
NaRnEA: An Information Theoretic Framework for Gene Set Analysis

Abstract
:1. Introduction
“We estimate the statistical significance (nominal P value) of the [GSEA enrichment score] by using an empirical phenotype-based permutation test procedure that preserves the complex correlation structure of the gene expression data. Specifically, we permute the phenotype labels and recompute the [GSEA enrichment score] of the gene set for the permuted data, which generates a null distribution for the [GSEA enrichment score]. The empirical, nominal P value of the observed [GSEA enrichment score] is then calculated relative to this null distribution. Importantly, the permutation of class labels preserves gene-gene correlations and, thus, provides a more biologically reasonable assessment of significance than would be obtained by permuting genes”.
“We show, in agreement with earlier observations, that the gene independence assumption is not realistic because gene correlations are non-trivial and produce a substantial amount of variance inflation in the global statistic that in turn produces a large number of false positive results”.
- A: Genes in a gene set are correlated when the gene set
- is not enriched in the gene expression signature.
- B: A gene set analysis method that assumes independence between
- genes will not control the Type I error rate of gene set analysis.
- ¬B: A gene set analysis method that assumes independence between
- genes adequately controls the Type I error rate of gene set analysis.
- ¬A: Genes in a gene set are not correlated when the gene set
- is not enriched in the gene expression signature.
“For each dataset in the benchmark, we randomized the phenotype labels 1000 times and ran both algorithms … The p values are computed using the areas under the empirical null histograms from GSEA and areas under the normal distribution for SEA”.
2. Materials and Methods
2.1. Nonparametric Analytical-Rank-Based Enrichment Analysis (NaRnEA)
- We assume that the differential gene expression signature value for the gth gene between the test phenotype () and the reference phenotype () is a real number that may be positive or negative.
- We assume that the differential gene expression signature value for the gth gene between the test phenotype () and the reference phenotype () is greater than zero if and only if the expression of the gth gene in the test phenotype () is greater than the expression of the gth gene in the reference phenotype (). More formally, this may be expressed by stating that the discrete random variable that represents the expression of the gth gene in the test phenotype () stochastically dominates the discrete random variable that represents the expression of the gth gene in the reference phenotype ().
- We assume that the differential gene expression signature value for the gth gene between the test phenotype () and the reference phenotype () is less than zero if and only if the expression of the gth gene in the test phenotype () is less than the expression of the gth gene in the reference phenotype (). More formally, this may be expressed by stating that the discrete random variable that represents the expression of the gth gene in the test phenotype () is stochastically dominated by the discrete random variable that represents the expression of the gth gene in the reference phenotype ().
- We assume that the differential gene expression signature value for the gth gene between the test phenotype () and the reference phenotype () will be equal in magnitude and opposite in sign if the ordering of the phenotypes were reversed; this follows naturally from the aforementioned definitions of positive differential gene expression (i.e., upregulation) and negative differential gene expression (i.e., downregulation).
- We assume that the magnitude of the differential gene expression signature value for the gth gene between the test phenotype () and the reference phenotype () should be greater than the magnitude of the differential gene expression signature value for the kth gene between the test phenotype () and the reference phenotype () if and only if the extent of differential expression for the gth gene is greater than the extent of differential expression for the kth gene between the two phenotypes; we formalize this notion here using the language of stochastic dominance as mentioned previously.
- The Association Weight between the rth regulator and the gth gene is strictly non-negative.
- The Association Weight between the rth regulator and the gth gene is greater than zero if and only if the expression of the gth gene exhibits a statistical dependency on the activity of the rth regulator; this may be formalized by stating that the mutual information between the discrete random variable representing the expression of the gth gene and the discrete random variable representing the activity of the rth regulator is nonzero.
- The Association Weight between the rth regulator and the gth gene is greater than the Association Weight between the rth regulator and the kth gene if and only if the expression of the gth gene exhibits a greater statistical dependency on the activity of the rth regulator than the expression of the kth gene as measured using the mutual information between the corresponding discrete random variables.
- The Association Mode between the rth regulator and the gth gene is a real number less than or equal to one and greater than or equal to negative one.
- The Association Mode between the rth regulator and the gth gene is greater than zero if and only if there is a positive monotonic relationship between the activity of the rth regulator and the expression of the gth gene; this may be formalized by stating that the Spearman correlation coefficient between the discrete random variable representing the expression of the gth gene and the discrete random variable representing the activity of the rth regulator is positive.
- The Association Mode between the rth regulator and the gth gene is less than zero if and only if there is a negative monotonic relationship between the activity of the rth regulator and the expression of the gth gene; this may be formalized by stating that the Spearman correlation coefficient between the discrete random variable representing the expression of the gth gene and the discrete random variable representing the activity of the rth regulator is negative.
- The magnitude of the Association Mode between the rth regulator and the gth gene is greater than the magnitude of the Association Mode between the rth regulator and the kth gene if and only if the Spearman correlation coefficient between the expression of the gth gene and the activity of the rth regulator is greater in magnitude than the Spearman correlation coefficient between the expression of the kth gene and the activity of the rth regulator quantified from the corresponding discrete random variables.
- In the first scenario, the activity of the rth regulator in the test phenotype () is greater than the activity of the rth regulator in the reference phenotype (). More formally, this may be expressed by stating that the discrete random variable that represents the activity of the rth regulator in the test phenotype () stochastically dominates the discrete random variable that represents the activity of the rth regulator in the reference phenotype ().
- In the second scenario, the activity of the rth regulator in the test phenotype () is less than the activity of the rth regulator in the reference phenotype (). More formally, this may be expressed by stating that the discrete random variable that represents the activity of the rth regulator in the test phenotype () is stochastically dominated by the discrete random variable that represents the activity of the rth regulator in the reference phenotype ().
- In the third scenario, the activity of the rth regulator in the test phenotype () is equal to the activity of the rth regulator in the reference phenotype (). More formally, this may be expressed by stating that the discrete random variable that represents the activity of the rth regulator in the test phenotype () neither stochastically dominates nor is stochastically dominated by the discrete random variable that represents the activity of the rth regulator in the reference phenotype ().
- The final Normalized Enrichment Score for the rth regulator is a standard normal random variable when the regulon gene set for the rth regulator is not enriched in the nonparametric differential gene expression signature computed between the test phenotype () and the reference phenotype (). Formally, the rate of this asymptotic convergence depends on the Association Weight and Association Mode values for the gene set members in accordance with the Berry–Esseen Theorem for non-identically distributed summands.
- The expected value of the final Normalized Enrichment Score for the rth regulator is positive when the regulon gene set for the rth regulator is positively enriched in the nonparametric differential gene expression signature computed between the test phenotype () and the reference phenotype ().
- The expected value of the final Normalized Enrichment Score for the rth regulator is negative when the regulon gene set for the rth regulator is negatively enriched in the nonparametric differential gene expression signature computed between the test phenotype () and the reference phenotype ().
- The Proportional Enrichment Score for the regulon gene set of the rth regulator is less than or equal to (1) and greater than or equal to (−1).
- The expected value of the Proportional Enrichment Score for the regulon gene set of the rth regulator is equal to (0) when the regulon gene set of the rth regulator is not enriched in the nonparametric differential gene expression signature computed between the test phenotype () and the reference phenotype ().
- The expected value of the Proportional Enrichment Score for the regulon gene set of the rth regulator is positive when the regulon gene set of the rth regulator is positively enriched in the nonparametric differential gene expression signature computed between the test phenotype () and the reference phenotype ().
- The expected value of the Proportional Enrichment Score for the regulon gene set of the rth regulator is negative when the regulon gene set of the rth regulator is negatively enriched in the nonparametric differential gene expression signature computed between the test phenotype () and the reference phenotype ().
2.2. The Algorithm for the Reconstruction of Accurate Cellular Networks 3 (ARACNe3)
2.3. Gene Set Enrichment Analysis (GSEA)
2.4. Analytical-Rank-Based Enrichment Analysis (aREA)
2.5. Clinical Proteomic Tumor Analysis Consortium (CPTAC) Differential Protein Abundance
2.6. Plotting and Visualization
2.7. Statistical Analysis
3. Results
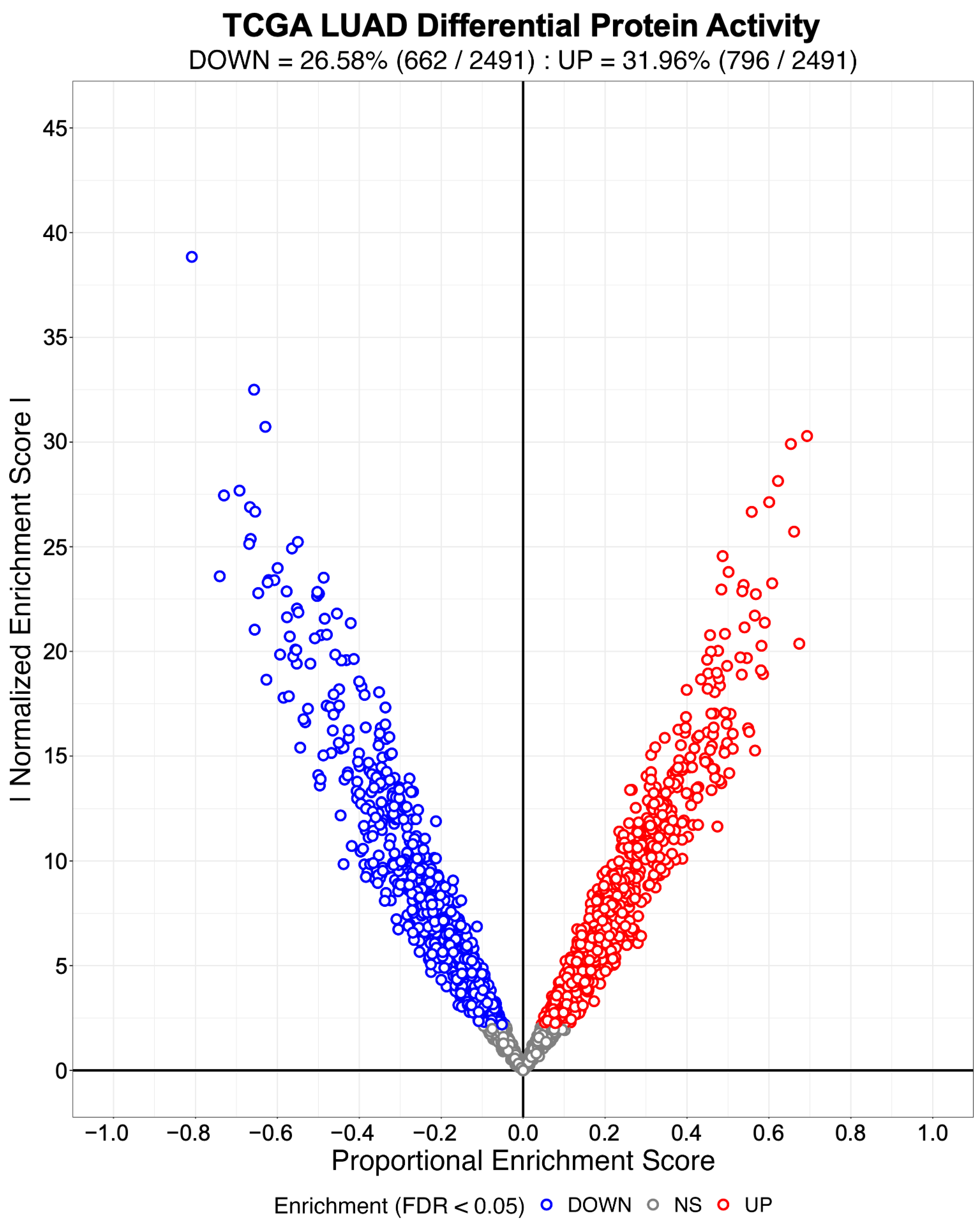
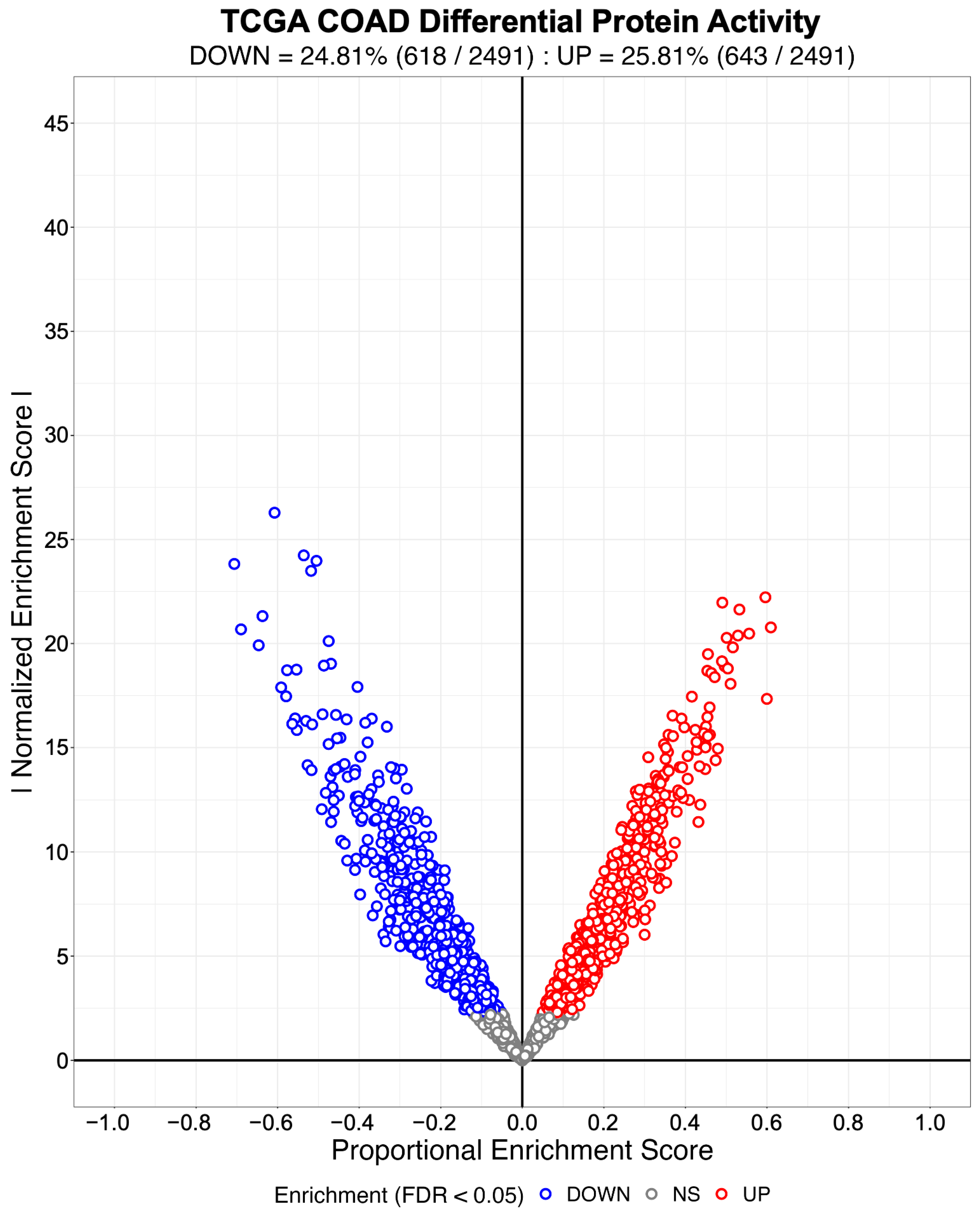
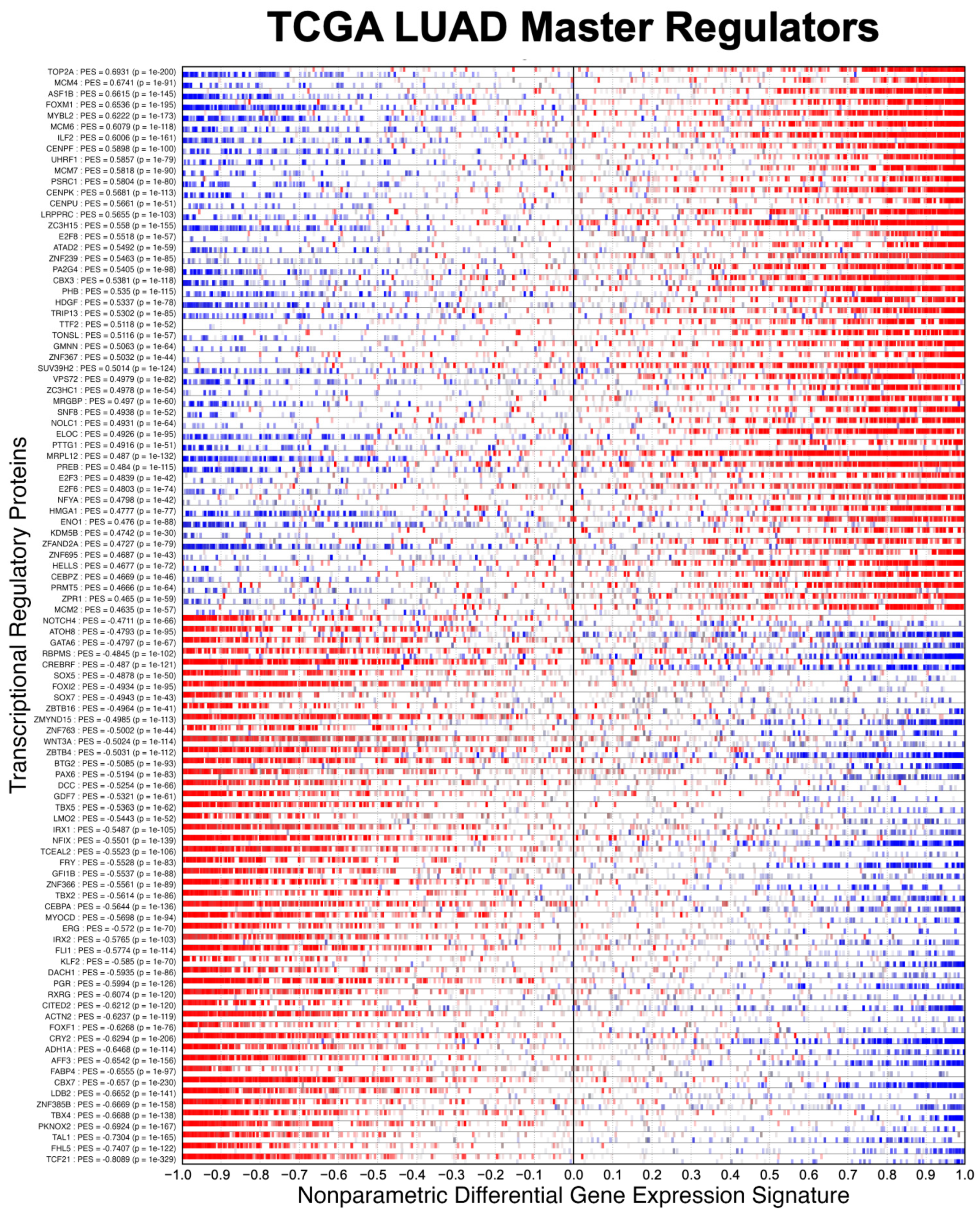
3.1. Evaluating the Sensitivity and Specificity of NaRnEA for Gene Set Analysis
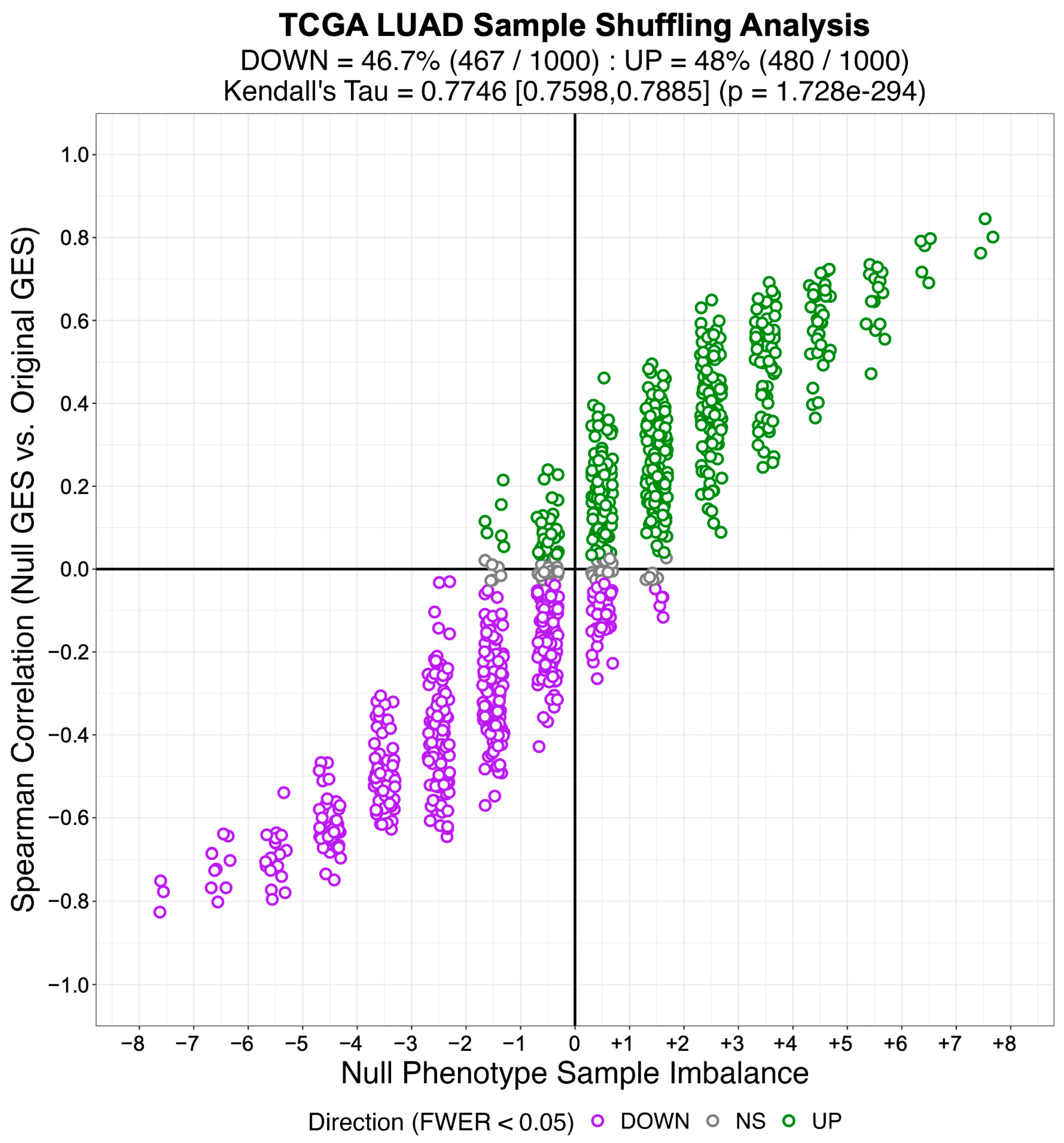
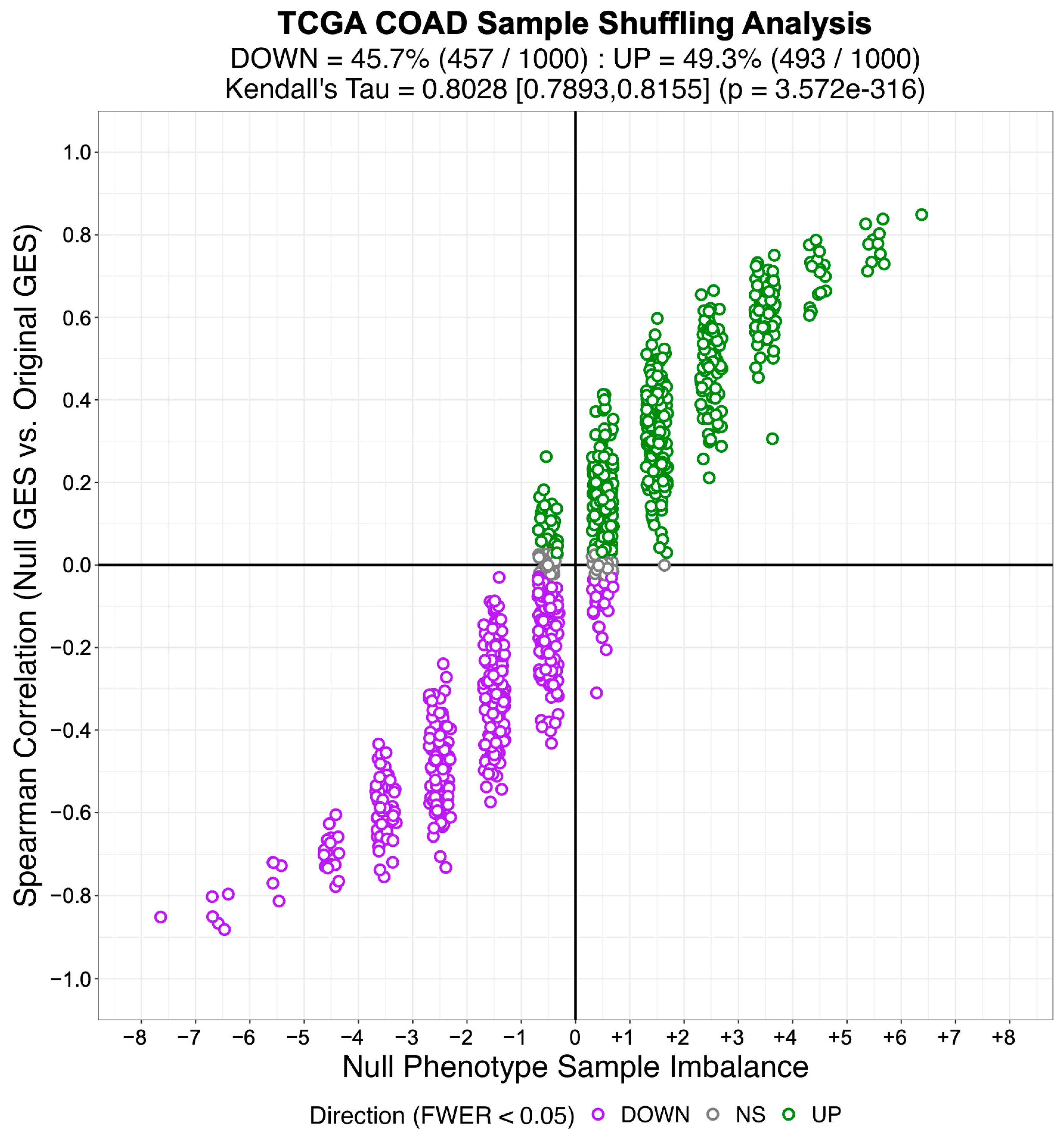
3.2. Identifying Systematic Biases in Phenotype-Based Permutation Null Models for Gene Set Enrichment
3.3. Examining the Alternative Null Model for GSEA
“… [GSEA] can be applied to ranked gene lists arising in other settings. Genes may be ranked based on the differences seen in a small data set, with too few samples to allow rigorous evaluation of significance levels by permuting the class labels. In these cases, a P value can be estimated by permuting the genes, with the result that genes are randomly assigned to the sets while maintaining their size. This approach is not strictly accurate: because it ignores gene-gene correlations, it will overestimate the significance levels and may lead to false positives. Nonetheless, it can be useful for hypothesis generation”.
“In the original implementation, the running-sum statistic used equal weights at every step, which yielded high scores for sets clustered near the middle of the ranked list … These sets do not represent biologically relevant correlation with the phenotype. We addressed this issue by weighting the steps according to each gene’s correlation with a phenotype”.
3.4. Examining the Alternative Null Model for aREA
“… the statistical significance for the enrichment score is estimated by comparison to a null model generated by permuting the samples uniformly at random or by an analytic approach equivalent to shuffle the genes in the signatures uniformly at random … Gene shuffling can be approximated analytically as follows: according to the central limit theorem, the mean of a sufficiently large number of independent random variables will be approximately normally distributed. The enrichment score of our null hypothesis fulfill this condition, and we ensure a mean of zero and variance equal to one for the enrichment score under the null hypothesis by applying a quantile transformation based on the normal distribution to the rank-transformed gene expression signature before computing the enrichment score”.
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marx, V. Biology: The big challenges of big data. Nature 2013, 498, 255–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Croft, D.; Mundo, A.F.; Haw, R.; Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.; Gillespie, M.; Kamdar, M.R.; et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014, 42, D472–D477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, M.J.; Shen, Y.; Giorgi, F.M.; Lachmann, A.; Ding, B.B.; Ye, B.H.; Califano, A. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 2016, 48, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Mangul, S.; Martin, L.S.; Hill, B.L.; Lam, A.K.-M.; Distler, M.G.; Zelikovsky, A.; Eskin, E.; Flint, J. Systematic benchmarking of omics computational tools. Nat. Commun. 2019, 10, 1393. [Google Scholar] [CrossRef] [Green Version]
- Maleki, F.; Ovens, K.; Hogan, D.J.; Kusalik, A.J. Gene Set Analysis: Challenges, Opportunities, and Future Research. Front. Genet. 2020, 11, 654. [Google Scholar] [CrossRef]
- Das, S.; McClain, C.J.; Rai, S.N. Fifteen Years of Gene Set Analysis for High-Throughput Genomic Data: A Review of Statistical Approaches and Future Challenges. Entropy 2020, 22, 427. [Google Scholar] [CrossRef] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide ex-pression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Tamayo, P.; Steinhardt, G.; Liberzon, A.; Mesirov, J.P. The limitations of simple gene set enrichment analysis assuming gene independence. Stat. Methods Med. Res. 2016, 25, 472–487. [Google Scholar] [CrossRef] [Green Version]
- Paull, E.O.; Aytes, A.; Jones, S.J.; Subramaniam, P.S.; Giorgi, F.M.; Douglass, E.F.; Tagore, S.; Chu, B.; Vasciaveo, A.; Zheng, S.; et al. A modular master regulator landscape controls cancer transcriptional identity. Cell 2021, 184, 334–351.e20. [Google Scholar] [CrossRef] [PubMed]
- Mercatelli, D.; Scalambra, L.; Triboli, L.; Ray, F.; Giorgi, F.M. Gene regulatory network inference resources: A practical overview. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194430. [Google Scholar] [CrossRef]
- Margolin, A.A.; Nemenman, I.; Basso, K.; Wiggins, C.; Stolovitzky, G.; Favera, R.D.; Califano, A. ARACNE: An algorithm for the reconstruction of gene regulatory networks in a mammalian cellu-lar context. BMC Bioinform. 2006, 7 (Suppl. S1), S7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lachmann, A.; Giorgi, F.M.; Lopez, G.; Califano, A. ARACNe-AP: Gene network reverse engineering through adaptive partitioning inference of mutual information. Bioinformatics 2016, 32, 2233–2235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, M.J.; Subramaniam, P.S.; Tang, L.H.; Grunn, A.; Aburi, M.; Rieckhof, G.; Komissarova, E.V.; Hagan, E.A.; Bodei, L.; Clemons, P.A.; et al. A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat. Genet. 2018, 50, 979–989. [Google Scholar] [CrossRef]
- Califano, A.; Alvarez, M.J. The recurrent architecture of tumour initiation, progression and drug sensitivity. Nat. Rev. Cancer 2017, 17, 116–130. [Google Scholar] [CrossRef] [Green Version]
- Jaynes, E.T.; Bretthorst, G.L. Probability Theory: The Logic of Science; Cambridge University Press: Cambridge, UK, 2003. [Google Scholar]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef] [Green Version]
- Cover, T.M.; Thomas, J.A. Elements of Information Theory; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005. [Google Scholar]
- Hu, C. Central limit theorems for sub-linear expectation under the Lindeberg condition. J. Inequalities Appl. 2018, 2018, 316. [Google Scholar] [CrossRef] [Green Version]
- Gut, A. The Multivariate Normal Distribution. In An Intermediate Course in Probability; Gut, A., Ed.; Springer: New York, NY, USA, 2009; pp. 117–145. [Google Scholar]
- DiCiccio, T.J.; Efron, B. Bootstrap Confidence Intervals. Stat. Sci. 1996, 11, 189–212. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Lukasz, K. mblm: Median-Based Linear Models; The R Foundation: Vienna, Austria, 2019. [Google Scholar]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phipson, B.; Smyth, G.K. Permutation P-values should never be zero: Calculating exact P-values when permutations are randomly drawn. Stat. Appl. Genet. Mol. Biol. 2010, 9, 39. [Google Scholar] [CrossRef]
- Welch, B.L. The Generalization Of ‘Students’ Problem when Several Different Population Variances Are Involved. Biometrika 1946, 34, 28–35. [Google Scholar] [CrossRef]
- Vasaikar, S.V.; Straub, P.; Wang, J.; Zhang, B. LinkedOmics: Analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018, 46, D956–D963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, H.B.; Whitney, D.R. On a Test of Whether one of Two Random Variables is Stochastically Larger than the Other. Ann. Math. Stat. 1947, 18, 50–60. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis. In Use R! Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate—A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Clopper, C.J.; Pearson, E.S. The Use of Confidence or Fiducial Limits Illustrated in the Case of the Binomial. Biometrika 1934, 26, 404–413. [Google Scholar] [CrossRef]
- Cui, X.; Churchill, G.A. Statistical tests for differential expression in cDNA microarray experiments. Genome Biol. 2003, 4, 210. [Google Scholar] [CrossRef] [Green Version]
- Bishop, D. Rein in the four horsemen of irreproducibility. Nature 2019, 568, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Leeuw, C.A.; Neale, B.M.; Heskes, T.; Posthuma, D. The statistical properties of gene-set analysis. Nat. Rev. Genet. 2016, 17, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Alvarez, M.J.; Bisikirska, B.C.; Lachmann, A.; Realubit, R.; Pampou, S.; Coku, J.; Karan, C.; Califano, A. Systematic, network-based characterization of therapeutic target inhibitors. PLoS Comput. Biol. 2017, 13, e1005599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obradovic, A.; Chowdhury, N.; Haake, S.M.; Ager, C.; Wang, V.; Vlahos, L.; Guo, X.V.; Aggen, D.H.; Rathmell, W.K.; Jonasch, E.; et al. Single-cell protein activity analysis identifies recurrence-associated renal tumor macrophages. Cell 2021, 184, 2988–3005.e16. [Google Scholar] [CrossRef]
- Ding, H.; Douglass, E.F., Jr.; Sonabend, A.M.; Mela, A.; Bose, S.; Gonzalez, C.; Canoll, P.D.; Sims, P.A.; Alvarez, M.J.; Califano, A. Quantitative assessment of protein activity in orphan tissues and single cells using the metaVIPER algorithm. Nat. Commun. 2018, 9, 1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chari, A.; Vogl, D.T.; Gavriatopoulou, M.; Nooka, A.K.; Yee, A.J.; Huff, C.A.; Moreau, P.; Dingli, D.; Cole, C.; Lonial, S.; et al. Oral Selinexor-Dexamethasone for Triple-Class Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 381, 727–738. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type I Error Rate | TCGA LUAD NaRnEA-Inferred NM Regulon Enrichment | TCGA COAD NaRnEA-Inferred NM Regulon Enrichment | TCGA HNSC NaRnEA-Inferred NM Regulon Enrichment |
|---|---|---|---|
| FPR < 0.05 | 4.86% [4.05%, 5.78%] | 5.50% [4.64%, 6.47%] | 5.34% [4.49%, 6.30%] |
| FDR < 0.05 | 0% [0%, 0.15%] | 0% [0%, 0.15%] | 0% [0%, 0.15%] |
| FWER < 0.05 | 0% [0%, 0.15%] | 0% [0%, 0.15%] | 0% [0%, 0.15%] |
| Type I Error Rate | TCGA LUAD NaRnEA-Inferred TS Regulon Enrichment | TCGA COAD NaRnEA-Inferred TS Regulon Enrichment | TCGA HNSC NaRnEA-Inferred TS Regulon Enrichment |
|---|---|---|---|
| FPR < 0.05 | 59.86% [57.90%, 61.79%] | 54.11% [52.13%, 56.09%] | 55.56% [53.58%, 57.52%] |
| FDR < 0.05 | 58.53% [56.57%, 60.47%] | 50.62% [48.64%, 52.60%] | 52.83% [50.85%, 54.81%] |
| FWER < 0.05 | 42.83% [40.88%, 44.80%] | 32.78% [30.92%, 34.64%] | 34.40% [32.54%, 36.31%] |
| TCGA LUAD vs. CPTAC LUAD Kendall’s Tau-B = 0.3832 [0.3393, 0.4271] Chi-Squared Test p = 7.481 × 10−51 | |||
|---|---|---|---|
| CPTAC DOWN (MWU Test) | CPTAC NS (MWU Test) | CPTAC UP (MWU Test) | |
| TCGA UP (NaRnEA) | Obs = 77 Exp = 154.35 Z = −9.891 FWER = 4.079 × 10−22 | Obs = 91 Exp = 106.92 Z = −2.158 FWER = 0.2782 | Obs = 279 Exp = 185.73 Z = 11.31 FWER = 1.037 × 10−28 |
| TCGA NS (NaRnEA) | Obs = 142 Exp = 159.88 Z = −2.160 FWER = 0.2768 | Obs = 131 Exp = 110.74 Z = 2.850 FWER = 3.393 × 10−2 | Obs = 190 Exp = 192.38 Z = −0.2241 FWER = 1.000 |
| TCGA DOWN (NaRnEA) | Obs = 204 Exp = 108.77 Z = 12.95 FWER = 2.179 × 10−37 | Obs = 71 Exp = 75.34 Z = −0.5846 FWER = 1.000 | Obs = 40 Exp = 130.89 Z = −12.67 FWER = 7.848 × 10−36 |
| TCGA COAD vs. CPTAC COAD Kendall’s Tau-B = 0.2913 [0.2333, 0.3492] Chi-Squared Test p = 1.456 × 10−18 | |||
|---|---|---|---|
| CPTAC DOWN (MWU Test) | CPTAC NS (MWU Test) | CPTAC UP (MWU Test) | |
| TCGA UP (NaRnEA) | Obs = 104 Exp = 147.20 Z = −6.260 FWER = 3.465 × 10−9 | Obs = 45 Exp = 54.40 Z = −1.722 FWER = 0.7656 | Obs = 167 Exp = 114.40 Z = 8.013 FWER = 1.006 × 10−14 |
| TCGA NS (NaRnEA) | Obs = 153 Exp = 149.06 Z = 0.6447 FWER = 1.000 | Obs = 66 Exp = 55.09 Z = 2.179 FWER = 0.2637 | Obs = 101 Exp = 115.85 Z = −2.170 FWER = 0.2701 |
| TCGA DOWN (NaRnEA) | Obs = 111 Exp = 71.74 Z = 7.230 FWER = 4.360 × 10−12 | Obs = 25 Exp = 26.51 Z = −0.2254 FWER = 1.000 | Obs = 18 Exp = 55.75 Z = −7.445 FWER = 8.757 × 10−13 |
| TCGA HNSC vs. CPTAC HNSC Kendall’s Tau-B = 0.3455 [0.2964, 0.3946] Chi-Squared Test p = 7.481 × 10−51 | |||
|---|---|---|---|
| CPTAC DOWN (MWU Test) | CPTAC NS (MWU Test) | CPTAC UP (MWU Test) | |
| TCGA UP (NaRnEA) | Obs = 43 Exp = 95.2 Z = −7.980 FWER = 1.320 × 10−14 | Obs = 97 Exp = 125.46 Z = −3.825 FWER = 1.179 × 10−3 | Obs = 270 Exp = 189.32 Z = 10.222 FWER = 1.429 × 10−23 |
| TCGA NS (NaRnEA) | Obs = 95 Exp = 104.51 Z = −1.311 FWER = 1.000 | Obs = 168 Exp = 137.70 Z = 4.085 FWER = 3.967 × 10−4 | Obs = 187 Exp = 207.79 Z = −2.500 FWER = 0.112 |
| TCGA DOWN (NaRnEA) | Obs = 117 Exp = 55.27 Z = 10.221 FWER = 1.433 × 10−23 | Obs = 71 Exp = 72.83 Z = −0.206 FWER = 1.000 | Obs = 50 Exp = 109.90 Z = −8.983 FWER = 2.364 × 10−18 |
| Type I Error Rate | TCGA LUAD GSEA-Inferred NM Regulon Enrichment | TCGA COAD GSEA-Inferred NM Regulon Enrichment | TCGA HNSC GSEA-Inferred NM Regulon Enrichment |
|---|---|---|---|
| FPR < 0.05 | 10.88% [9.58%, 12.17%] | 13.01% [11.71%, 14.39%] | 1.49% [1.05%, 2.04%] |
| FDR < 0.05 | 0% [0%, 0.15%] | 0% [0%, 0.15%] | 0% [0%, 0.15%] |
| FWER < 0.05 | 0% [0%, 0.15%] | 0% [0%, 0.15%] | 0% [0%, 0.15%] |
| Type I Error Rate | TCGA LUAD aREA-Inferred NM Regulon Enrichment | TCGA COAD aREA-Inferred NM Regulon Enrichment | TCGA HNSC aREA-Inferred NM Regulon Enrichment |
|---|---|---|---|
| FPR < 0.05 | 3.41% [2.73%, 4.20%] | 4.18% [3.42%, 5.04%] | 5.14% [4.30%, 6.08%] |
| FDR < 0.05 | 0% [0%, 0.15%] | 0% [0%, 0.15%] | 0% [0%, 0.15%] |
| FWER < 0.05 | 0% [0%, 0.15%] | 0% [0%, 0.15%] | 0% [0%, 0.15%] |
| Type I Error Rate | TCGA LUAD GSEA-Inferred TS Regulon Enrichment | TCGA COAD GSEA-Inferred TS Regulon Enrichment | TCGA HNSC GSEA-Inferred TS Regulon Enrichment |
|---|---|---|---|
| FPR < 0.05 | 7.51% [6.50%, 8.61%] | 6.62% [5.68%, 7.67%] | 5.26% [4.42%, 6.21%] |
| FDR < 0.05 | 0% [0%, 0.15%] | 0% [0%, 0.15%] | 0% [0%, 0.15%] |
| FWER < 0.05 | 0% [0%, 0.15%] | 0% [0%, 0.15%] | 0% [0%, 0.15%] |
| Type I Error Rate | TCGA LUAD aREA-Inferred TS Regulon Enrichment | TCGA COAD aREA-Inferred TS Regulon Enrichment | TCGA HNSC aREA-Inferred TS Regulon Enrichment |
|---|---|---|---|
| FPR < 0.05 | 10.88% [9.68%, 12.17%] | 3.49% [2.81%, 4.29%] | 6.62% [5.68%, 7.67%] |
| FDR < 0.05 | 0% [0%, 0.15%] | 0% [0%, 0.15%] | 0% [0%, 0.15%] |
| FWER < 0.05 | 0% [0%, 0.15%] | 0% [0%, 0.15%] | 0% [0%, 0.15%] |
| Type I Error Rate | TCGA LUAD NaRnEA-Inferred TN Regulon Enrichment | TCGA COAD NaRnEA-Inferred TN Regulon Enrichment | TCGA HNSC NaRnEA-Inferred TN Regulon Enrichment |
|---|---|---|---|
| FPR < 0.05 | 0% [0%, 0.15%] | 0% [0%, 0.15%] | 0% [0%, 0.15%] |
| FDR < 0.05 | 0% [0%, 0.15%] | 0% [0%, 0.15%] | 0% [0%, 0.15%] |
| FWER < 0.05 | 0% [0%, 0.15%] | 0% [0%, 0.15%] | 0% [0%, 0.15%] |
| Type I Error Rate | TCGA LUAD GSEA-Inferred TN Regulon Enrichment | TCGA COAD GSEA-Inferred TN Regulon Enrichment | TCGA HNSC GSEA-Inferred TN Regulon Enrichment |
|---|---|---|---|
| FPR < 0.05 | 100% [99.85%, 100%] | 100% [99.85%, 100%] | 100% [99.85%, 100%] |
| FDR < 0.05 | 100% [99.85%, 100%] | 100% [99.85%, 100%] | 100% [99.85%, 100%] |
| FWER < 0.05 | 0% [0%, 0.15%] | 0% [0%, 0.15%] | 0% [0%, 0.15%] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Griffin, A.T.; Vlahos, L.J.; Chiuzan, C.; Califano, A. NaRnEA: An Information Theoretic Framework for Gene Set Analysis. Entropy 2023, 25, 542. https://doi.org/10.3390/e25030542
Griffin AT, Vlahos LJ, Chiuzan C, Califano A. NaRnEA: An Information Theoretic Framework for Gene Set Analysis. Entropy. 2023; 25(3):542. https://doi.org/10.3390/e25030542
Chicago/Turabian StyleGriffin, Aaron T., Lukas J. Vlahos, Codruta Chiuzan, and Andrea Califano. 2023. "NaRnEA: An Information Theoretic Framework for Gene Set Analysis" Entropy 25, no. 3: 542. https://doi.org/10.3390/e25030542