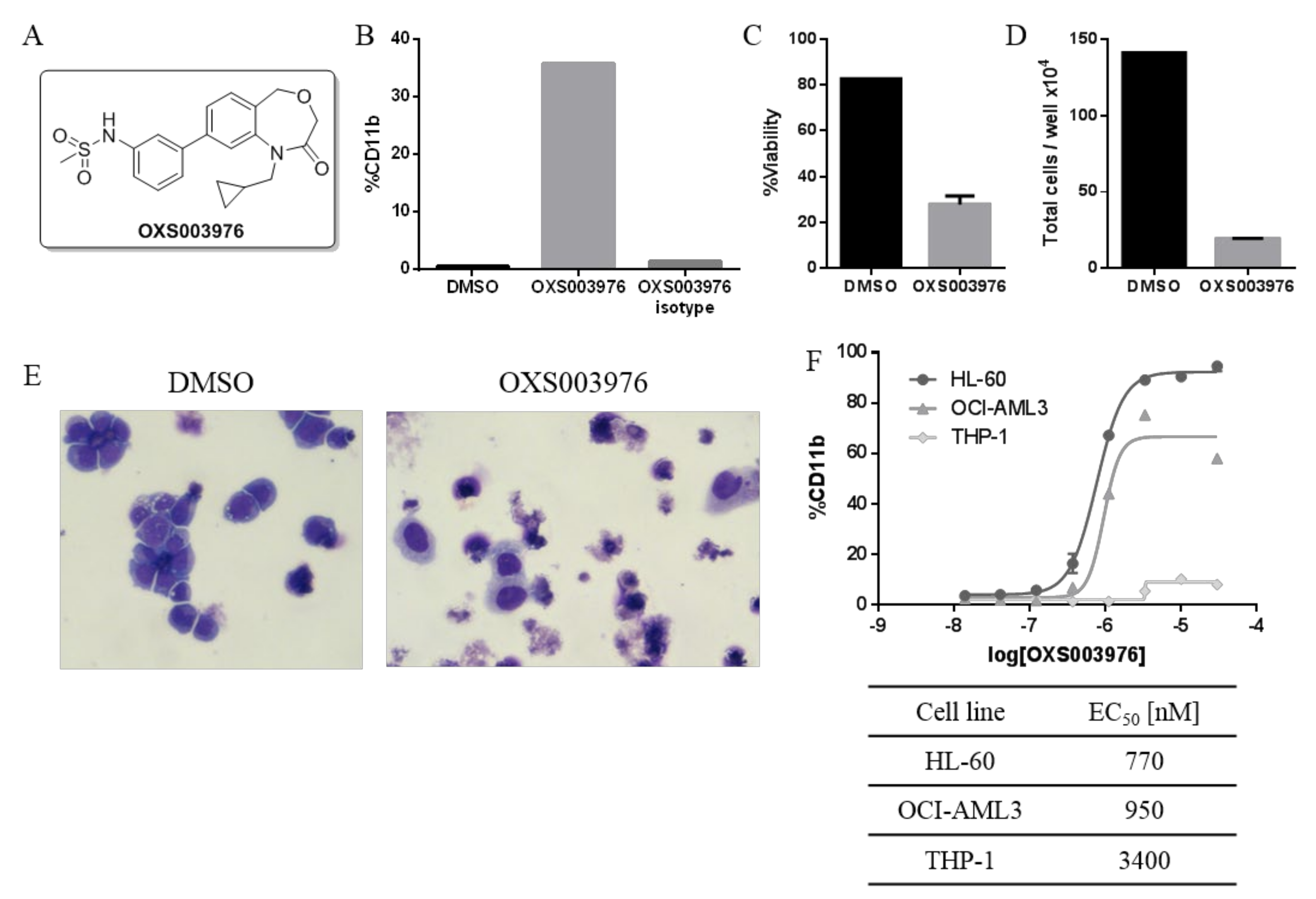
4.14.4. N-(3-(1-(Cyclopropylmethyl)-2-oxo-1,2,3,5-tetrahydrobenzo[e][1,4]oxazepin-8-yl)phenyl)methanesulfonamide (OXS003976 5)
General procedure D from 4 (50 mg, 0.17 mmol); yield 84% (55 mg, 0.14 mmol); white solid. 1H NMR (400 MHz, CDCl3) δ 7.49 (qd, J = 3.5, 1.9 Hz, 4H), 7.46–7.43 (m, 1H), 7.41 (dt, J = 7.8, 1.4 Hz, 1H), 7.29–7.24 (m, 1H), 6.94 (s, 1H), 4.75 (s, 2H), 4.02 (s, 2H), 3.89 (d, J = 7.2 Hz, 2H), 3.08 (s, 3H), 1.16–1.04 (m, 1H), 0.49–0.42 (m, 2H), 0.32–0.24 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 168.3, 143.4, 142.3, 141.8, 137.7, 131.1, 130.5, 129.5, 125.5, 124.2, 120.6, 120.1, 119.5, 67.6, 67.6, 51.7, 39.8, 10.4, 4.1; m/z (ESI+) 387.1 ([M+H]+, 70%); HRMS (ESI+) C20H23N2O4S+ ([M+H]+) requires 387.13730; found 387.13728.
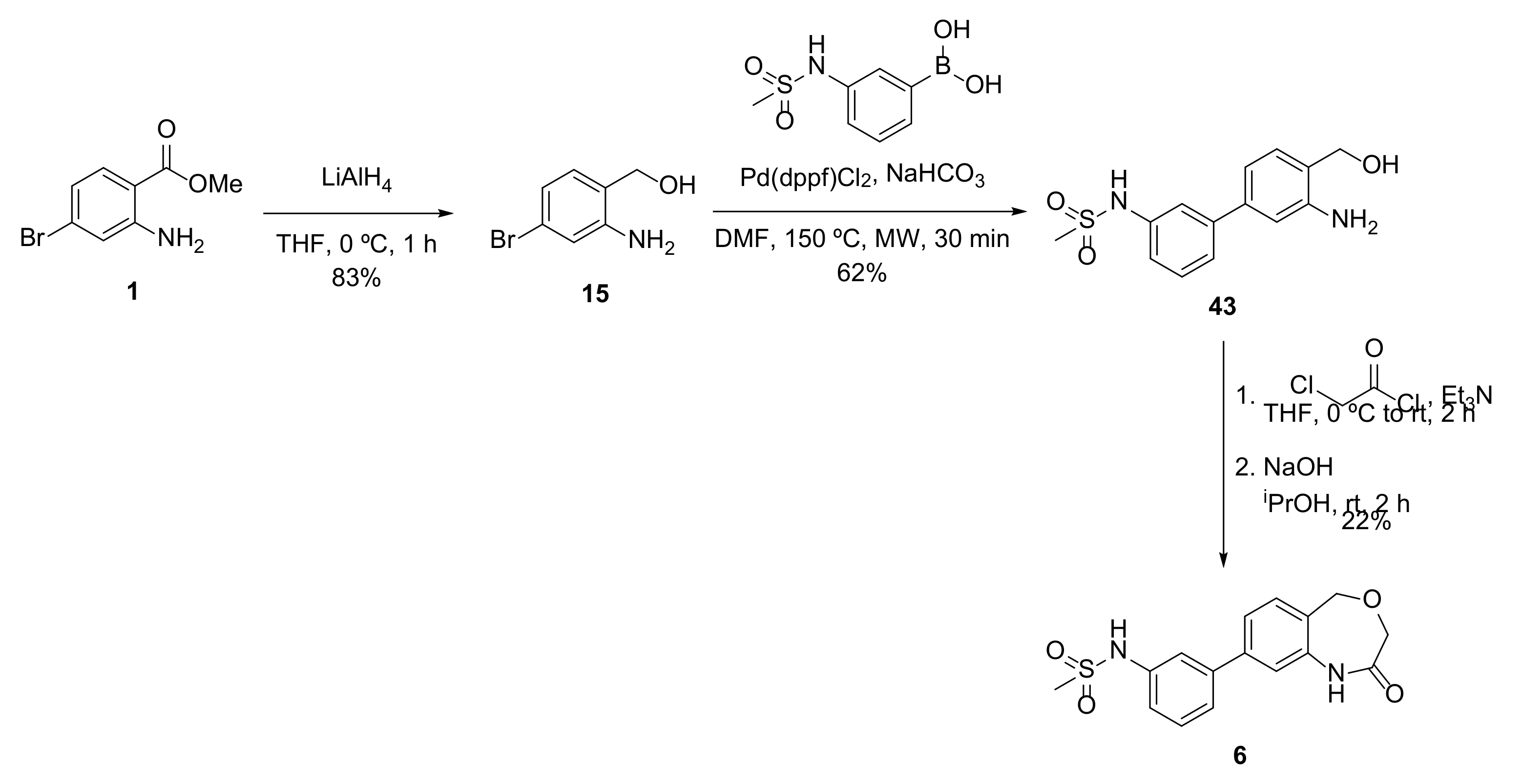
Compound
6 was prepared as shown in
Scheme 4.
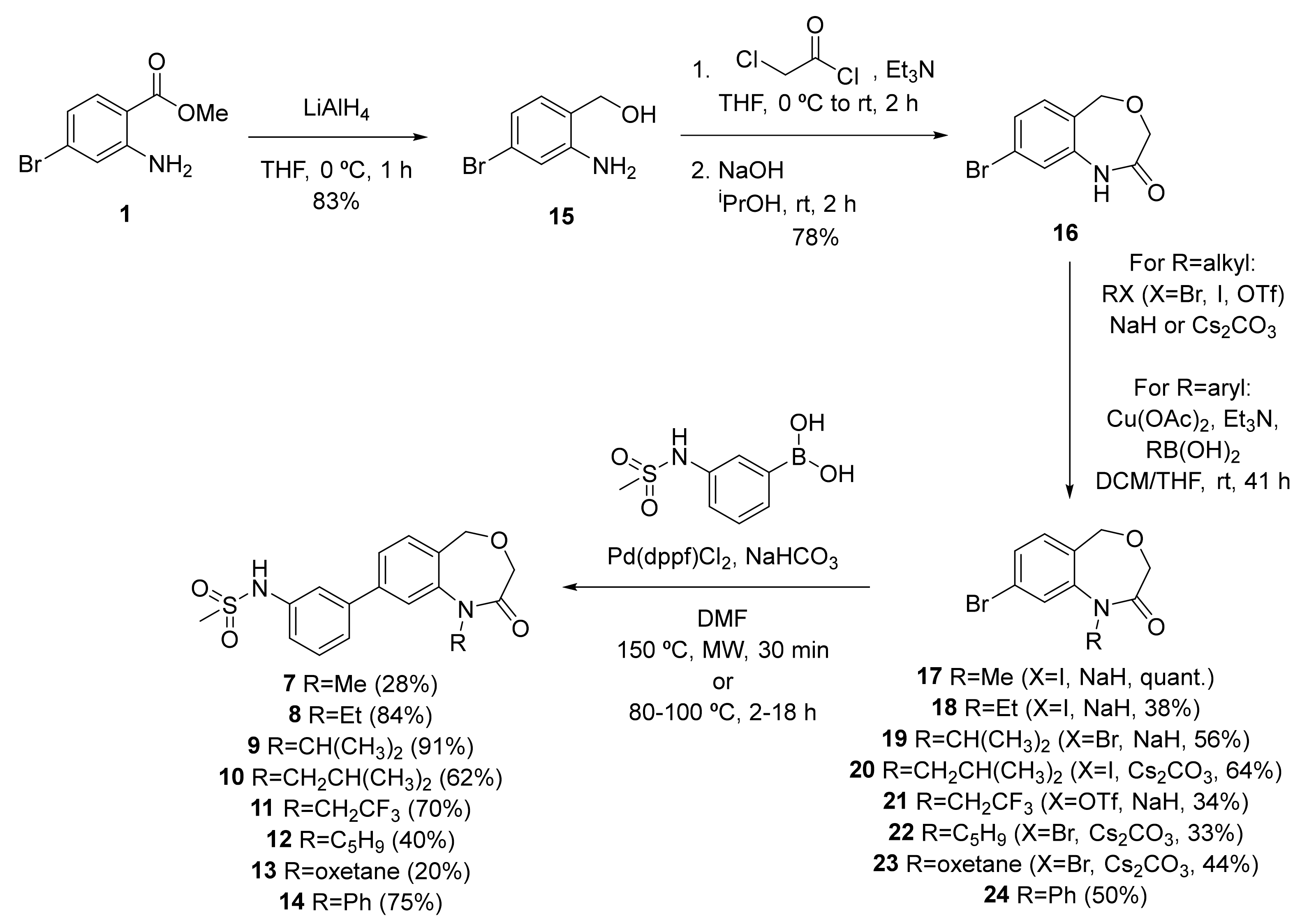
4.14.5. (2-Amino-4-bromophenyl)methanol (15)
General procedure B from methyl 2-amino-4-bromobenzoate (500 mg, 2.17 mmol); yield 83% (363 mg, 1.80 mmol); orange solid. 1H NMR (400 MHz, CD3OD) δ 6.97 (d, J = 8.0 Hz, 1H), 6.89 (d, J = 2.0 Hz, 1H), 6.75 (dd, J = 8.0, 2.0 Hz, 1H), 4.51 (s, 2H), NH2 and OH not visible; 13C NMR (100 MHz, CD3OD) δ 149.2, 131.1, 125.6, 123.0, 121.1, 119.1, 62.9; HRMS (ESI-) C7H7NOBr- ([M-H]−) requires 199.97165; found 199.97135.
4.14.6. N-(3′-Amino-4′-(hydroxymethyl)-[1,1′-biphenyl]-3-yl)methanesulfonamide (43)
General procedure D from 15 (100 mg, 0.495 mmol); yield 62% (90 mg, 0.31 mmol); yellow solid. 1H NMR (500 MHz, CD3OD) δ 7.45 (t, J = 2.0 Hz, 1H), 7.35 (q, J = 7.6 Hz, 1H), 7.32 (dt, J = 7.7, 1.6 Hz, 1H), 7.21–7.18 (m, 1H), 7.17 (d, J = 7.9 Hz, 1H), 7.01 (d, J = 1.9 Hz, 1H), 6.92 (dd, J = 7.7, 1.8 Hz, 1H), 4.62 (s, 2H), 2.96 (s, 3H); 13C NMR (125 MHz, CD3OD) δ 146.4, 142.6, 141.0, 139.6, 129.2, 128.9, 125.0, 122.2, 118.9, 118.7, 116.3, 114.2, 61.9, 37.7; m/z (ESI+) 293.1 ([M+H]+, 15%); HRMS (ESI+) C14H17N2O3S+ ([M+H]+) requires 293.09544; found 293.09542.
4.14.7. N-(3-(2-Oxo-1,2,3,5-tetrahydrobenzo[e][1,4]oxazepin-8-yl)phenyl)methanesulfonamide (6)
General procedure C.1 from 43 (79 mg, 0.27 mmol); yield 22% (20 mg, 0.060 mmol); white solid. 1H NMR (500 MHz, CDCl3) δ 7.86 (d, J = 1.9 Hz, 1H), 7.51 (d, J = 8.0 Hz, 1H), 7.45—7.40 (m, 3H), 7.35 (d, J = 7.8 Hz, 1H), 7.22 (ddd, J = 7.8, 2.2, 1.0 Hz, 1H), 5.48 (t, J = 5.4 Hz, 1H), 4.56 (d, J = 5.2 Hz, 2H), 4.37 (s, 2H), 3.01 (s, 3H), 1 NH is not visible; 13C NMR (125 MHz, CDCl3) δ 165.0, 140.8, 139.3, 138.8, 135.5, 133.9, 129.9, 128.2, 123.4, 122.0, 121.7, 118.8, 117.8, 60.1, 43.2, 39.4; m/z (ESI−) 331.1 ([M-H]−, 12%); HRMS (ESI−) C16H15N2O4S- ([M-H]−) requires 331.07580; found 331.07550.
4.14.8. 2-Bromo-N-(5-bromo-2-(hydroxymethyl)phenyl)acetamide (44)
To a mixture of 15 (1.0 g, 4.9 mmol) in DCM (40 mL) was added 2-bromoacetyl bromide (0.47 mL, 5.4 mmol) at 0 °C. After stirring for 5 min at 0 °C, 2 M aqueous Na2CO3 (2.5 mL) was added over a period of 10 min. The cooling bath was removed and stirring was continued for 3 h. The reaction mixture was concentrated under vacuum and the aqueous residue was extracted with EtOAc. The combined organic layers were washed with brine, dried and concentrated under vacuum. Purification by recrystallisation using a mixture of EtOAC and pentane produced the title compound 44 (1.06 g, 3.28 mmol, 67%) as an off white solid. 1H NMR (500 MHz, CDCl3 9.53 (brs, 1H), 8.33 (d, J = 2.0 Hz, 1H), 7.25 (dd, J = 8.0, 2.0 Hz, 1H), 7.07 (d, J = 8.1 Hz, 1H), 4.73 (s, 2H), 4.03 (s, 2H), 2.07 (brs, 1H); 13C NMR (125 MHz, CDCl3) 164.4, 138.3, 130.1, 128.3, 128.0, 125.2, 123.0, 64.0, 29.5; m/z (ESI+) 345.8 ([M+Na]+, 20%); HRMS (ESI+) C9H9Br2NO2Na+ ([MNa]+) requires 345.88718; found 345.88718.
4.14.9. 8-Bromo-1,5-dihydrobenzo[e][1,4]oxazepin-2(3H)-one (16)
To a suspension of 44 (0.90 g, 2.8 mmol) in 2-propanol (28 mL) was added, portion-wise, potassium tert-butoxide (0.81 g, 7.2 mmol) at 0 °C. The reaction mixture was stirred for 3 h and subsequently poured on ice/water. The resulting precipitate was collected by filtration, washed with water, dissolved in MeOH, dried, filtered and concentrated under vacuum to produce the cyclised product 16 (0.61 g, 2.5 mmol, 90%) as a light-yellow solid. 1H NMR (500 MHz, CDCl3) 8.83 (brs, 1H), 7.18 (dd, J = 8.1, 1.8 Hz, 1H), 7.11 (d, J = 1.8 Hz, 1H), 6.99 (d, J = 8.1 Hz, 1H), 4.69 (s, 2H), 4.59 (s, 2H); 13C NMR (125 MHz, CDCl3) 173.4, 137.3, 130.0, 127.8, 126.7, 122.5, 122.2, 73.7, 72.6; HRMS (ESI+) C9H10BrNO2+ ([MH]+) requires 241.98112; found 241.98129.
4.14.10. 8-Bromo-1-methyl-1,5-dihydrobenzo[e][1,4]oxazepin-2(3H)-one (17)
To a solution of amide 16 (50 mg, 0.21 mmol) in anhydrous DMF (1 mL) were added iodomethane (25 μL, 0.41 mmol) and sodium hydride (12 mg, 0.31 mmol) at 0 °C. The mixture was stirred at room temperature overnight then quenched with saturated NH4Cl, extracted with 3xEtOAc, dried over anhydrous Na2SO4 and concentrated in vacuo to produce methylated 17 (53 mg, 0.21 mmol, quant.) as a yellow solid, which was taken to the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 7.42–7.36 (m, 2H), 7.22 (d, J = 8.6 Hz, 1H), 4.59 (s, 2H), 4.01 (s, 2H), 3.41 (s, 3H); m/z (ESI+) 256.1 ([M+H]+, 100%); HRMS (ESI+) C10H11NO2Br+ ([M+H]+) requires 255.99677; found 255.99686.
4.14.11. N-(3-(1-Methyl-2-oxo-1,2,3,5-tetrahydrobenzo[e][1,4]oxazepin-8-yl)phenyl)methanesulfonamide (7)
General procedure D from 17 (53 mg, 0.21 mmol); yield 28% (20 mg, 0.058 mmol); yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.50 (t, J = 1.9 Hz, 1H), 7.48–7.37 (m, 6H), 7.29 (ddd, J = 7.9, 2.2, 1.2 Hz, 1H), 4.68 (s, 2H), 4.05 (s, 2H), 3.49 (s, 3H), 3.06 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 168.7, 144.4, 142.5, 141.8, 137.6, 131.1, 130.5, 128.5, 125.3, 124.3, 120.2, 119.6, 119.5, 67.7, 67.7, 39.8, 34.9; m/z (ESI+) 347.1 ([M+H]+, 100%); HRMS (ESI+) C17H19N2O4S+ ([M+H]+) requires 347.10600; found 347.10634.
4.14.12. 8-Bromo-1-ethyl-1,5-dihydrobenzo[e][1,4]oxazepin-2(3H)-one (18)
To a solution of amide 16 (64 mg, 0.26 mmol) in anhydrous DMF (1.3 mL) were added iodoethane (40 μL, 0.53 mmol) and sodium hydride (16 mg, 0.40 mmol) at 0 °C. The mixture was stirred at room temperature overnight, then quenched with saturated NH4Cl, extracted with 3xEtOAc, dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by flash column chromatography (5% to 20% EtOAc in pentane) to produce ethyl-substituted 18 (27 mg, 0.10 mmol, 38%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.44—7.37 (m, 2H), 7.23 (d, J = 8.0 Hz, 1H), 4.57 (s, 2H), 3.96 (s, 2H), 3.95 (q, J = 7.1 Hz, 2H), 1.22 (t, J = 7.1 Hz, 3H); m/z (ESI+) 270.0 ([M+H]+, 100%); HRMS (ESI+) C11H13NO2Br+ ([M+Na]+) requires 270.01242; found 270.01266.
4.14.13. N-(3-(1-Ethyl-2-oxo-1,2,3,5-tetrahydrobenzo[e][1,4]oxazepin-8-yl)phenyl)methanesulfonamide (8)
General procedure D from 18 (26 mg, 0.096 mmol); yield 84% (29 mg, 0.081 mmol); yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.50–7.43 (m, 5H), 7.41 (ddd, J = 7.8, 1.9, 0.9 Hz, 1H), 7.29–7.27 (m, 1H), 6.97 (s, 1H), 4.67 (s, 2H), 4.06 (q, J = 7.1 Hz, 2H), 4.02 (s, 2H), 3.07 (s, 3H), 1.26 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 168.0, 143.1, 142.5, 141.8, 137.7, 131.2, 130.5, 129.3, 125.5, 124.3, 120.1, 120.1, 119.5, 67.8, 67.6, 42.7, 39.8, 13.71; m/z (ESI+) 361.1 ([M+H]+, 100%); HRMS (ESI+) C18H21N2O4S+ ([M+H]+) requires 361.12165; found 361.12165.
4.14.14. 8-Bromo-1-isopropyl-1,5-dihydrobenzo[e][1,4]oxazepin-2(3H)-one (19)
To a suspension of 16 (100 mg, 0.413 mmol) in dry DMF (2.8 mL) was added NaH (60% w/w, 20 mg, 0.50 mmol) at 0 °C under an inert atmosphere. After the reaction mixture was stirred at 0 °C for 30 min, 2-bromopropane (58 µL, 0.62 mmol) was added. The reaction was stirred at 50 °C for 18 h and then quenched with water at 0 °C. The aqueous layer was extracted with EtOAc and the combined organic layers were washed with brine, dried and concentrated under vacuum. The residue was purified by flash column chromatography (15–20% EtOAc in pentane) to produce the desired product 19 (66 mg, 0.23 mmol, 56%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.42 (dt, J = 4.5, 2.0 Hz, 2H), 7.22 (d, J = 8.5 Hz, 1H), 4.64 (hept, J = 7.0 Hz, 1H), 4.56 (s, 2H), 3.88 (s, 2H), 1.38 (d, J = 7.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) 167.7, 143.4, 131.7, 130.2, 129.2, 126.3, 123.1, 68.1, 67.2, 51.1, 21.3; m/z (ESI+) 347.1 ([M+H]+, 100%); HRMS (ESI+) C17H19N2O4S+ ([M+H]+) requires 347.10600; found 347.10634.
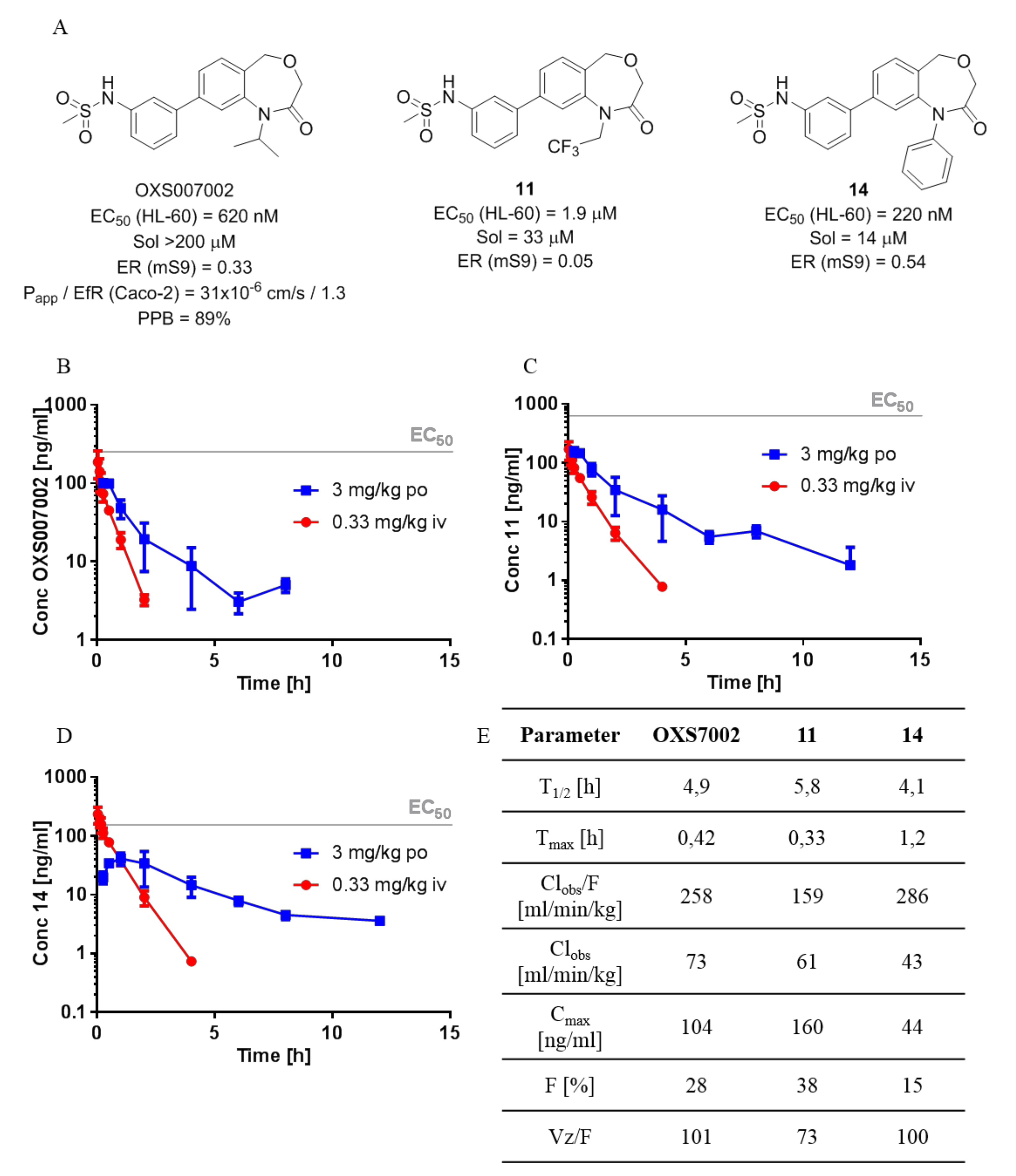
4.14.15. N-(3-(1-Isopropyl-2-oxo-1,2,3,5-tetrahydrobenzo[e][1,4]oxazepin-8-yl)phenyl)methanesulfonamide (OXS007002 9)
General procedure D from 19 (20 mg, 0.070 mmol); yield 91% (24 mg, 0.064 mmol); yellow solid. 1H NMR (500 MHz, CDCl3) δ 7.53–7.46 (m, 2H), 7.48–7.43 (m, 2H), 7.46–7.38 (m, 2H), 7.24 (ddd, J = 7.9, 2.4, 1.0 Hz, 1H), 6.47 (s, 1H), 4.75 (hept, J = 6.9 Hz, 1H), 4.67 (s, 2H), 3.95 (s, 2H), 3.07 (s, 3H), 1.44 (d, J = 6.9 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 168.0, 142.6, 141.9, 141.9, 137.5, 131.0, 130.6, 129.9, 125.9, 124.3, 121.9, 120.1, 119.5, 68.2, 67.5, 50.8, 39.9, 21.6, 1.2; m/z (ESI+) 375.1 ([M+H]+, 100%); HRMS (ESI+) C19H23N2O4S+ ([M+H]+) requires 375.13730; found 375.13681.
4.14.16. 8-Bromo-1-isobutyl-1,5-dihydrobenzo[e][1,4]oxazepin-2(3H)-one (20)
To a suspension of 16 (0.30 g, 1.2 mmol) in dry DMF (8.3 mL) were added 1-iodo-2-methylpropane (0.29 mL, 2.5 mmol) and Cs2CO3 (0.81 g, 2.48 mmol). The reaction was stirred at room temperature for 48 h and then quenched with water. The aqueous layer was extracted with EtOAc and the combined organic layers were washed with brine, dried and concentrated under vacuum. The residue was purified by flash column chromatography (25–30% EtOAc in pentane), to produce the desired compound 20 (0.24 g, 0.80 mmol, 64%) as a white solid. 1H NMR (500 MHz, CDCl3) 7.41 (d, J = 1.9 Hz, 1H), 7.39 (dd, J = 7.9 Hz, 1.9, 1H), 7.23 (d, J = 8.0 Hz, 1H), 4.65 (s, 2H), 3.97 (s, 2H), 3.77 (d, J = 7.5 Hz, 2H), 1.94 (tsep, J = 13.7, 6.9 Hz, 1H), 0.88 (d, J = 6.5 Hz, 6H); 13C NMR (125 MHz, CDCl3) 168.3, 144.1, 132.2, 129.5, 128.4, 124.4, 123.7, 67.8, 67.7, 53.7, 27.4, 20.6; m/z (ESI+) 298.1 ([M+H]+, 100%); HRMS (ESI+) C13H17BrNO2+ ([M+H]+) requires 298.04372; found 298.04383.
4.14.17. N-(3-(1-Isobutyl-2-oxo-1,2,3,5-tetrahydrobenzo[e][1,4]oxazepin-8-yl)phenyl)methanesulfonamide (10)
General procedure D from 20 (30 mg, 0.10 mmol; yield 62% (24 mg, 0.062 mmol); white solid. 1H NMR (500 MHz, CDCl3) 7.50–7.37 (m, 6H), 7.24 (ddd, J = 8.0, 2.3, 1.1 Hz, 1H) 6.64 (s, 1H), 4.47 (s, 2H), 4.03 (s, 2H), 3.87 (d, J = 7.3 Hz, 2H), 3.07 (s, 3H), 1.99 (tsep, J = 13.7, 6.8 Hz, 1H), 0.90 (d, J = 6.7 Hz, 6H); 13C NMR (125 MHz, CDCl3) 168.6, 143.4, 142.3, 141.9, 137.6, 131.5, 130.5, 129.0, 125.3, 124.3, 120.1, 119.9, 119.5, 67.9 (2×C), 53.8, 39.9, 27.5, 20.7; m/z (ESI+) 389.1 ([M+H]+, 100%); HRMS (ESI+) C20H25N2O4S+ ([M+H]+) requires 389.15295; found 389.15300.
4.14.18. 8-Bromo-1-(2,2,2-trifluoroethyl)-1,5-dihydrobenzo[e][1,4]oxazepin-2(3H)-one (21)
To a suspension of 16 (0.20 g, 0.83 mmol) in dry DMF (5.5 mL) was added NaH (60% w/w in mineral oil, 40 mg, 0.99 mmol) at 0 °C under an inert atmosphere. After the reaction mixture was stirred at 0 °C for 30 min, 2,2,2-trifluoroethyl trifluoromethanesulfonate (0.23 g, 0.99 mmol) was added. The reaction was stirred at room temperature for 18 h and then quenched with water at 0 °C. The aqueous layer was extracted with EtOAc and the combined organic layers were washed with brine, dried and concentrated under vacuum. The residue was purified by flash column chromatography (10% EtOAc in pentane) to produce the desired product 21 (91 mg, 0.28 mmol, 34%) as a white solid. 1H NMR (500 MHz, CDCl3) 7.50 (dd, J = 8.1, 1.8 Hz, 1H), 7.46 (d, J = 1.8 Hz, 1H), 7.30 (d, J = 8.1 Hz, 1H), 4.68 (s, 2H), 4.59 (q, J = 8.5 Hz, 2H), 4.04 (s, 2H); 13C NMR (125 MHz, CDCl3) 168.1, 142.4, 132.3, 130.8, 128.6, 127.1, 124.9, 124.3, 123.8, 122.7, 120.4, 67.51, 66.5, 46.9, 46.7, 46.4, 46.1; m/z (ESI+) 323.9 ([M+H]+, 100%); HRMS (ESI+) C11H10BrF3NO2+ ([M+H]+) requires 323.98415; found 323.98420.
4.14.19. N-(3-(2-Oxo-1-(2,2,2-trifluoroethyl)-1,2,3,5-tetrahydrobenzo[e][1,4]oxazepin-8-yl)phenyl)methanesulfonamide (11)
General procedure D from 21 (30 mg, 0.10 mmol); yield 70% (28 mg, 0.068 mmol); white solid. 1H NMR (500 MHz, CDCl3) δ 7.53 (dd, J = 7.8, 1.7 Hz, 1H), 7.46 (ddd, J = 7.9, 3.4, 1.6 Hz, 4H), 7.39 (dt, J = 7.8, 1.3 Hz, 1H), 7.28–7.25 (m, 1H), 4.73 (s, 2H), 4.66 (q, J = 8.6 Hz, 2H), 4.06 (s, 2H), 3.07 (s, 3H), NH not visible; 13C NMR (125 MHz, CDCl3) δ 168.7, 142.9, 142.0, 141.5, 138.0, 131.7, 130.7, 129.2, 126.6, 124.3, 124.2 (q, J = 281 Hz), 120.4, 119.9, 119.6, 67.5, 66.8, 46.9 (q, J = 34 Hz), 39.8; m/z (ESI+) 415.0 ([M+H]+, 100%); HRMS (ESI+) C18H18F3N2O4S + ([M+H]+) requires 415.09339; found 415.09328.
4.14.20. 8-Bromo-1-cyclopentyl-1,5-dihydrobenzo[e][1,4]oxazepin-2(3H)-one (22)
To a solution of 16 (100 mg, 0.41 mmol) in DMF (2 mL) were added Cs2CO3 (404 mg, 1.24 mmol) and bromocyclopentane (0.13 mL, 1.24 mmol). The mixture was stirred at 50 °C for 16 h, cooled to room temperature, diluted with EtOAc washed with brine (3×), dried and concentrated in vacuo. The crude product was purified by flash column chromatography (10–40% EtOAc in pentane) to produce 22 (42 mg, 0.14 mmol, 33%) as a yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.45 (dd, J = 8.0, 1.9 Hz, 1H), 7.42 (d, J = 1.9 Hz, 1H), 7.24 (d, J = 8.0 Hz, 1H), 4.61 (s, 2H), 4.49 (p, J = 8.8 Hz, 1H), 3.91 (s, 2H), 2.05–1.96 (m, 4H), 1.96–1.85 (m, 2H), 1.68–1.59 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 167.7, 144.4, 131.5, 129.9, 128.9, 126.2, 123.1, 68.1, 67.1, 61.5, 29.8 (2 × C), 24.6 (2 × C); HRMS (ESI+) C14H17BrNO2+ ([M+H]+) requires 310.0437; found 310.0438.
4.14.21. N-(3-(1-Cyclopentyl-2-oxo-1,2,3,5-tetrahydrobenzo[e][1,4]oxazepin-8-yl)phenyl)methanesulfonamide (12)
General procedure D from 22 (30 mg, 0.10 mmol); yield 40% (16 mg, 0.04 mmol); white solid. 1H NMR (500 MHz, CDCl3) δ 7.51–7.44 (m, 3H), 7.44–7.37 (m, 3H), 7.24 (ddd, J = 7.4, 2.5, 1.0 Hz, 1H), 6.69 (s, 1H), 4.67 (s, 2H), 4.60 (p, J = 8.8 Hz, 1H), 3.94 (s, 2H), 3.07 (s, 3H), 2.06–1.97 (m, 4H), 1.92–1.84 (m, 2H), 1.65–1.59 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 168.1, 143.5, 141.9, 141.7, 137.5, 130.8, 130.4, 129.5, 125.7, 124.1, 121.9, 119.9, 119.3, 68.2, 67.4, 61.2, 39.7, 30.0 (2 × C), 24.7 (2 × C); HRMS (ESI+) C21H23N2O4S+ ([M+H]+) requires 401.1530; found 401.1530.
4.14.22. 8-Bromo-1-(oxetan-3-yl)-1,5-dihydrobenzo[e][1,4]oxazepin-2(3H)-one (23)
To a suspension of 16 (100 mg, 0.41 mmol) in dry DMF (2.7 mL) were added Cs2CO3 (269 mg, 0.830 mmol) and 3-bromooxetane (51 µL, 0.62 mmol). The reaction was stirred at 50 °C for 3 days and then quenched with water. The aqueous layer was extracted with EtOAc and the combined organic layers were washed with brine, dried and concentrated under vacuum. The residue was purified by flash column chromatography (10–25% EtOAc in pentane) to produce the desired product 23 (54 mg, 0.18 mmol, 44%) which was used in the next step without additional purification. 1H NMR (400 MHz, CDCl3) δ 7.43 (dd, J = 8.1, 1.8 Hz, 1H), 7.29 (d, J = 8.1 Hz, 1H), 6.90 (d, J = 1.8 Hz, 1H), 5.20 (p, J = 7.3 Hz, 1H), 4.89 (t, J = 7.5 Hz, 2H), 4.76 (s, 2H), 4.55 (t, J = 7.4 Hz, 2H), 3.97 (s, 2H); m/z (ESI+) 297.9 ([M+H]+, 100%).
4.14.23. N-(3-(1-(Oxetan-3-yl)-2-oxo-1,2,3,5-tetrahydrobenzo[e][1,4]oxazepin-8-yl)phenyl)methanesulfonamide (13)
General procedure D from 23 (30 mg, 0.10 mmol); yield 20% (7.7 mg, 0.020 mmol); white solid. 1H NMR (500 MHz, CDCl3) 7.50–7.45 (m, 3H), 7.42 (d, J = 2.0 Hz, 1H), 7.34 (d, J = 7.8 Hz, 1H), 7.22 (dd, J = 7.9, 2.3 Hz, 1H), 6.92 (s, 1H), 5.32 (p, J = 7.2 Hz, 1H), 4.92 (t, J = 7.2 Hz, 2H), 4.84 (s, 2H), 4.61 (t, J = 7.1 Hz, 2H), 4.02 (s, 2H), 3.07 (s, 3H), NH not visible; 13C NMR (125 MHz, CDCl3) 168.2, 142.4, 141.4, 140.8, 137.6, 131.5, 130.6, 129.2, 125.7, 124.2, 120.1, 119.3, 119.1, 75.7, 67.9, 67.5, 51.7, 39.9; m/z (ESI+) 389.1 ([M+H]+, 100%); HRMS (ESI+) C19H21N2O5S+ ([M+H]+) requires 389.11657; found 389.11688.
4.14.24. 8-Bromo-1-phenyl-1,5-dihydrobenzo[e][1,4]oxazepin-2(3H)-one (24)
To a solution of 16 (100 mg, 0.41 mmol) in THF (2.8 mL) and DCM (2.8 mL) was added Et3N (0.43 mL, 3.07 mmol). This was followed by the addition of Cu(OAc)2·H2O (131 mg, 0.66 mmol) and phenylboronic acid (101 mg, 0.83 mmol). The reaction mixture was stirred at room temperature for 41 h. Upon completion, it was filtrated through Celite and concentrated under vacuum. The crude was purified by flash column chromatography (15–20% EtOAc in pentane) to produce 24 as an off-white solid (65 mg, 0.20 mmol, 50%). 1H NMR (500 MHz, CDCl3) 7.46–7.42 (m, 2H), 7.40 (dd, J = 8.1, 1.9 Hz, 1H), 7.38–7.33 (m, 1H), 7.29 (d, J = 8.1 Hz, 1H), 7.22–7.17 (m, 2H), 7.01 (d, J = 1.9 Hz, 1H), 4.80 (s, 2H), 4.16 (s, 2H); 13C NMR (125 MHz, CDCl3) 167.9, 144.9, 140.3, 131.9, 129.9, 129.7, 128.3, 128.0, 127.8, 126.8, 123.6, 68.2, 67.7; m/z (ESI+) 318.0 ([M+H]+, 100%); HRMS (ESI+) C15H13BrNO2+ ([M+H]+) requires 318.01242; found 318.01260.
4.14.25. N-(3-(2-Oxo-1-phenyl-1,2,3,5-tetrahydrobenzo[e][1,4]oxazepin-8-yl)phenyl)methanesulfonamide (14)
General procedure D from 24 (30 mg, 0.09 mmol); yield 75% (29 mg, 0.071 mmol); white solid. 1H NMR (500 MHz, CDCl3) 7.51–7.32 (m, 7H), 7.23–7.17 (m, 4H), 7.02 (d, J = 1.7 Hz, 1H), 6.43 (brs, 1H), 4,81 (s, 2H), 4.21 (s, 2H), 3.01 (s, 3H); 13C NMR (125 MHz, CDCl3) 168.2, 144.2, 142.3, 141.6, 140.8, 137.4, 131.1, 130.4, 129.5, 129.0, 127.8, 127.7, 125.7, 124.3, 122.5, 119.9, 119.4, 68.3, 67.9, 39.8; m/z (ESI+) 409.1 ([M+H]+, 100%); HRMS (ESI+) C22H21N2O4S+ ([M+H]+) requires 409.12165; found 409.12159.
4.14.26. N-(2-(1-Isopropyl-2-oxo-1,2,3,5-tetrahydrobenzo[e][1,4]oxazepin-8-yl)phenyl)methanesulfonamide (25)
General procedure D from 19 (20 mg, 0.070 mmol); yield 87% (23 mg, 0.061 mmol); a yellow solid. 1H NMR (700 MHz, CDCl3) 7.66 (d, J = 8.2 Hz, 1H), 7.54–7.45 (m, 2H), 7.32–7.30 (m, 4H), 6.38 (s, 1H), 4.70–4.65 (m, 3H), 3.99 (s, 2H), 3.03 (s, 3H), 1.45 (d, J = 6.9 Hz, 6H); 13C NMR (176 MHz, CDCl3) 167.9, 143.4, 139.3, 134.1, 132.0, 131.5, 131.0, 130.4, 129.8, 127.6, 125.2, 123.9, 120.0, 68.4, 67.5, 51.5, 40.6, 21.5; m/z (ESI+) 375.1 ([M+H]+, 100%); HRMS (ESI+) C19H23N2O4S+ ([M+H]+) requires 375.13730; found 375.13744.
4.14.27. N-(4-(1-Isopropyl-2-oxo-1,2,3,5-tetrahydrobenzo[e][1,4]oxazepin-8-yl)phenyl)methanesulfonamide (26)
General procedure D from 19 (30 mg, 0.11 mmol); yield 88% (35 mg, 0.093 mmol); white solid. 1H NMR (500 MHz, CDCl3) 7.60–7.56 (m, 2H), 7.48 (dd, J = 7.7, 1.8 Hz, 1H), 7.44–7.40 (m, 2H), 7.36–7.32 (m, 2H), 6.62 (s, 1H), 4.75 (h, J = 6.9 Hz, 1H), 4.66 (s, 2H), 3.94 (s, 2H), 3.08 (s, 3H), 1.43 (d, J = 6.9 Hz, 6H); 13C NMR (125 MHz, CDCl3) 168.1, 142.6, 141.9, 137.2, 136.8, 131.0, 129.5, 128.6, 125.6, 121.6, 121.2, 68.2, 67.5, 50.8, 39.8, 21.6; m/z (ESI+) 375.1 ([M+H]+, 100%); HRMS (ESI+) C19H23N2O4S+ ([M+H]+) requires 375.13730; found 375.13742.
Compound
27 was prepared as shown in
Scheme 5.
4.14.28. 1-Isopropyl-8-(3-(methylthio)phenyl)-1,5-dihydrobenzo[e][1,4]oxazepin-2(3H)-one (45)
General procedure D from 19 (80 mg, 0.28 mmol); yield 98% (91 mg, 0.28 mmol); pale yellow solid. 1H NMR (500 MHz, CDCl3) 7.42 (dd, J = 7.8, 1.7 Hz, 1H), 7.38–7.32 (m, 4H), 7.27 (ddd, J = 7.7, 1.8, 1.2 Hz, 1H), 7.22 (ddd, J = 7.7, 1.9, 1.2 Hz, 1H), 4.67 (h, J = 6.9 Hz, 1H), 4.59 (s, 2H), 3.87 (s, 2H), 2.48 (s, 3H), 1.36 (d, J = 6.9 Hz, 6H); 13C NMR (125 MHz, CDCl3) 168.0, 142.6, 142.4, 140.7, 139.6, 130.9, 129.6, 129.5, 126.0, 125.9, 125.4, 124.1, 121.9, 68.2, 67.5, 50.8, 21.6, 16.0; m/z (ESI+) 328.1 ([M+H]+, 100%); HRMS (ESI+) C19H22NO2S+ ([M+H]+) requires 328.13767; found 328.13655.
4.14.29. 1-Isopropyl-8-(3-(S-methylsulfonimidoyl)phenyl)-1,5-dihydrobenzo[e][1,4]oxazepin-2(3H)-one (27)
To a solution of 45 (30 mg, 0.12 mmol) in MeOH (0.25 mL) were added (diacetoxyiodo)benzene (74 mg, 0.23 mmol) and ammonium carbamate (14 mg, 0.23 mmol). The reaction was stirred at room temperature for 1 h. Upon completion the reaction was concentrated under vacuum and purified by flash column chromatography (0–1% MeOH in EtOAc) to produce the desired product 27 (26 mg, 0.073 mmol, 80%) as pale-yellow oil. 1H NMR (500 MHz, CDCl3) 8.22 (t, J = 1.9, 1H), 8.05 (ddd, J = 7.8, 1.9, 1.1 Hz, 1H), 7.82 (ddd, J = 7.8, 1.9, 1.1 Hz, 1H), 7.68 (t, J = 1.9 Hz, 1H), 7.54 (dd, J = 7.7, 1.8 Hz, 1H), 7.47 (s, 1H), 7.45 (d, J = 6, 1H), 4.72 (h, J = 6.9 Hz, 1H), 4.67 (s, 2H), 3.93 (s, 2H), 3.17 (s, 3H), 1.43 (d, J = 6.9 Hz, 6H), NH not visible; 13C NMR (125 MHz, CDCl3) 168.0, 144.7, 142.8, 141.5, 141.2, 131.8, 131.2, 130.3, 130.2, 127.2, 126.4, 126.1, 121.9, 68.2, 67.4, 51.1, 46.4, 21.6; m/z (ESI+) 359.1 ([M+H]+, 100%); HRMS (ESI+) C19H23N2O3S+ ([M+H]+) requires 359.14239; found 359.14225.
4.14.30. 8-(3-Aminophenyl)-1-isopropyl-1,5-dihydrobenzo[e][1,4]oxazepin-2(3H)-one (29)
General procedure D from 19 (1.50 g, 5.28 mmol); yield 70% (1.10 g, 3.71 mmol); orange solid. 1H NMR (400 MHz, DMSO-d6) δ 7.51 (m, 2H), 7.45 (s, 1H), 7.15–7.11 (m, 1H), 6.9–6.89 (m, 1H), 6.84–6.82 (m, 1H), 6.63–6.60 (m, 1H), 5.16 (br s, 2H), 4.62–4.55 (m, 3H), 3.75 (s, 2H), 1.38 (s, 3H), 1.37 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.6, 149.7, 143.2, 142.7, 140.3, 131.1, 130.0, 129.1, 125.4, 121.3, 114.9, 114.2, 112.7, 68.0, 66.8, 50.9, 21.5; m/z (ESI+) 297.2 ([M+H]+, 100%); HRMS (ESI+) C18H21N2O2+ ([M+H]+) requires 297.1598; found 297.1598.
4.14.31. Methyl (3-(1-isopropyl-2-oxo-1,2,3,5-tetrahydrobenzo[e][1,4]oxazepin-8-yl)phenyl)carbamate (28)
A solution of amine 29 (97 mg, 0.33 mmol), triethylamine (68 μL, 0.49 mmol) and methyl chloroformate (25 μL, 0.33 mmol) in DCM (4 mL) was stirred at room temperature for 18 h. LCMS showed 75% starting material and 14% product. More methyl chloroformate (75 μL, 0.98 mmol) and triethylamine (204 μL, 1.47 mmol) were added and stirred at room temperature for 18 h. More methyl chloroformate (100 μL, 1.31 mmol) and triethylamine (204 μL, 1.47 mmol) were added and stirred at room temperature for 1.5 h. The solution was washed with 1 M HCl (6 mL), concentrated in vacuo, and purified by flash column chromatography (0.5% to 3% MeOH in DCM) to produce carbonate 28 (63 mg, 0.18 mmol, 54%) as an off white solid. 1H NMR (400 MHz, CDCl3) δ 7.13 (s, 1H), 7.51–7.35 (m, 5H), 7.28 (d, J = 7.6 Hz, 1H), 6.77 (s, 1H), 4.74 (quin, J = 6.8 Hz, 1H), 4.65 (s, 2H), 3.94 (s, 2H), 3.80 (s, 3H), 1.43 (d, J = 6.8 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 167.9, 154.0, 142.5, 142.2, 140.9, 138.6, 137.0, 129.7, 129.3, 125.8, 122.2, 121.8, 118.2, 117.4, 68.0, 67.3, 52.4, 50.7, 21.4; m/z (ESI+) 355.1 ([M+H]+, 100%); HRMS (ESI+) C20H23N2O4+ ([M+H]+) requires 355.1652; found 355.1653.
4.14.32. 8-(3-(Dimethylamino)phenyl)-1-isopropyl-1,5-dihydrobenzo[e][1,4]oxazepin-2(3H)-one (30)
General procedure D from 19 (150 mg, 0.528 mmol); yield 60% (103 mg, 0.317 mmol); orange solid. 1H NMR (400 MHz, CDCl3) δ δ 7.51 (dd, J = 1.6, 7.6 Hz, 1H), 7.47 (d, J = 1.6 Hz, 1H), 7.39 (d, J = 8.0 Hz, 1H), 7.35 (t, J = 7.8 Hz, 1H), 6.92 (d, J = 7.6 Hz, 1H), 6.89 (s, 1H), 6.79 (dd, J = 2.4, 8.0 Hz, 1H), 4.75 (quin, J = 6.8 Hz, 1H), 4.66 (s, 2H), 3.95 (s, 2H), 3.03 (s, 6H), 1.43 (s, 3H), 1.42 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 168.0, 151.0, 144.0, 142.1, 141.0, 130.5, 129.7, 128.9, 125.9, 122.0, 115.6, 112.2, 111.2, 68.1, 67.4, 50.6, 40.6, 21.4; m/z (ESI-) 309.1 ([M-Me]-, 18%); HRMS (ESI+) C20H25N2O2+ ([M+H]+) requires 325.1911; found 325.1911.
4.14.33. 1-Isopropyl-8-(3-methoxyphenyl)-1,5-dihydrobenzo[e][1,4]oxazepin-2(3H)-one (31)
General procedure D from 19 (30 mg, 0.11 mmol); yield 67% (22 mg, 0.07 mmol); white solid. 1H NMR (500 MHz, CDCl3) δ 7.51 (dd, J = 7.8, 1.7 Hz, 1H), 7.46 (d, J = 1.7 Hz, 1H), 7.44–7.37 (m, 2H), 7.26 (s, 1H), 7.17 (ddd, J = 7.6, 1.7, 0.9 Hz, 1H), 7.11 (t, J = 2.1 Hz, 1H), 6.95 (ddd, J = 8.2, 2.6, 0.9 Hz, 1H), 4.74 (p, J = 6.9 Hz, 1H), 4.66 (s, 2H), 3.94 (s, 2H), 3.89 (s, 3H), 1.43 (d, J = 6.9 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 168.0, 160.1, 142.8, 142.3, 141.4, 130.7, 130.1, 129.3, 125.8, 121.8, 119.6, 113.3, 113.1, 68.1, 67.4, 55.4, 50.7, 21.5 (2 × C); HRMS (ESI+) C19H22NO3+ ([M+H]+) requires 312.1594; found 312.1590.
4.14.34. 3-(1-Isopropyl-2-oxo-1,2,3,5-tetrahydrobenzo[e][1,4]oxazepin-8-yl)benzoic acid (32)
General procedure D from 19 (120 mg, 0.422 mmol); yield 33% (46 mg, 0.141 mmol); beige solid. 1H NMR (400 MHz, CDCl3) δ 13.03 (bs, 1H), 8.21 (t, J = 1.6 Hz, 1H), 7.99 (dd, J = 1.6, 7.6 Hz, 2H), 7.68–7.59 (m, 2H), 7.57 (d, J = 2.0 Hz, 2H), 4.62 (s, 2H), 4.61–4.54 (m, 1H), 3.77 (s, 2H), 1.39 (d, J = 6.8 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 167.6, 149.7, 143.2, 142.7, 140.3, 131.1, 130.0, 129.1, 125.4, 121.3, 114.9, 114.2, 112.7, 68.0, 66.8, 50.9, 21.5; m/z (ESI−) 324.2 ([M-H]−, 100%); HRMS (ESI+) C19H20NO4+ ([M+H]+) requires 326.1387; found 326.1387.
4.14.35. N-(3-(1-Isopropyl-2-oxo-1,2,3,5-tetrahydrobenzo[e][1,4]oxazepin-8-yl)phenyl)propane-1-sulfonamide (33)
Aniline 29 (120 mg, 0.405 mmol) was stirred with triethylamine (68 μL, 0.49 mmol) in DCM (4 mL) and propanesulfonyl chloride (46 μL, 0.40 mmol). The solution was stirred at room temperature for 1 h, then washed with water, dried over anhydrous MgSO4 and concentrated in vacuo. The resulting residue was passed down a silica plug with 0.5% MeOH in DCM, then stirred overnight in pentane filtered and dried to produce sulfonamide 33 (17 mg, 0.042 mmol, 10%) as a beige solid. 1H NMR (400 MHz, CDCl3) δ 7.51–7.39 (m, 6H), 7.37–7.22 (m, 1H), 6.73 (bs, 1H), 4.76 (quin, J = 6.9 Hz, 1H), 4.67 (s, 2H), 3.95 (s, 2H), 3.15–3.11 (m, 2H), 1.90 (td, J = 15.2 Hz, 7.6 Hz, 2H), 1.44 (d, J = 6.8 Hz, 6H), 1.05 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 168.0, 142.4, 141.9, 141.6, 137.7, 130.9, 130.3, 129.7, 125.8, 123.8, 121.8, 119.6, 119.0, 68.1, 67.3, 53.7, 50.7, 21.5, 17.3, 12.9; m/z (ESI+) 403.1 ([M+H]+, 100%); HRMS (ESI+) C21H27N2O4S+ ([M+H]+) requires 403.1686; found 403.1687.
4.14.36. 1,1,1-Trifluoro-N-(3-(1-isopropyl-2-oxo-1,2,3,5-tetrahydrobenzo[e][1,4]oxazepin-8-yl)phenyl)methanesulfonamide (34)
Aniline 29 (120 mg, 0.40 mmol) in DCM (4.0 mL) was cooled in an ice bath. Triflic anhydride (0.068 mL, 0.41 mmol) was added followed by triethylamine (0.056 mL, 0.40 mmol) and the reaction mixture was stirred at room temperature overnight. TLC showed that the reaction was not complete. The mixture was cooled and more triflic anhydride (0.068 mL, 0.41 mmol) was added. After 6 h, more triflic anhydride (0.068 mL, 0.41 mmol) was added and stirred at room temperature overnight. The reaction mixture was quenched with saturated aqueous NH4Cl, separated and dried with Na2SO4. The crude residue was purified by flash column chromatography (0–35% EtOAc in hexane) to produce 34 (17 mg, 0.040 mmol, 10%) as a pale orange glassy oil. 1H NMR (400 MHz, DMSO-d6) δ 7.65–7.51 (m, 6H), 7.32–7.30 (m, 1H), 4.64–4.57 (m, 3H), 3.77 (s, 2H), 1.38 (s, 3H), 1.37 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.6, 143.0, 141.2, 140.9, 136.6, 131.5, 130.8, 129.9, 125.6, 125.4, 122.5, 121.9, 121.6, 121.4, 68.0, 66.8, 51.0, 21.4; m/z (ESI+) 429.1 ([M+H]+, 100%).
Compound
35 was prepared as shown in
Scheme 6.
4.14.37. 8-(3-Amino-4-fluorophenyl)-1-isopropyl-1,5-dihydrobenzo[e][1,4]oxazepin-2(3H)-one (46)
General procedure D from 19 (34 mg, 0.12 mmol); yield 95% (36 mg, 0.11 mmol); white solid. 1H NMR (600 MHz, CDCl3) δ 7.43 (dd, J = 7.7, 1.8 Hz, 1H), 7.40–7.36 (m, 2H), 7.07 (dd, J = 10.7, 8.4 Hz, 1H), 6.97 (dd, J = 8.4, 2.3 Hz, 1H), 6.88 (ddd, J = 8.4, 4.4, 2.3 Hz, 1H), 4.73 (hept, J = 6.9 Hz, 1H), 4.65 (s, 2H), 3.93 (s, 2H), 3.85 (s, 2H), 1.43 (d, J = 6.9 Hz, 6H); 13C NMR (151 MHz, CDCl3) δ 168.0, 151.7 (d, J = 241 Hz), 142.3 (d, J = 30 Hz), 136.6 (d, J = 3 Hz), 134.9 (d, J = 13 Hz), 130.7, 129.0, 125.6, 121.6, 117.4 (d, J = 7 Hz), 115.8, 115.7, 115.5 (d, J = 4 Hz), 68.1, 67.4, 50.7, 24.9, 21.5 (2 × C); HRMS (ESI+) C18H20FN2O2+ ([M+H]+) requires 315.1503; found 315.1502.
4.14.38. N-(2-Fluoro-5-(1-isopropyl-2-oxo-1,2,3,5-tetrahydrobenzo[e][1,4]oxazepin-8-yl)phenyl)methanesulfonamide (35)
To a solution of 46 (15 mg, 0.048 mmol) in DCM (0.5 mL) at 0 °C were added pyridine (4 µL, 0.05 mmol) and methanesulfonyl chloride (4 µL, 0.05 mmol). The mixture was warmed to room temperature for 1 h, then diluted with DCM, washed with 1 M HCl, dried and concentrated in vacuo. The crude product was purified by flash column chromatography (10–60% EtOAc in pentane) to produce 35 (10 mg, 0.025 mmol, 53%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.81 (dd, J = 7.6, 2.3 Hz, 1H), 7.47 (dd, J = 7.8, 1.7 Hz, 1H), 7.45–7.35 (m, 3H), 7.26 (s, 2H), 6.66–6.62 (m, 1H), 4.75 (p, J = 6.9 Hz, 1H), 4.66 (s, 2H), 3.94 (s, 2H), 3.08 (s, 3H), 1.43 (d, J = 6.9 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 167.9, 153.9 (d, J = 246 Hz), 142.5, 141.1, 137.4 (d, J = 4 Hz), 131.0, 129.7, 125.7, 125.2, 125.13 (d, J = 8 Hz), 125.12, 122.0 (d, J = 61 Hz), 116.4 (d, J = 20 Hz), 68.1, 67.3, 50.7, 40.0, 21.5 (2 × C); HRMS (ESI+) C19H22FN2O4S+ ([M+H]+) requires 393.1279; found 393.1276.
Compound
36 was prepared as shown in
Scheme 7.
4.14.39. 1-Isopropyl-8-(4-methyl-3-nitrophenyl)-1,5-dihydrobenzo[e][1,4]oxazepin-2(3H)-one (47)
General procedure D from 19 (58 mg, 0.20 mmol); yield 69% (48 mg, 0.14 mmol); white solid. 1H NMR (500 MHz, CDCl3) δ 8.19 (d, J = 2.0 Hz, 1H), 7.71 (dd, J = 8.0, 2.0 Hz, 1H), 7.52 (dd, J = 7.8, 1.7 Hz, 1H), 7.49–7.43 (m, 3H), 4.74 (hept, J = 6.9 Hz, 1H), 4.67 (s, 2H), 3.94 (s, 2H), 2.67 (s, 3H), 1.44 (d, J = 6.9 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 167.9, 149.7, 142.7, 140.3, 139.1, 133.6, 133.2, 131.3, 131.2, 130.1, 125.6, 123.1, 121.6, 68.1, 67.3, 50.9, 21.5, 20.2; HRMS (ESI+) C19H21N2O4+ ([M+H]+) requires 341.1496; found 341.1495.
4.14.40. 8-(3-Amino-4-methylphenyl)-1-isopropyl-1,5-dihydrobenzo[e][1,4]oxazepin-2(3H)-one (48)
To a solution of 47 (40 mg, 0.12 mmol) and ammonium formate (8 mg, 0.12 mmol) in MeOH (3 mL) was added palladium on carbon (10% w/w, 4 mg). The flask was purged with hydrogen and the mixture stirred at room temperature for 2 h, then concentrated in vacuo. The crude product was purified by flash column chromatography (10–60% EtOAc in pentane) to produce 48 (27 mg, 0.087 mmol, 72%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.47 (dd, J = 7.8, 1.7 Hz, 1H), 7.43 (d, J = 1.7 Hz, 1H), 7.37 (d, J = 7.8 Hz, 1H), 7.15 (d, J = 7.6 Hz, 1H), 6.92 (dd, J = 7.6, 1.9 Hz, 1H), 6.88 (d, J = 1.9 Hz, 1H), 4.74 (hept, J = 6.9 Hz, 1H), 4.65 (s, 2H), 3.94 (s, 2H), 3.74 (s, 2H), 2.23 (s, 3H), 1.42 (d, J = 6.9 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 168.0, 145.1, 143.2, 142.1, 138.9, 131.1, 130.6, 128.8, 125.6, 122.4, 121.6, 117.4, 113.4, 68.1, 67.4, 50.6, 21.5 (2 × C), 17.1; HRMS (ESI+) C19H23N2O2+ ([M+H]+) requires 311.1754; found 311.1753.
4.14.41. N-(5-(1-Isopropyl-2-oxo-1,2,3,5-tetrahydrobenzo[e][1,4]oxazepin-8-yl)-2-methylphenyl)methanesulfonamide (36)
To a solution of 48 (19 mg, 0.061 mmol) in DCM (0.5 mL) at 0 °C were added pyridine (5 µL, 0.06 mmol) and methanesulfonyl chloride (5 µL, 0.06 mmol). The mixture was warmed to room temperature for 3 h, then diluted with DCM, washed with 1 M HCl, dried and concentrated in vacuo. The crude product was purified by flash column chromatography (10–60% EtOAc in pentane) to yield 36 (18 mg, 0.046 mmol, 76%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.73 (d, J = 1.7 Hz, 1H), 7.50 (dd, J = 7.8, 1.7 Hz, 1H), 7.45 (d, J = 1.7 Hz, 1H), 7.41 (d, J = 7.8 Hz, 1H), 7.39–7.31 (m, 2H), 7.26 (s, 1H), 6.52 (s, 1H), 4.75 (hept, J = 6.9 Hz, 1H), 4.66 (s, 2H), 3.94 (s, 2H), 3.06 (s, 3H), 2.39 (s, 3H), 1.42 (d, J = 6.9 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 168.0, 142.3, 141.8, 139.3, 135.4, 131.8, 130.8, 129.8, 129.4, 125.6, 124.5, 121.7, 121.5, 68.0, 67.3, 50.6, 40.1, 21.5 (2 × C), 17.7; HRMS (ESI+) C20H25N2O4S+ ([M+H]+) requires 389.1530; found 389.1523.
4.14.42. N-(5-(1-Isopropyl-2-oxo-1,2,3,5-tetrahydrobenzo[e][1,4]oxazepin-8-yl)-2-methoxyphenyl)methanesulfonamide (37)
General procedure D from 19 (30 mg, 0.11 mmol); yield 75% (32 mg, 0.079 mmol); off white solid. 1H NMR (500 MHz, CDCl3) 7.80 (d, J = 2.3 Hz, 1H), 7.48 (dd, J = 1.8, 7.8 Hz, 1H), 7.42 (d, J = 1.7 Hz, 1H), 7.38 (d, J = 8.0 Hz, 1H), 7.37 (dd, J = 2.0, 8.5 Hz, 1H), 7.02 (d, J = 8.5 Hz, 1H), 6.86 (brs, 1H), 4.75 (h, J = 7.0 Hz, 1H), 4.65 (s, 2H), 3.95 (s, 3H), 3.94 (s, 2H), 3.00 (s, 3H), 1.43 (d, J = 6.9 Hz, 6H); 13C NMR (125 MHz, CDCl3) 168.1, 149.6, 142.5, 141.7, 133.5, 130.9, 129.2, 126.7, 125.6, 124.2, 121.6, 119.9, 111.3, 68.2, 67.5, 56.2, 50.7, 39.4, 21.6; m/z (ESI+) 405.1 ([M+H]+, 100%); HRMS (ESI+) C20H25N2O5S+ ([M+H]+) requires 405.14787; found 405.14784.
4.14.43. N-(3-(1-Isopropyl-2-oxo-1,2,3,5-tetrahydrobenzo[e][1,4]oxazepin-8-yl)-5-methoxyphenyl)methanesulfonamide (38)
General procedure D from 19 (30 mg, 0.11 mmol); yield 84% (36 mg, 0.089 mmol); white solid. 1H NMR (500 MHz, CDCl3) 7.48 (dd, J = 1.8, 7.7 Hz, 1H), 7.42 (s, 1H), 7.41 (d, J = 6.5 Hz, 1H), 6.99 (t, J = 1.7 Hz, 1H), 6.92 (dd, J = 2.3, 1.5 Hz, 1H), 6.85 (t, J = 2.1 Hz, 1H), 6.65 (s, 1H), 4.74 (h, J = 6.9 Hz, 1H), 4.66 (s, 2H), 3.94 (s, 2H), 3.89 (s, 3H), 3.08 (s, 3H), 1.43 (d, J = 6.9 Hz, 6H); 13C NMR (125 MHz, CDCl3) 168.1, 161.3, 142.9, 142.5, 142.0, 138.7, 131.0, 130.0, 125.9, 121.9, 111.5, 110.2, 105.3, 68.2, 67.5, 55.8, 50.9, 39.7, 21.6; m/z (ESI+) 405.1 ([M+H]+, 100%); HRMS (ESI+) C20H25N2O5SNa+ ([M+Na]+) requires 427.12981; found 427.12980.
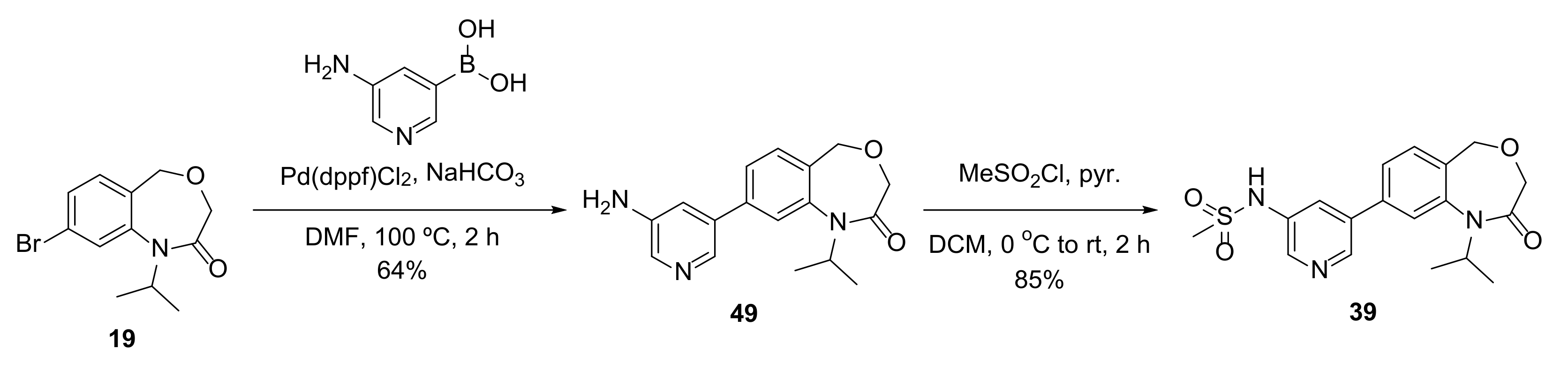
Compound
39 was prepared as shown in
Scheme 8.
4.14.44. 8-(5-Aminopyridin-3-yl)-1-isopropyl-1,5-dihydrobenzo[e][1,4]oxazepin-2(3H)-one (49)
General procedure D from 19 (30 mg, 0.11 mmol); yield 64% (20 mg, 0.07 mmol); yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.25 (d, J = 1.9 Hz, 1H), 8.13 (d, J = 2.6 Hz, 1H), 7.47 (dd, J = 7.8, 1.7 Hz, 1H), 7.45–7.42 (m, 2H), 7.14 (t, J = 2.3 Hz, 1H), 4.74 (hept, J = 6.9 Hz, 1H), 4.66 (s, 2H), 3.94 (s, 2H), 3.84 (s, 2H), 1.43 (d, J = 6.9 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 167.9, 142.54, 142.51, 139.9, 138.5, 137.0, 135.8, 131.0, 129.9, 125.8, 121.7, 119.6, 68.1, 67.3, 50.8, 21.5 (2 × C); HRMS (ESI+) C17H19N3O2+ ([M+H]+) requires 298.1550; found 298.1549.
4.14.45. N-(5-(1-Isopropyl-2-oxo-1,2,3,5-tetrahydrobenzo[e][1,4]oxazepin-8-yl)pyridin-3-yl)methanesulfonamide (39)
To a solution of 49 (15 mg, 0.050 mmol) in DCM (0.5 mL) at 0 °C was added pyridine (4 µL, 0.05 mmol) and methanesulfonyl chloride (4 µL, 0.05 mmol). The mixture was warmed to room temperature for 2 h, then diluted with DCM, washed with 1 M HCl, dried and concentrated in vacuo. The crude product was purified by flash column chromatography (10–60% EtOAc in pentane) to produce 39 (16 mg, 0.043 mmol, 85%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 8.69 (s, 1H), 8.49 (s, 1H), 7.93 (t, J = 2.3 Hz, 1H), 7.55–7.44 (m, 3H), 7.07 (s, 1H), 4.76 (hept, J = 6.9 Hz, 1H), 4.68 (s, 2H), 3.95 (s, 2H), 3.12 (s, 3H), 1.44 (d, J = 6.9 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 167.9, 145.0, 142.8, 141.3, 138.4, 136.4, 133.9, 131.3, 130.6, 126.4, 125.9, 121.9, 68.1, 67.3, 50.8, 40.3, 21.5 (2 × C); HRMS (ESI+) C18H22N3O4S+ ([M+H]+) requires 376.1326; found 376.1325.
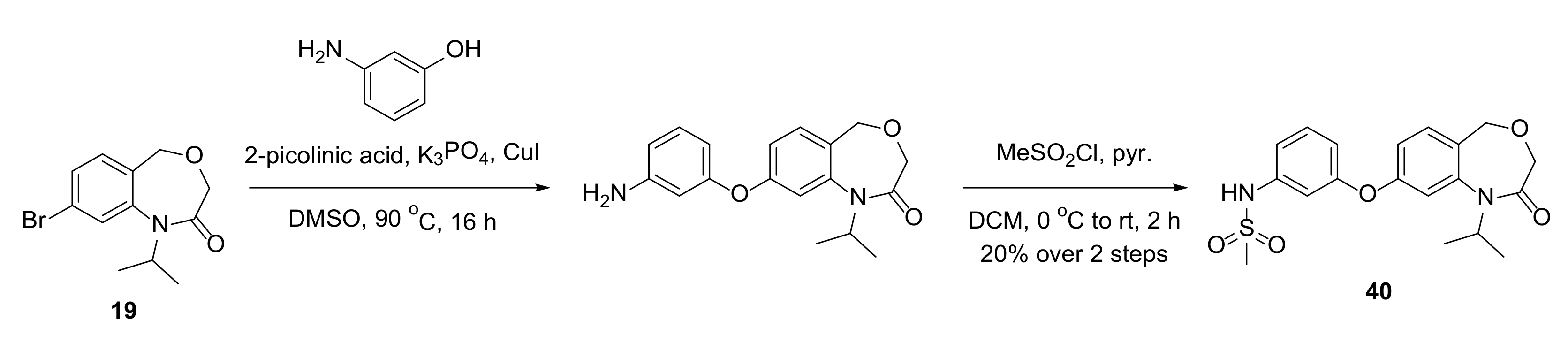
Compound
40 was prepared as shown in
Scheme 9.
4.14.46. N-(3-((1-Isopropyl-2-oxo-1,2,3,5-tetrahydrobenzo[e][1,4]oxazepin-8-yl)oxy)phenyl)methanesulfonamide (40)
To a flame-dried vial under argon were added 19 (47 mg, 0.17 mmol), 3-aminophenol (22 mg, 0.20 mmol), 2-picolinic acid (4 mg, 0.03 mmol), potassium phosphate (70 mg, 0.33 mmol) and copper iodide (3 mg, 0.02 mmol), followed by DMSO (2 mL). The mixture was heated to 90 °C for 16 h, then diluted with EtOAc, washed with water, NaHCO3, then brine, and the organic phase dried and concentrated in vacuo. The crude product was purified by flash column chromatography (20–100% EtOAc in pentane) to produce an inseparable mixture of products which was used without further purification.
To a solution of the resulting mixture (23 mg) in DCM (0.5 mL) at 0 °C was added pyridine (4 µL, 0.05 mmol) and methanesulfonyl chloride (4 µL, 0.05 mmol). The mixture was warmed to room temperature for 2 h, then diluted with DCM, washed with 1 M HCl, dried and concentrated in vacuo. The crude product was purified by flash column chromatography (20–100% EtOAc in pentane) to produce 40 (13 mg, 0.033 mmol, 20% over two steps) as a white solid.
1H NMR (500 MHz, CDCl3) δ 7.35 (t, J = 8.0 Hz, 1H), 7.31 (d, J = 8.3 Hz, 1H), 7.00–6.96 (m, 2H), 6.95 (d, J = 2.4 Hz, 1H), 6.91 (dd, J = 8.3, 2.4 Hz, 1H), 6.83 (ddd, J = 8.3, 2.3, 1.0 Hz, 1H), 6.77 (s, 1H), 4.68 (hept, J = 6.9 Hz, 1H), 4.60 (s, 2H), 3.92 (s, 2H), 3.05 (s, 3H), 1.35 (d, J = 6.9 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 167.9, 157.6, 157.5, 143.3, 138.5, 131.6, 131.0, 125.7, 116.8, 115.38, 115.39, 113.6, 111.0, 68.0, 67.1, 50.5, 39.7, 21.3 (2 × C); HRMS (ESI+) C19H23N2O5S+ ([M+H]+) requires 391.1322; found 391.1314.
Compound
41 was prepared as shown in
Scheme 10.
4.14.47. 7-Bromo-1-(cyclopropylmethyl)-1,4-dihydro-2H-benzo[d][1,3]oxazin-2-one (50)
DIPEA (200 μL, 1.17 mmol) was added to a solution of 3 (100 mg, 0.390 mmol) and triphosgene (46 mg, 0.16 mmol) in THF (1.3 mL) at 0 °C. The mixture was stirred at room temperature for 3 days, then diluted with Et2O, filtered and diluted with EtOAc. The organic layer was washed with 1xNH4Cl and 1xbrine, dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was purified by flash column chromatography (5% to 15% EtOAc in pentane) to produce cyclised 50 (87 mg, 0.31 mmol, 79%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.23–7.18 (m, 2H), 6.99 (d, J = 8.1 Hz, 1H), 5.15 (d, J = 0.8 Hz, 2H), 3.80 (d, J = 6.9 Hz, 2H), 1.23–1.12 (m, 1H), 0.62–0.53 (m, 2H), 0.48–0.41 (m, 2H); 13C NMR (100 MHz, CDCl3) 152.8, 139.1, 125.9, 125.8, 122.8, 119.7, 116.9, 67.1, 48.5, 9.5, 4.3; m/z (ESI+) 282.0 ([M+H]+, 100%); HRMS (ESI+) C12H13NO2Br+ ([M+H]+) requires 282.01242; found 282.01251.
4.14.48. N-(3-(1-(Cyclopropylmethyl)-2-oxo-1,4-dihydro-2H-benzo[d][1,3]oxazin-7-yl)phenyl)methanesulfonamide (41)
General procedure D from 50 (13 mg, 0.046 mmol); yield 76% (13 mg, 0.035 mmol); white solid. 1H NMR (500 MHz, CDCl3) δ 7.46 (t, J = 7.8 Hz, 1H), 7.43 (t, J = 2.0 Hz, 1H), 7.39 (dt, J = 7.7, 1.4 Hz, 1H), 7.26 (s, 1H), 7.25–7.22 (m, 2H), 7.20 (d, J = 7.7 Hz, 1H), 6.61 (s, 1H), 5.26 (s, 2H), 3.91 (d, J = 6.8 Hz, 2H), 3.07 (s, 3H), 1.34–1.17 (m, 1H), 0.62–0.56 (m, 2H), 0.49 (dt, J = 6.4, 4.6 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 153.3, 142.5, 141.6, 138.3, 137.5, 130.5, 125.2, 124.3, 122.0, 120.5, 119.9, 119.5, 112.6, 67.3, 48.5, 39.8, 9.7, 4.5; m/z (ESI+) 373.1 ([M+H]+, 100%); HRMS (ESI+) C19H21N2O4S+ ([M+H]+) requires 373.12165; found 373.12177.
Compound
42 was prepared as shown in
Scheme 11.
4.14.49. Methyl 3-amino-5-bromopicolinate (51)
To a solution of 3-amino-5-bromo-pyridine-2-carboxylic acid (600 mg, 2.76 mmol) in MeOH (28 mL) was added 4 M HCl in 1,4-dioxane (2 mL). The reaction was refluxed for 18 h. Upon completion, the reaction was concentrated under vacuum and diluted with water. The aqueous layer was extracted with a mixture of CHCl3/IPA (3:1) and the combined organic layers were dried, filtered and concentrated under vacuum. The desired product 51 (512 mg, 2.22 mmol, 80%) was isolated as an off white solid and used in the following step without any further purification. 1H NMR (500 MHz, CDCl3) 8.06 (d, J = 2.0 Hz, 1H), 7.24 (d, J = 2.0 Hz, 1H), 5.82 (s, 2H), 3.97 (s, 3H); 13C NMR (125 MHz, CDCl3) 167.7, 147.6, 139.5, 126.6, 126.4, 125.4, 52.7; m/z (ESI+) 231.0 ([M+H]+, 100%); HRMS (ESI+) C7H8BrN2O2+ ([M+H]+) requires 230.97637; found 230.97668.
4.14.50. (3-Amino-5-bromopyridin-2-yl)methanol (52)
General procedure B from 51 (250 mg, 1.08 mmol) with 1.2 equivalents of LiAlH4; yield 61% (135 mg, 0.660 mmol). The product was isolated with some minor impurities, but was taken to the following step and further purified thereafter. 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 1.9 Hz, 1H), 7.13 (d, J = 1.9 Hz, 1H), 4.64 (d, J = 4.0 Hz, 2H), 3.91 (br s, 2H), 3.44 (t, J = 4.4 Hz, 1H); m/z (ESI+) 203.0 ([M+H]+, 25%).
4.14.51. 8-Bromo-1,5-dihydropyrido[3,2-e][1,4]oxazepin-2(3H)-one (53)
General procedure C.2 from 52 (100 mg, 0.49 mmol); yield 37% (45 mg, 0.19 mmol); pale yellow solid. 1H NMR (500 MHz, CDCl3) 8.30 (d, J = 1.9 Hz, 1H), 8.02 (brs, 1H), 7.39 (d, J = 1.9 Hz, 1H), 4.90 (s, 2H), 4.59 (s, 2H); 13C NMR (125 MHz, CDCl3) 173.7, 146.4, 144.5, 133.2, 128.7, 119.5, 75.6, 74.0; m/z (ESI+) 243.0 ([M+H]+, 100%); HRMS (ESI+) C8H8BrN2O2+ ([M+H]+) requires 242.97637; found 242.97659.
4.14.52. 8-Bromo-1-isopropyl-1,5-dihydropyrido[3,2-e][1,4]oxazepin-2(3H)-one (54)
To a suspension of 53 (42 mg, 0.17 mmol) in dry DMF (1.2 mL) were added Cs2CO3 (110 mg, 0.34 mmol) and 2-bromopropane (49 µL, 0.52 mmol). The mixture was stirred at room temperature for 2 h and then the reaction quenched with water. The aqueous layer was extracted with EtOAc and the combined organic layers were washed with brine, dried and concentrated under vacuum. The residue was purified by flash column chromatography (7–15% EtOAc in pentane) to produce the desired product 54 (10 mg, 0.035 mmol, 20%) as a pale-yellow solid. 1H NMR (500 MHz, CDCl3) 8.59 (d, J = 2.0 Hz, 1H), 7.74 (d, J = 2.0 Hz, 1H), 4.77 (s, 2H), 4.67 (hept, J = 6.9 Hz, 1H), 3.96 (s, 2H), 1.40 (d, J = 6.9 Hz, 6H); 13C NMR (125 MHz, CDCl3) 167.5, 148.9, 148.6, 139.0, 133.0, 120.6, 69.2, 68.5, 51.4, 21.4; m/z (ESI+) 285.0 ([M+H]+, 100%); HRMS (ESI+) C11H14BrN2O2+ ([M+H]+) requires 285.02332; found 285.02332.
4.14.53. N-(3-(1-Isopropyl-2-oxo-1,2,3,5-tetrahydropyrido[3,2-e][1,4]oxazepin-8-yl)phenyl)methanesulfonamide (42)
General procedure D from 54 (10 mg, 0.04 mmol); yield 46% (6 mg, 0.02 mmol); white solid. 1H NMR (500 MHz, CDCl3) 8.73 (d, J = 2.1 Hz, 1H), 7.72 (d, J = 2.0 Hz, 1H) 7.52 (t, J = 7.9 Hz, 1H), 7.47 (t, J = 2.0 Hz, 1H), 7.41 (dt, J = 7.8, 1.3 Hz, 1H), 7.30 (ddd, J = 8.1, 2.3, 1.0 Hz, 1H), 4.86 (s, 2H), 4.77 (h, J = 6.9 Hz, 1H), 4.01 (s, 2H), 3.08 (s, 3H), 1.44 (dd, J = 6.9 Hz, 6H); 13C NMR (125 MHz, CDCl3) 167.8, 149.4, 146.3, 138.5, 138.2, 137.9, 136.8, 130.9, 128.9, 124.3, 120.6, 119.4, 69.5, 68.6, 50.9, 40.0, 21.7; m/z (ESI+) 376.1 ([M+H]+, 100%); HRMS (ESI+) C18H22N3O4S+ ([M+H]+) requires 376.13255; found 376.13254.


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





























